
Abstract
Immunotherapy, especially immune checkpoint inhibitors (ICIs), has revolutionized the treatment of many types of cancer, particularly advanced-stage cancers. Nevertheless, although a subset of patients experiences dramatic and long-term disease regression in response to ICIs, most patients do not benefit from these treatments. Some may even experience cancer progression. Immune escape by tumor cells may be a key reason for this low response rate. N6-methyladenosine (m6A) is the most common type of RNA methylation and has been recognized as a critical regulator of tumors and the immune system. Therefore, m6A modification and related regulators are promising targets for improving the efficacy of tumor immunotherapy. However, the association between m6A modification and tumor immune escape (TIE) has not been comprehensively summarized. Therefore, this review summarizes the existing knowledge regarding m6A modifications involved in TIE and their potential mechanisms of action. Moreover, we provide an overview of currently available agents targeting m6A regulators that have been tested for their elevated effects on TIE. This review establishes the association between m6A modifications and TIE and provides new insights and strategies for maximizing the efficacy of immunotherapy by specifically targeting m6A modifications involved in TIE.
Similar content being viewed by others


Introduction
According to the concept of cancer immunoediting, the immune system has dual roles in preventing and shaping tumors. In immunocompetent hosts, tumor cells obtain three fates: elimination, equilibrium, and escape. Tumor immune escape (TIE) is a process in which the immunologic microenvironment of sculpted tumors can expand in an uncontrolled pathway [1]. Antitumor immunity is mainly mediated by CD8+ T cells that specifically recognize antigenic peptides presented by the major histocompatibility complex (MHC, in vertebrates) or class I human leukocyte antigens (HLA-I, in humans) and are activated to kill tumor cells [2]. However, tumor cells can produce inhibitory signals that suppress T-cell function. Immune checkpoint inhibition (ICI) therapy can effectively suppress these signals and reactivate T cells in only a minority of patients. Its limited effectiveness has been demonstrated to be due to reduced antigen presentation, increased immunosuppressive factors, and abnormally activated signaling pathways.
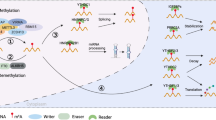
N 6-methyladenosine (m6A) is the most prevalent RNA modification in mammalian cells and was first discovered in 1974 [3]. m6A modification sites are evolutionally conserved within a consensus motif DRACH (D = A, G or U; H = A, C, or U), in which A is converted to m6A and generally occurs in the coding sequence, 3′ untranslated region (3′ UTR) proximal to the stop codon, and 5′ untranslated region (5′ UTR) of mRNAs [4,5,6]. It is a potentially reversible and dynamic post-transcriptional modification of RNA molecules that is regulated by methyltransferases (writers) and demethylases (erasers) and recognized by specific binding proteins (readers). The m6A modification has significant functions in regulating alternative splicing of pre-mRNAs [5], mRNA degradation [7], mRNA stabilization [8], miRNA processing [9], and cap-independent translation [10] (Fig. 1).

The molecular mechanism of N6-methyladenosine (m6A) modification. m6A modification is a dynamic and reversible epigenetic modification that is regulated by “writers” and “erasers.” It is primarily catalyzed by the m6A methyltransferase complex comprising the main components METTL3/METTL14/WTAP and other regulatory proteins (RBM15/15B, KIAA1429, ZC3H13, CBLL1, and VIRMA). In addition, METTL16, METTL5, ZCCHC4, and PCIF1 are methyltransferases that directly catalyze m6A modifications in RNA molecules. The erasers mainly consist of FTO, ALKBH5, and ALKBH3. The “readers” are binding proteins that recognize m6A modifications in the RNA. m6A modification can affect alternative splicing of pre-mRNA, mRNA degradation, mRNA stabilization, miRNA processing, and translation
Currently, m6A modification involved in TIE is emerging and is expected to be a potential target for improving the limited antitumor immunotherapy response rates. This lack of responsiveness suggests the need to expand the knowledge of m6A modifications in TIE regulation. Furthermore, an in-depth understanding of the underlying mechanism of action will contribute to developing new combination therapies to promote antitumor immunotherapy. This review describes the existing evidence regarding m6A modifications, their influence on TIE, and their underlying molecular regulatory mechanisms. Moreover, we discuss available agents targeting m6A modifications to improve the efficacy of immunotherapy. We hope this review will broaden our understanding of the association between m6A modifications and TIE and provide new insights into therapeutic strategies targeting m6A involvement in TIE.
N 6-methyladenosine (m6A) regulators
Writers
The deposition of m6A is mainly catalyzed by a methyltransferase complex (MTC) comprising numerous components. As a core component of MTC, methyltransferase-like protein 3 (METTL3) is an S-adenosyl methionine (SAM)-binding protein that catalyzes the transfer of methyl groups in SAM to adenine bases in RNA, METTL14 stabilizes the structure of MTC and identifies the consensus motif DRACH, and Wilms tumor 1-associated protein (WTAP) promotes the recruitment of METTL3 and METTL14 [11, 12]. In addition to the MTC, several other enzymes act as independent methyltransferases: METTL16 catalyzes m6A on U6 snRNA, ncRNAs, and pre-RNAs [13], zinc finger CCHC-type containing 4 (ZCCHC4) deposits m6A on 28S rRNA [14], METTL5 adds m6A to 18S rRNA [15, 16] and phosphorylated CTD interacting factor 1 (PCIF1) catalyzes both m6A and a different RNA modification: N6,2′-O-dimethyladenosine (m6Am) [17,18,19]. However, with the advent of m6A-Crosslinking-Exonuclease-sequencing (m6ACE-seq), a newly developed technique for quantitative single-base-resolution sequencing of m6A and m6Am, which quantitatively map precise locations of transcriptome-wide m6A/m6Am in cells [20]. This technique can compensate for the shortcomings of past techniques, such as the poor resolution (~ 150 nt) of methylated RNA immunoprecipitation sequencing (MeRIP-seq), the time-consuming and inconvenient procedures (such as radioactive gel electrophoresis) of m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP), and their inability to detect m6A between different samples. It is argued that METTL16 does not methylate m6A directly at its specific ‘UACm6AGAGAA’ motif but controls intracellular SAM levels [20]. Knockdown of METTL16 resulted in the loss of m6A in the transcriptome, except for METTL16 directly methylated sites. Motif analysis also indicated that sequences located at the METTL16-dependent m6A sites delineate the METTL3-dependent “DRm6ACH” motif. METTL16 knockdown also reduced m6A at all 5 ‘UACAGAGAA’ sites and ‘UACAGAAAA’ sites within the 3′ UTR of Mat2a transcript, which encodes a SAM synthetase. All these results suggest that METTL16 may catalyze m6A through an indirect mechanism, likely via regulating MAT2A expression to control intracellular SAM levels.
Erasers
Fat mass and obesity-associated protein (FTO) is the first demethylase discovered in 2011, and another demethylase, Alkb homolog 5 (ALKBH5), was discovered 2 years later [21, 22]. Recent studies in 2017 have identified ALKBH3 as a demethylase responsible for removing m6A on transfer RNA (tRNA) [23]. All three demethylases belong to the alpha-ketoglutarate-dependent dioxygenase family and catalyze m6A demethylation in an Fe (II)- and α-ketoglutaric acid-dependent manner. With the discovery of these demethylases, m6A is considered a dynamic reversible process; however, questioned in further studies. FTO is primarily and potentially exclusively located in the nucleus [21, 24]. Therefore, FTO can demethylate m6A only within a short window after biogenesis in the nucleus, while it cannot regulate m6A of cytosolic mRNAs. Thus, FTO-mediated demethylation cannot be dynamic for cytoplasmic mRNAs. Furthermore, individual depletion of ALKBH5 and using m6ACE-seq to estimate m6A accumulation showed increased m6A accumulation in the DRACH motifs. They found that ALKBH5 maintains the DRACH sites constantly and completely unmethylated rather than reverse methylation. Thus, FTO keeps m6Am sites unmethylated and suppresses m6A of mRNA before it shuttles out of the nucleus [20]. This indicated that m6A is not reversible in the presence of demethylases. More importantly, several studies have argued that the physiologic target of FTO is not m6A but m6Am [25]. First, several studies found that once the m6A is deposited, it cannot be removed [26,27,28]. Second, the reaction rates of FTO toward m6A are low [21]. Third, the m6A demethylation function of FTO is not sequence-specific [29,30,31,32,33]. Lastly, transcriptome-wide analysis of FTO knockdown does not show a robust increase in m6A [34]. A detailed analysis of m6A peak intensities in wild-type and FTO knockdown mice to investigate the reasons for the lack of the global increase in m6A levels when FTO was knocked down. The results showed that the m6A peak exhibits a 5′ UTR increase which is the site of m6Am. Further study also proved that FTO had nearly 100 times higher catalytic activity against m6Am compared to m6A, suggesting m6Am as the indeed substrate [25, 35]. Furthermore, FTO is demonstrated to demethylate m6Am of snRNA which may influence mRNA splicing [36]. This finding is confirmed by an antibody-independent technique MAZTER-seq, that neither knockdown nor overexpression of FTO impact the m6A levels in human embryonic stem cells and HEK293T cells. However, overexpressed ALKBH5 induces a subtle decrease in methylation levels [37]. However, many studies have demonstrated that FTO acts on specific m6A sites and impacts targeted genes in cancer. This discrepancy may be due to the limitations of the detection and statistical methods used in previous studies, which could not distinguish between m6A and m6Am. In contrast, demethylases may be recruited to specific transcripts or induced cytosolic translocation to activate the demethylation program in some cancers [38, 39]. However, FTO does not affect global m6A, possibly because it affects certain gene expressions that promote m6A. Overall, m6A demethylation is rare, and the reversibility is unlikely to be a major mechanism in most cells.
Readers
The m6A recognized by different readers can execute diverse downstream biological functions (Fig. 1). m6A is mainly recognized by the YT521-B homology domain-containing proteins (including YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3), which has an aromatic cage to specifically accommodate methyl mark [40, 41]. YTHDC1 is broadly expressed and is predominantly localized in the nucleus, which regulates RNA export and pre-mRNA splicing [42, 43]. In contrast, the YTHDC2 expression is tissue-restricted and located in both nuclear and cytosolic, regulating mRNA stabilization and translation [7]. YTHDC2 also has an N-terminal R3H domain, a C-terminal YTH domain, and a helicase core module with sequence motifs characteristic of superfamily 2 DExHbox helicases, an ankyrin repeat pair insertion, and two C-terminal HA2 and OB domain extensions, which confers YTHDC2 helicase activity. Studies have shown that YTHDC2 plays a critical role in the meiotic entry process by interacting with m6A-modified transcripts and reducing their stability or enhancing translation [44,45,46,47]. However, Jain et al. argued that the helicase activity of YTHDC2 plays a role in spermatogenesis rather than recognizing m6A. To lay down the principles underlying its molecular function, they found that mutation of the m6A binding pocket of YTHDC2 has no significant effect on gametogenesis and mouse fertility. Cross-linking and immunoprecipitation (CLIP) data suggested that YTHDC2 binds to the site containing U-rich and UG-rich motifs and affects their steady-state levels rather than binds to m6A-modified sites to regulate translation. Mutation of the ATPase motif in the helicase domain of YTHDC2 did not affect meiotic entry. However, it blocked the progression of meiotic prophase I, causing catastrophic failure of spermatogenesis and leading to sterility [48]. It may be consistent that YTHDC2 recruits 5′-3′ RNA exonuclease XRN1 as a partner to reduce the target RNA stability [47]. This provides insight into how gene expression regulation has diversified over the course of evolution.
The YTHDF family contains three paralogs, YTHDF1, YTHDF2, and YTHDF3, each of which has been reported to have different functions with limited redundancy. YTHDF1 promotes mRNA translation, YTHDF2 enhances mRNA degradation, and YTHDF3 cooperates with YTHDF1 and YTHDF2 to accelerate translation and degradation [49,50,51,52]. However, the latest research proved that the YTHDF proteins enhance mRNA degradation in a redundant manner rather than translation efficiency [53]. Zaccara et al. proposed a distinctly unified model that all m6A sites bind all three YTHDF proteins to promote mRNA degradation in an m6A-dependent manner in Hela cell and leukemia cell line models rather than enhance translation. This redundancy may be due to the similar characteristics of YTHDF proteins, including RNA-binding properties, m6A binding preferences and affinities in transcriptome, high confidence with RNA degradation machinery, and intracellular localization. The mRNA degradation efficiency was highest when all three proteins were present, and compensation cannot occur when all these proteins are depleted; the mRNA is the most stable. Notably, the previous studies that YTHDF paralogs promote translation, which was affected by bioinformatic and technical issues, may be incorrect [53]. Together, these diverse lines of evidence suggest that YTHDF proteins do not promote translation; however, we cannot exclude their role in promoting translation in other cell lines or conditions. Because YTHDF proteins show significantly different expression levels in different tissues, their ability may also be affected by different phosphorylation modes, specific stimuli, and cellular contexts [49, 54,55,56,57,58]. Lasman et al. found that this functional compensation is context-dependent, which affects mRNA stability via an m6A-dependent manner rather than translation [59]. They explored the effects of YTHDF proteins in vivo in mouse gametogenesis, postnatal viability, and in vitro in mouse embryonic stem cells (mESCs). Only YTHDF2 is crucial for mouse gametogenesis, which may be due to the different expression spatial or temporal space, cell types, and intracellular localization. YTHDF proteins show compensation for one another in a dosage-dependent manner during embryonic development and gestation. In mESCs, only triple knockout of YTHDF paralogs shows the effect on mRNA decay and differentiation.
In addition to these canonical readers, other proteins also act as indirect m6A readers with a distinct binding mechanism, including insulin-like growth factor 2 mRNA binding proteins (IGF2BPs) (including IGF2BP1, IGF2BP2, and IGF2BP3) and RNA binding proteins (RBPs) (including HNRNPA2B1, HNRNPC, and HNRNPG) [60,61,62]. Instead of directly recognizing and binding to m6A, these proteins bind to target RNAs via an indirect mechanism termed “m6A-switch”, that m6A can alter its local RNA structure and enhance the accessibility of its base-paired residues or nearby regions to modulate protein binding. The m6A-switch is proven to regulate HNRNPC binding activities, affecting target mRNAs’ abundance and alternative splicing, demonstrating the regulatory role of m6A-switch on gene expression and RNA maturation [60]. The HNRNPA2B1 protein was initially proposed as a direct m6A mark reader interacting with the consensus motif via its RNA-recognition motifs (RRMs) and regulating alternative splicing [63]. However, Wu et al. found that HNRNPA2B1 may mediate the effects of m6A via the m6A-switch mechanism [62]. Their structural study showed no aromatic cage-like surface in crystal structures. HNRNPA2B1 does not recognize or show enhanced binding to the m6A-modified RNA substrates in vitro. In vivo study showed that only a small fraction of the m6A nuclear sites is located near HNRNPA2B1. Therefore, the m6A-switches may account for the enhanced HNRNPA2B1 binding to m6A [61]. Moreover, HNRNPG is also proved an indirect reader to use the m6A-switch mechanism, which binds to m6A-modified RNAs by a low-complexity region [62].
The three IGF2BP paralogs are highly similar, with two N-terminal RRMs and four C-terminal K-Homology (KH) domains, characterized by a conserved αβ-topology that can be structurally and functionally included in two di-domains (KH1-2 and KH3-4) [64]. Functionally, IGF2BP1-3 fortifies the stability and increases m6A-modified mRNAs translation efficiency [8]. Nevertheless, the latest research takes a different view of their roles as m6A readers. Sun et al. used the in vivo click selective 2-hydroxyl acylation and profiling experiments (icSHAPE), a technique they developed to map RNA structure in three compartments – chromatin, nucleoplasm, and cytoplasm, to help determine the precise relationship between RNA structure and cellular processes, including transcription, translation, RNA decay, RBP interaction, and RNA modification. They used the cytotopic RNA structurome data to filter published CLIP-seq data and calculated m6A modification effect on protein binding. The results showed that while the canonical readers bind most strongly to the m6A sites, HNRNPC and IGF2BP protein binding peaks at a distance. RNA pull-down experiment showed that IGF2BP3 exhibits enhanced binding to the m6A-modified RNAs and uracil mutations compared to the unmethylated RNAs. This suggests that IGF2BP proteins may also be able to read the structural changes induced by m6A-switch [65].
Impaired antigen processing presentation and tumor immune escape (TIE)
Tumor antigens (tAgs) are expressed on the surface of cells by HLA-I and undergo two distinct pathways to induce an effective antitumor response. First, tAgs are taken up by dendritic cells (DCs) and cross-presented to prime naive CD8+ T cells [66]. Second, tAgs are directly presented by tumor cells to be recognized and killed by primed CD8+ T cells. Under constant T-cell selection, tumor cells can avoid immune recognition via different evasion mechanisms [67]. In both pathways, tumors impair antigen presentation by disrupting DC function or reducing HLA-I expression to escape immune recognition, eventually leading to TIE.
Dendritic cell defects and TIE
DCs take up dying tumor cells that release dangerous signals, undergo maturation, migrate to the draining lymph nodes, and process and load tAgs onto HLA-I to CD8+ T cells (Fig. 2). METTL3-mediated m6A modification was demonstrated to promote DC maturation and activation in an innate immune response model. Mechanistically, CD40, CD80, and TLR4 signaling adaptor Tirap transcripts are methylated by METTL3, promoting their translation in DC to enhance T cell activation and cytokine production [68]. In contrast, m6A modification generally displays the opposite role in antitumor immunity models. In melanoma and hepatocellular carcinoma (HCC), activating the β-catenin signaling pathway can prevent the infiltration of DCs and CD8+ T cells within the tumor microenvironment (TME) by inhibiting the secretion of the CC chemokine ligands CCL4 or CCL5, which directly impairs the therapeutic benefit of ICI [69, 70]. Numerous studies have demonstrated that m6A modifications can activate the β-catenin signaling pathway in different cancers (Table 1). For example, m6A modification promotes the stability of circRNA-SORE mRNA, which enhances sorafenib resistance through the circRNA-SORE/miR-103a-2-5p/miR-660-3p/Wnt2b/β-catenin pathway in HCC [72]. YTHDF2 decreases lncAY levels in an m6A-dependent manner, which promotes the proliferation and migration of HCC cells via the lncAY/BMI1/Wnt/β-catenin signaling pathway [85]. These studies have demonstrated that m6A modification promotes progression and drug resistance by activating the β-catenin signaling pathway in various cancer types. We envision that m6A may promote TIE by activating the β-catenin signaling pathway, which needs further studies. Prostaglandin E2 (PGE2) secretion in a cyclooxygenase 2 (COX2)-dependent manner can impair the accumulation and viability of DCs in tumors and suppress DC maturation [87,88,89]. In lung cancer (LC), HNRNPA2B1 upregulates COX2 expression, PGE2 production, and promotes tumor growth [90]. Moreover, m6A modification induces TIE by increasing antigen degradation in DCs [91]. m6A modifies transcripts encoding lysosomal proteases to promote lysosomal cathepsins translation in DCs, which is associated with enhanced ingested antigens degradation [92, 93]. In summary, m6A modification plays a vital role in regulating the cross-presentation function of DCs and priming of CD8+ T cells, including maturation, migration, activity, and antigen degradation of DCs.

Dendritic cells (DCs) in antitumor immunity. DCs are recruited into the tumor bed by chemokines, such as CC chemokine ligands 4 (CCL4), CCL5, and XC-chemokine ligand 1 (XCL1). FMS-like tyrosine kinase 3 ligand (FLT3L) promotes the differentiation and survival of DCs. Immature DCs take up dying tumor cells that release damage-associated molecular patterns, migrate to the draining lymph nodes, and process and load cancer antigens onto human leukocyte antigen (HLA)-I and HLA-II for presentation to CD8+ and CD4+ T cells, respectively. Naive CD4+ T cells are primed first, which allows DCs to prime CD8+ T cells via CD40-CD40L signaling. Moreover, intratumoral DCs generate chemokines CXC-chemokine ligand 9 (CXCL9) and CXCL10 to recruit effector CD8+ T cells from draining lymph nodes. Tumors can change DC functions to achieve tumor immune escape. Vascular endothelial growth factor (VEGF) prevents DC differentiation and maturation. Activation of β-catenin signaling and expression of prostaglandin E2 (PGE2) prevent the recruitment of DCs to the tumor bed by blocking chemokine secretion, including CCL4, CCL5, and XCL1. PGE2 prevents the recruitment and maturation of DCs. Tumor cells, CD4+ regulatory T cells (Treg), myeloid-derived suppressor cells (MDSCs), and M2 macrophages produce cytokines, including tumor growth factor-β (TGFβ), interleukin (IL)-6, IL-10, PGE2, and VEGF, to prevent DC maturation. CCL4, CC-chemokine ligands 4; CXCL9, CXC-chemokine ligand 9; DAMP, damage-associated molecular pattern; DC, dendritic cell; FLT3L, FMS-like tyrosine kinase 3 ligand; HLA-I, class I human leukocyte antigen; IL-6, interleukin-6; MDSC, myeloid-derived suppressor cell; PGE2, prostaglandin E2; TGFβ, transforming growth factor-β; Treg cell, regulatory T cell; VEGF, vascular endothelial growth factor; XCL1, XC-chemokine ligand 1.
Defective class I human leukocyte antigen presentation in tumors
After priming with DCs, CD8+ T cells migrate from the draining lymph nodes to the tumor and recognize the tAgs present on HLA-I by the tumor cells for elimination. In addition to the failure in the cross-presentation function of DCs, malfunctions also occur in the tAg presentation pathway of tumor cells. The antigen processing and presenting machinery (APM) in tumor cells mainly consists of the ubiquitin-protease system and HLA-I complex. The presentation processes are divided into four steps: (i) processing proteins into peptides, (ii) peptide transportation, (iii) installation of peptides on the HLA-I complex, and (iv) peptide-HLA-I complex translocation and presentation (Fig. 3). m6A modification has been reported to be correlated with APM in tumors, such as HCC, pancreatic cancer (PC), esophageal cancer (EC), and breast cancer (BC) [94,95,96,97]. In BC, higher m6A modification levels are associated with elevated expression of HLA-A and more tumor-infiltrating CD8+ T cells, helper T cells, and natural killer cells, but decreased expression of programmed cell death protein ligand 1 (PD-L1), PD-L2, T-cell immunoglobulin and mucin-domain containing 3 (TIM3), and CCR4. Lower m6A modification is related to the hallmarks of PI3K/AKT signaling in cancer, KRAS signaling, angiogenesis, and shorter overall survival (OS) [97]. In HCC, downregulated METTL3 expression is related to increased MHC, co-stimulatory, and adhesion molecules [94]. However, current studies are based on the analysis of public databases that require further experimental verification.

Overview of human leukocyte antigen (HLA)-I antigen processing and presentation machinery. Peptides are generated by the degradation of endogenous proteins via the proteasomal pathway. The peptides are then translocated by transporters associated with the antigen processing (TAP) in the endoplasmic reticulum (ER). In the ER lumen, peptides can be further trimmed by ER aminopeptidase 1 (ERAP1) and ERAP2. The peptide-loading complex, comprising ERp57 and calreticulin (CALR), helps the loading and folding of HLA-I molecules with peptide and β2-microglobulin (β2M). Tapasin assesses peptides for stable binding to the complex formed by the HLA-I heavy chain α1 and α2 domains. CALR, calreticulin; ER, endoplasmic reticulum; ERAP1, ER aminopeptidase 1; HLA-I, human leukocyte antigen-I; TAP, transporters associated with antigen processing; β2M, β2-microglobulin
Immunosuppressive cells and TIE
CD4+ regulatory T cells (Tregs) and TIE
Based on different antigen signals and cytokine stimulations, CD4+ T cells can differentiate into numerous subtypes, such as Tregs and helper T cells 1, 2, and 17 (Th1, Th2, and Th17). Tregs generally play pro-tumor roles, are immunosuppressive, and are associated with TIE [98]. Tregs inhibit the function of antitumor T cells by producing inhibitory cytokines, including interleukin (IL)-10, IL-35, and TGF-β [99]. Tregs can also inhibit the activation of CD8+ T cells by expressing cytotoxic T lymphocyte antigen 4 (CTLA4), an inhibitory molecule [98]. Tregs inhibit T-cell activation by blocking the maturation and function of DCs [99]. Studies have shown that METTL3 regulates T-cell homeostasis and differentiation in mouse T cells. METTL3-meditated m6A modification promotes mRNA degradation of SOCS family genes and enhances T-cell differentiation and proliferation via the IL7R/JAK/STAT5 pathway [100]. In a further study, they demonstrated in vivo that METTL3-mediated m6A modification promotes the IL2-STAT5 pathway and immunosuppressive functions of Tregs [101].
Myeloid-derived suppressor cells (MDSCs) and TIE
MDSCs are potent immunosuppressive cells in cancer, promote tumor initiation and metastasis, and have attracted attention as targeted therapeutic interventions [102]. MDSCs induce TIEs via various pathways. First, MDSCs significantly inhibit MHC II expression, cross-presentation, DC activation, and T-cell stimulation by upregulating indoleamine 2,3-dioxygenase (IDO) and myeloperoxidase. Second, increased IDO levels enhance the differentiation and suppressive functions of Tregs. Moreover, the IDO-induced depletion of tryptophan attenuates cytotoxic T lymphocyte (CTL) proliferation. Furthermore, upregulated arginase 1 causes CTL disability and apoptosis [103]. In CC, the expression of METTL3 and CD33+ MDSCs in tumor tissues is higher than that in adjacent normal tissues, and their expression is positively correlated. Their levels in tumor samples are significantly related to poor disease-free survival (DFS) and OS. More importantly, the expression of METTL3 is an independent factor for DFS and OS in patients, whereas the number of CD33+ MDSCs is an independent predictor of DFS. Furthermore, knockdown of METTL3 potently reduces CD33+ CD11b+ HLA-DR− MDSCs and tumor-derived MDSCs in CD33+ or HeLa cells [104].
Tumor-associated macrophages (TAMs) and TIE
Macrophages are myeloid cells with various phenotypes, of which M1 and M2 subtypes are extreme. M1 can kill tumor cells by emerging as antigen-presenting cells (APCs) or generating nitric oxide, type 1 cytokines, and chemokines. M2 macrophages are activated by IL-4, IL-13, TGF-β, and/or glucocorticoids. M2 macrophages produce cytokines and type II chemokines to accelerate tumor growth. In turn, stromal and tumor-associated factors in the TME can polarize macrophages to the M2 type, particularly the TAM type, to drive TIE. In BC, M2 macrophages produce high levels of IL-10, which effectively attenuate CD8+ T cell-dependent responses to paclitaxel by downregulating IL-12 in intratumoral DCs [105]. FTO has been demonstrated to promote both M1 and M2 polarizations. Gu et al. found that FTO knockdown reduced the mRNA stability of STAT1 in M1 and PPAR-γ in M2 in a YTHDF2-dependent manner [106]. Furthermore, IGF2BP2 has been reported to shift M1 macrophages to M2 activation by stabilizing TSC1 and PPAR-γ in an m6A-dependent manner [107]. ALKBH5 was demonstrated to promote macrophage recruitment, and M2 polarization and phagocytosis in glioblastoma multiforme cells and induce immunosuppression in allograft tumors. ALKBH5-mediated m6A demethylation stabilizes NEAT1, leading to TAM recruitment and TIE via ALKBH5/NEAT1/CXCL8/IL-8 pathway [108].
In summary, immunosuppressive Tregs, MDSCs, and M2 macrophages can help tumors obtain TIE in different ways, which prevents the immune system from recognizing and destroying the tumor. These immunosuppressive cells are regulated by m6A modification to obtain TIE, which may be a promising target for antagonizing TIE and reinforcing the effect of antitumor immunotherapy.
Immune checkpoint molecules and TIE
Immune checkpoints have emerged as a large number of inhibitory pathways in the immune system that are critical for maintaining self-tolerance and regulating the duration and magnitude of the peripheral tissue physiological immune response to reduce collateral tissue damage. Tumors acquire TIE through these immune checkpoint pathways [109]. Co-inhibitory and co-stimulatory receptor signaling pathways play crucial roles in T cell activation, differentiation, effector function, and survival. Therefore, promising therapeutic approaches to reinvigorate a T-cell response can be achieved using inhibitors of co-inhibitory factors or agonists of co-stimulatory factors. The currently known co-inhibitory receptors include CTLA4, programmed cell death protein 1 (PD-1), PD-L1, lymphocyte activation gene 3, TIM3, and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT). In addition, the co-stimulatory factors include B- and T-cell lymphocyte attenuator, glucocorticoid-induced tumor necrosis factor (TNF) receptor family-related protein, OX40, 41BB, and inducible T cell co-stimulatory. Next, we review the research progress on m6A in immune checkpoint regulation.
The discovery and clinical implementation of ICIs targeting CTLA4, PD-1, and PD-L1 have revolutionized cancer treatment and have been recognized by the 2018 Nobel Prize for Medicine and Physiology. PD-L1, also known as B7 homolog 1 or cluster of differentiation 274, is the first functionally characterized ligand of co-inhibitory PD-1. PD-L1 is a transmembrane protein expressed in various tissues, but it is primarily found in T cells, B cells, DCs, monocytes, and various tumor cells. However, PD-1 is mainly expressed on the surfaces of activated T cells, B cells, and DCs. T cells are the basis of the immune response, and their activation requires the interaction of two signals: (i) via the CD3 complex upon the binding of the T-cell receptor (TCR) expressed on the surface of T cells with HLA-I and its cognate peptide antigen on APCs or a target cell and (ii) another co-stimulatory receptor, CD28, which binds to the B7 family of co-stimulatory molecules mainly expressed on APCs. T cells differentiate, proliferate, produce cytokines, and subsequently form memory T cells [110]. PD-1 interacts with its ligand PD-L1, leading to the dephosphorylation of downstream TCR signaling molecules and inhibition of TCR-mediated IL2 production and T-cell proliferation, which causes TIE [111]. Currently, anti-PD-1/PD-L1 antibodies are approved for use in patients with a wide range of tumor types [112]. Antibodies against PD-1 include nivolumab, pembrolizumab, and cemiplimab. The PD-L1 antibodies include durvalumab, atezolizumab, and avelumab. m6A modification has been found to modulate PL-L1/PD-1 expression and promote immunosuppression, thus contributing to TIE [38, 113,114,115]. Knockdown of m6A regulators or targeted inhibitors can potently enhance the immunotherapeutic effects of these antibodies. ALKBH5 was verified to promote PD-L1 expression, consequently reshaping TME and affecting immunotherapy efficacy in intrahepatic cholangiocarcinoma. Knockdown of ALKBH5 increases m6A modification in the 3’UTR of PD-L1 mRNA, thereby promoting its degradation in a YTHDF2-dependent manner. Moreover, ALKBH5 inhibits the expansion and cytotoxicity of T cells by promoting PD-L1 expression. Single-cell mass cytometry analysis showed that ALKBH5 promotes PD-L1 expression in monocytes/macrophages and reduces the infiltration of MDSCs. Analysis of samples from patients receiving anti-PD1 immunotherapy showed that tumors with strong nuclear expression patterns of ALKBH5 are more sensitive to PD-L1 immunotherapy [115]. FTO promotes melanoma tumorigenesis and inhibits anti-PD-1 blockade immunotherapy. FTO knockdown increases m6A modification of PD-1 (PDCD1), CXCR4, and SOX10, contributing to enhanced RNA decay via the m6A reader YTHDF2 in melanoma cells. It has been consistently proven that loss of FTO sensitizes melanoma cells to interferon-γ and sensitizes melanoma to anti-PD-1 treatment in mice, which depends on adaptive immunity [38]. In addition to PD-1/PD-L1, other immune checkpoint molecules are also associated with m6A modifications and related regulators, such as CTLA-4, TIGIT, and TIM-3 [116, 117].
Speculative functions of m6A modification in TIE
In addition to the mechanisms discussed above, many other signaling pathways can modulate TIE, including metabolic alterations, acquisition of stemness, and epithelial-mesenchymal transition (EMT). m6A modification has been proven to play crucial roles in these pathways. Therefore, we speculate that m6A may also impact TIE via these pathways, which are discussed in the following section.
Aerobic glycolysis and TIE
Despite aerobic conditions, tumor cells prefer to produce energy through glycolysis rather than aerobic oxidation, along with more lactic acid generation, known as the Warburg effect [118]. Excess lactic acid acidifies the TME, endows tumor cells with stronger viability and aggressiveness, and has been identified as an important therapeutic target [119]. More importantly, the acidic TME impairs the immune response by weakening cytotoxic T cell function, blocking DC maturation, and enhancing helper cell activities [120]. In melanoma, ALKBH5 knockdown effectively promotes sensitivity to anti-PD-1 treatment. Mechanistically, during anti-PD-1 treatment, ALKBH5 promotes lactate generation and consequent accumulation of Tregs and MDSCs in TME by stabilizing Mct4/Slc16a3 mRNA, which is a pivotal enzyme mediating the transmembrane transport of lactate [121]. Many studies have found that m6A modifications play critical roles in regulating glycolysis.
The glycolysis process in tumor cells and the regulation of glycolysis-related enzymes by m6A are summarized in Fig. 4 and Table 2. Glucose, the feedstock for glycolysis, is transported into the cells by glucose transporters (GLUTs), which are membrane proteins on the cell surface. Abnormal expression of GLUTs promotes glucose intake and glycolysis. In colorectal cancer (CRC), METTL3 promotes glycolysis and cancer progression in an m6A-dependent manner. METTL3-mediated m6A modification promotes SLC2A1 (GLUT1) and hexokinase 2 expression through IGF2BP2/3-dependent mRNA stability regulation, and activates glycolysis [122]. METTL3 was also shown to promote CRC progression by activating the m6A/GLUT1/mTORC1 pathway. Mechanistically, METTL3 promoted GLUT1 translation in an m6A-dependent manner, increasing glucose uptake and lactate production and further activating the mTORC1 signaling pathway [123].

Overview of the regulation of tumor immune escape-associated glycolytic enzymes by N6-methyladenosine modification in tumor cells. The afferent blood delivers glucose to tissues, where it reaches the cells by diffusion. Glucose is taken up by specific glucose transporters (GLUTs), which are first converted to glucose-6-phosphate by hexokinase (HK) and then to pyruvate by various enzymes, including glucose phosphate isomerase (GPI), phosphofructokinase (PFK), aldolase (ALDO), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGM), enolase (Eno), pyruvate kinase (PK), pyruvate dehydrogenase (PDH), and lactate dehydrogenase (LDH). ALDO, aldolase; Eno, enolase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GLUT, glucose transporter; GPI, glucose phosphate isomerase; HK, hexokinase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; PFK, phosphofructokinase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PK, pyruvate kinase
Glycolysis is triggered after glucose is taken into cells. Its progression is maintained by several enzymes, such as hexokinase (HK), aldose enzyme, glucose phosphate isomerase (GPI), phosphofructokinase (PFK), aldolase (ALDO), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGM), enolase (Eno), pyruvate kinase (PK), pyruvate dehydrogenase (PDH), and lactate dehydrogenase (LDH), which have been reported to be regulated by m6A modification (Fig. 4, Table 2). In cervical cancer (CC), METTL3 promotes proliferation and aerobic glycolysis of CC cells. METTL3 binds to the 3′-UTR of HK2 mRNA to catalyze m6A modification, which is recognized by YTHDF1 to enhance HK2 stability [126]. In general, m6A is shown to enhance glycolysis by regulating the expression of key enzymes. METTL3 promotes glucose uptake, lactate generation, and ATP level, which can be reversed by METTL3 knockdown. Downregulation of METTL3 suppresses tumor growth and glycolysis progression in tumor cells and xenograft mouse models. Since m6A has the function of reprogramming energy metabolism, we hypothesized that this process is accompanied by reprogramming TME. Consequently, targeting m6A may effectively overcome TIE and effectively treat a variety of cancers in combination with immunotherapy.
In tumor cells, hypoxia-inducible factor-1α (HIF-1α) has been reported to enhance glycolysis by upregulating glycolysis-related enzymes, inhibiting PDH and mitochondrial oxidative phosphorylation, and subsequently leading to TME acidification and TIE [173,174,175]. METTL3 promotes glycolysis and malignant biological behaviors of HCC cells by methylating HIF-1α [133]. Moreover, the m6A reader YTHDC2 can promote metastasis by enhancing HIF-1α translation in CRC [134]. However, further studies are required to determine whether this occurs by regulating m6A modification. Although m6A modification has been shown to affect glycolysis from multiple pathways, its effect on TIE deserves further exploration.
Cancer stem cell-like characteristics and TIE
Cancer stem cells (CSCs) are subpopulations with strong self-renewal ability, pluripotency, and tumorigenicity that are closely related to tumor initiation, metastasis, drug resistance, and recurrence. They can also obtain TIE through their immunomodulatory properties [176]. Aberrant m6A modifications are related to the initiation and maintenance of CSC-like phenotypes in tumor cells (Table 2). FTO promotes the self-renewal of leukemia stem cells (LSCs) and TIE, which can be reversed by small-molecule FTO inhibitors (FTOis) [39]. FTOis (CS1 and CS2) impairs LSC self-renewal properties by occupying the catalytic pocket of FTO to inhibit its demethylase activity, which leads to increased m6A abundance and decreased expression of MYC and CEBPA. In contrast, FTO promotes LILRB4 expression, an immune checkpoint gene, by demethylating LILRB4 to inhibit the m6A-mediated degradation. FTOis effectively overcomes TIE and sensitizes acute myeloid leukemia (AML) cells to T cell cytotoxicity. More importantly, LSCs share numerous characteristics with hematopoietic stem cells (HSCs). Therefore, eliminating LSCs without damaging HSCs as much as possible is the key to treating leukemia. Knockdown of YTHDF2 promotes HSC self-renewal to enhance HSC ex vivo expansion without any noticeable lineage bias or leukemic potential by stabilizing Tal1 mRNA [177]. This may help overcome the limitations of umbilical cord blood in treating leukemia, as there are insufficient HSCs in a single human umbilical cord blood unit. METTL3 promotes glioblastoma stem cell (GSC) self-renewal and tumorigenesis by enhancing m6A modifications to stabilize transcripts of some CSC-related genes, such as AMAD19, SOX2, SRSF3/6/11, NOTCH1, and HES1 [136,137,138,139]. Furthermore, YTHDF2 promotes glioblastoma stemness by stabilizing MYC and VEGFA in an m6A-dependent manner [153]. In a word, m6A promotes the initiation and maintenance of CSC-like phenotypes in tumor cells. Inhibitors targeting m6A can effectively reverse stem-like phenotype and block TIE. Further studies are needed to dissect whether m6A reverses TIE by affecting CSCs. What is more, none of the existing immunotherapy approaches selectively targets CSCs. Therefore, understanding this mechanism will provide a solution for optimizing or identifying new immunotherapy strategies and their combinations.
Epithelial-mesenchymal transition and TIE
EMT is a cellular reprogramming process that detaches epithelial cells from each other and the underlying basement membrane, eventually transforming them into mesenchymal cells. EMT increases the developmental and metastatic potential of cancer cells and drug resistance [178]. An increasing number of studies have shown that EMT can regulate antitumor immunity. For example, in melanoma cells, SNAIL-induced EMT stimulates the secretion of TGF-β and thrombospondin 1, which promotes the formation of Treg cells and impairs the antigen-presenting capacity of DCs [179]. This attenuates the immunogenicity of melanoma cells and their sensitivity to immunotherapy, which can be restored by inhibiting SNAIL [179]. Furthermore, EMT promotes immunosuppression and TIE in BC cells. Tumors derived from more mesenchymal carcinoma cell lines express lower MHC-I and higher PD-L1 and contain within their stroma Treg cells, M2 macrophages, and exhausted CD8+ T cells than tumors derived from more epithelial carcinoma cell lines [180]. The above evidence indicates that EMT can promote immunosuppression of tumor cells and obtain TIE.
EMT promotes the expression of the EMT-inducing transcription factors (TFs) ZEB, SNAIL, and TWIST, which activate mesenchymal state-related genes (N-cadherin, vimentin, fibronectin, β1 and β3 integrins, and matrix metalloproteinases) and resilient epithelial state-related genes (E-cadherin, epithelial cell adhesion molecule, occludins, claudins, α6β4 integrins, and cytokeratins). Studies have demonstrated that upregulated m6A levels promote EMT in cancer cells, such as HeLa cells (CC), HepG2 cells (liver cancer), Huh7 cells (liver cancer), and A549 cells (LC). Increased m6A modification promotes SNAIL mRNA translation, which can be enhanced or inhibited by ALKBH5 or METTL3 knockdown. Depletion of METTL3 blocks invasion, migration, and EMT of cancer cells and tumor metastasis [181]. Furthermore, METTL3-mediated m6A also potentiates EMT by regulating integrin-β1 and ZMYM1/E-cadherin pathway [159, 169]. In addition to these EMT-inducing TFs, m6A also induces EMT by regulating the expression of other genes, such as MALAT1, HMGA1, circ1662, miR-20a-5p, FOXM1, DUXAP9, YAP, and ZBTB4 [160,161,162,163, 165, 166, 171, 172]. In addition to its roles in regulating RNA stability and translation processes, m6A has been found to promote EMT by promoting the splicing of precursor miRNAs. For example, METTL3-mediated m6A modification promotes the splicing of precursor miR-143-3p to produce mature miRNAs, thus contributing to enhanced EMT via the METTL3/miR-143-3p/VASH1 pathway in LC cells [164]. In liver cancer, METTL3 determines the fate of the HSP5 transcript to process it into circHSP5 rather than mRNA. Increased circHSP5 acts as a miR-370 sponge to promote HMGA2 expression and potentiate EMT [167]. Abnormal m6A promotes EMT by regulating EMT-inducing TFs and other genes; however, its role in TIE remains unclear. Whether regulation of EMT through m6A modification can attenuate TIE and promote immunotherapy efficacy remains to be further explored.
Targeting m6A modification in cancer therapy
The m6A levels of specific RNA transcripts have been shown to influence tumor development [182]. Therefore, inhibitors targeting m6A regulators may be effective new approaches for tumor therapy (Table 3). Rhein derived from the rhizome of Rheum palmatum, which was identified as the first competitive inhibitor target Alkb subfamily by structure-based in silico high-throughput screening and further structural optimization [183]. It competitively bounds to FTO or AlkB catalytic domain to form a complex and prevent the recognition of m6A substrates inside cells, which can increase the cellular m6A on mRNA. Rhein shows several bioactivities; however, its cellular targets remain largely unknown [196, 197]. Rhein also inhibits ALKBH2 activity, which is responsible for demethylating N1-methyladenosine modification in vitro. Furthermore, rhein inhibits other Fe2+- and 2OG-dependent hydroxylases by high-throughput screening in the NIH Molecular Libraries Probe Development Center Network (MLPCN) program, including the Jumonji domain containing 2A and 2E (JMJD2) histone demethylases and prolyl-4-hydroxylase. Rhein binding to FTO still has the possibility of multiple orientations. The crystal structure of small molecules in complexes with FTO has not been determined. In addition, increased m6A distributions due to rhein in cells may result from the direct inhibition of cellular demethylation via FTO or other members of nucleic acid demethylases. Characterizing the cell specificity of rhein will also be important to demonstrate its use as a cellular probe for nucleic acid demethylase in future studies. Rhein has been shown to have anticancer activity against various cancers. For example, rhein inhibited tumor growth in 4 T1 BC xenografts [184]. Another inhibitor, meclofenamic acid (MA), was identified by a high-throughput fluorescence polarization assay and selectively inhibited FTO demethylation of m6A over ALKBH5 [186]. This slightly higher selectivity for FTO in the Alkb subfamily mainly depends on its structure. MA is neither a mimic of 2OG nor a chelator of iron, and the structural complex of MA bound to FTO is a β-hairpin motif which is a part of the nucleotide recognition lid (NRL) for providing hydrophobic interactions with MA. In contrast to FTO, ALKBH5 lacks this part of NRL loop, resulting in leakage upon MA binding. In addition, MA could not inhibit ALKBH2 and ALKBH3 which have this region of NRL. The existence of the hydrophilic and bulky residues in the part of the NRL might significantly disturb the inhibitor MA binding to ALKBH2 and ALKBH3. Based on this structural complex, it should be possible to design more optimized analogs for FTO specificity and potency. In addition, MA increases m6A abundance in HeLa cells in an FTO activity-dependent manner. In glioblastoma, MA2 (an ethyl ester form of MA) significantly inhibits GSC growth and self-renewal, effectively suppresses GSC-induced tumorigenesis, and prolongs the lifespan of GSC-grafted mice [137]. In later studies, MA2 was shown to enhance the effect of the chemotherapeutic drug temozolomide on the suppression of glioma cell proliferation [187]. Subsequently, many FTOis can effectively inhibit the proliferation, promote the apoptosis of AML cells in vitro and suppress tumor growth in vivo, including FB23-2, R-2HG, CS1, and CS2 [39, 191, 192].
In addition to FTO, other m6A regulators are key targets for treating m6A-associated tumors. BTYNB is screened as a potent and selective inhibitor among 16,000 small molecules via the fluorescence anisotropy-based assay, which inhibits IGF2BP1 binding to a specific high-affinity binding site in the coding region stability determinant of c-Myc mRNA. BTYNB effectively reduces the expression of c-Myc mRNA and protein. It inhibits proliferation and anchorage-independent growth of IGF2BP1-positive cancer cells [193]. Furthermore, IGF2BP1 acts as the dependent E2F-transcription super-enhancer. The E2F pathway is regulated by IGF2BP1 in an m6A-dependent manner. BTYNB blocks E2F1 expression at both mRNA and protein levels, as well as inhibits E2F/IGF2BP1-driven gene expression by reducing the binding of IGF2BP1 to E2F1 mRNA. BTYNB effectively blocks tumor cells’ growth and spread in xenograft tumors. Moreover, BTYNB synergizes with palbociclib at low concentrations of both compounds, suggesting that BTYNB is beneficial for combination therapy to impinge tumor cell proliferation [194]. However, BTYNB showed the ability to reduce IGF2BP1-dependent stabilization of mRNAs, and its putative off-target effects are unknown. Therefore, we hypothesized that BTYNB might interfere with the interaction between IGF2BP1 and m6A sites. STM2457 is a highly potent and selective inhibitor of METTL3 without disrupting the METTL3–METTL14 complex, identified by high-throughput screening. STM2457 effectively inhibits AML cell growth and promotes differentiation and apoptosis while reducing m6A levels of known AML-related mRNAs to block their translation and expression. In in vivo studies, STM2457 disrupted engraftment and prolonged survival in AML mouse models [195]. In addition to these competitive inhibitors, there are other ways to inhibit the functions of m6A regulators. First, the catalytic capacity of METTL3 depends on the heterodimer structure formed with METTL14; hence, it would be reasonable to design inhibitors based on protein-protein interaction strategies. Second, structural analysis of METTL3 suggested that the binding sites of the substrate SAM are merged into a large pocket; therefore, the development of bi-substrate inhibitors occupying both binding sites may be another effective strategy. Third, the proteolysis-targeting chimera strategy is a promising technology to degrade target proteins via proteasomes. Targeted m6A modification for clinical application is still in the initial stage and has not yet entered clinical trials. However, with increasing knowledge of the function and mechanism of m6A modification in cancer, it is expected that drugs targeting m6A modification will be developed and applied to clinical treatment in the near future.
Targeting m6A modification in TIE
Regulation of the antitumor immune response by m6A RNA methylation is still in its infancy. However, recent studies have shown the possibility of combining immunotherapy with newly developed m6A regulator inhibitors for cancer therapy. Rhein significantly enhances the antiproliferative effects of atezolizumab (an anti-PD-L1 antibody) in 4 T1 BC xenografts. Moreover, the proportion of CD8+ T cells in the spleen and tumor is significantly increased in the combination therapy group and is significantly different from that in the monotherapy groups. Serum levels of TNF-α and IL-6 are significantly elevated in the rhein and combination therapy groups. In addition, the levels of various apoptotic factors in the tumor tissues are significantly higher in the combination treatment group [185]. Two FTOis are identified by structure-based virtual screening, CS1 and CS2, which show strong antitumor activity in leukemia. Inhibition of FTO by CS1 or CS2 significantly suppresses LSC self-renewal and promotes the immune response by suppressing the expression of the immune checkpoint gene LILRB4. They reverse TIE by abolishing the FTO-induced stability of LILRB4 mRNA and enhancing the sensitivity of AML cells to T-cell cytotoxicity [39]. ALKBH5 promotes lactate generation and Tregs and MDSC accumulation by stabilizing Mct4/Slc16a3 mRNA in melanoma cells. A specific ALKBH5 inhibitor, ALK-04, is identified by in silico screening of compounds using the X-ray crystal structure of ALKBH5 and by performing structure-activity relationship studies on a library of synthesized compounds. ALK-04 potently enhances anti-PD-1 therapy response in in vivo experiments [121]. These studies show not only the inhibition of m6A demethylases as a potential anticancer target but also their potential to reverse TIE. Although inhibitors of m6A modification regulators have been demonstrated to have anticancer roles by modulating tumor immunity, none have been tested in a clinical setting.
Conclusions and perspectives
In this review, we outline the different TIE mechanisms and summarize the increasing excitement surrounding the development of the regulatory roles of m6A modification involved in these mechanisms. Due to many new related discoveries in recent years, updating the academic progress of m6A modification is still necessary. Here, we summarize the research progress of m6A modification and the core function of m6A in TIE. Although m6A modification is directly related to TIE, the exact molecular mechanism underlying its regulation remains unclear. The complex TIE mechanism in tumor cells is the cause of the low response rate to immunotherapy. Therefore, m6A modification may be considered a potential candidate target for targeting the TIE mechanism and is expected to be vital to overcoming immunotherapy-related challenges.
In the pathogenesis of tumors, m6A modification regulates RNA splicing, decay, nuclear export, stability, and translation, promotes the expression of oncogenes, or inhibits the expression of tumor suppressor genes, thereby inducing TIE. The putatively dynamic and reversible characteristics of m6A modifications make them attractive in the field of anticancer therapy. In light of new findings on the physiological roles of m6A regulators. At present, it is more important to clarify the ‘real’ regulator and their specific physiological function in certain cancers. This will help to explore the clear mechanism of m6A in TIE and target it more accurately. m6A RNA demethylase inhibitors have shown the potential to enhance immunotherapy [39, 121, 185]. The m6A methyltransferases METTL3 and METTL14 also show antitumor functions by reprogramming macrophages [198, 199]. SAM, the methyl donor for RNA, has shown anticancer activity in various cancer types by targeting histone methylation and DNA hypomethylation. It is unknown whether SAM inhibits tumor growth and metastasis by upregulating m6A RNA methylation levels or promoting immunotherapy. This means that m6A agonists, not just inhibitors, may also inhibit the growth of certain tumors. In addition, m6A modification exhibits cellular heterogeneity; that is, the same writer, eraser, and reader proteins may have different biological functions in different cells. This may cause it to act in the opposite manner in tumor or immune-related cells. Furthermore, other hypotheses could explain the paradox. First, m6A regulators may function independently of their m6A catalytic activity. Second, as recognized by different readers, genes modified by m6A modification can undergo different fates. Third, the location of m6A modifications in different regions of the same mRNA transcript may lead to different results. Therefore, additional studies are required to clarify the roles of m6A modifications in TIE, including the contributions of specific regulators, targets, modes of action, and TME.
Further studies are required to determine whether m6A modification is likely to provide new insights into identifying patients with tumors susceptible to specific drugs, identifying prognostic indicators, and developing targeted drugs. We believe that this approach is highly personalized. Utilizing a detailed understanding of m6A levels and each patient’s immune status will be the basis for the next step in immunotherapy. In addition, m6A status, the TME-infiltration characteristics, and the immune system can change over time, especially during treatment. Therefore, continuous monitoring of m6A regulators and immune markers is essential for developing and adjusting treatment regimens.
Both DNA and histone methylation inhibitors have been shown to enhance the efficacy of immunotherapy. Therefore, the mechanism by which m6A modification interacts with DNA and histone epigenetics to regulate gene expression and whether there is a potential association between m6A modification and other types of methylation remains unclear. In addition, based on the theories and mechanisms obtained from these studies, we explored whether m6A inhibitors combined with other epigenetic drugs can synergistically promote immunotherapy. Moreover, dual inhibitors targeting both m6A regulators and immune checkpoints may achieve better efficacy and fewer side effects.
With the continuous development of this field, it will be necessary to continue to study the mechanism of m6A modifications leading to TIE as a theoretical basis and simultaneously accelerate clinical trials to translate the theories obtained. In this way, the outcomes of patients with cancer can be further improved.
Availability of data and materials
Not applicable.
Abbreviations
- 3′ UTR:
-
3′ untranslated region
- 5′ UTR:
-
5′ untranslated region
- ALKBH5:
-
Alkb homolog 5
- ALDO:
-
Aldolase
- AML:
-
Acute myeloid leukemia
- APC:
-
Antigen-presenting cell
- APM:
-
Antigen processing and presenting machinery
- BC:
-
Breast cancer
- CC:
-
Cervical cancer
- CLIP:
-
Cross-linking and immunoprecipitation
- COX2:
-
Cyclooxygenase 2
- CRC:
-
Colorectal cancer
- CTL:
-
Cytotoxic T lymphocyte
- CTLA4:
-
Cytotoxic T lymphocyte antigen 4
- CSC:
-
Cancer stem cell
- DC:
-
Dendritic cell
- DFS:
-
Disease-free survival
- EC:
-
Esophageal cancer
- EMT:
-
Epithelial-mesenchymal transition
- Eno:
-
Enolase
- FTO:
-
Fat mass and obesity-associated protein
- FTOi:
-
FTO inhibitor
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GLUT:
-
Glucose transporter
- GPI:
-
Glucose phosphate isomerase
- GSC:
-
Glioblastoma stem cell
- HCC:
-
Hepatocellular carcinoma
- HIF-1α:
-
Hypoxia-inducible factor-1α
- HK:
-
Hexokinase
- HLA-I:
-
Class I human leukocyte antigens
- HSC:
-
Hematopoietic stem cell
- ICI:
-
Immune checkpoint inhibition
- icSHAPE:
-
In vivo click selective 2-hydroxyl acylation and profiling experiment
- IDO:
-
Indoleamine 2,3-dioxygenase
- IGF2BP:
-
Insulin-like growth factor 2 mRNA binding protein
- IL-10:
-
Interleukin-10
- JMJD2:
-
Jumonji domain containing 2
- KH:
-
K-homology
- LC:
-
Lung cancer
- LDH:
-
Lactate dehydrogenase
- LSC:
-
Leukemia stem cell
- m6A:
-
N 6-methyladenosine
- m6ACE-seq:
-
m6A-Crosslinking-Exonuclease-sequencing
- m6Am:
-
N 6,2′-O-dimethyladenosine
- MA:
-
Meclofenamic acid
- MDSC:
-
Myeloid-derived suppressor cell
- MeRIP-seq:
-
Methylated RNA immunoprecipitation sequencing
- mESC:
-
Mouse embryonic stem cell
- METTL3:
-
Methyltransferase-like protein 3
- MHC:
-
Major histocompatibility complex
- miCLIP:
-
m6A individual-nucleotide-resolution cross-linking and immunoprecipitation
- MLPCN:
-
Molecular Libraries Probe Development Center Network
- MTC:
-
Methyltransferase complex
- NRL:
-
Nucleotide recognition lid
- OS:
-
Overall survival
- PC:
-
Pancreatic cancer
- PCIF1:
-
Phosphorylated CTD interacting factor 1
- PDH:
-
Pyruvate dehydrogenase
- PD-L1:
-
Programmed death ligand 1
- PD-1:
-
Programmed cell death protein 1
- PGE2 :
-
Prostaglandin E2
- PFK:
-
Phosphofructokinase
- PGK:
-
Phosphoglycerate kinase
- PGM:
-
Phosphoglycerate mutase
- PK:
-
Pyruvate kinase
- RBP:
-
RNA binding protein
- RRM:
-
RNA-recognition motif
- SAM:
-
S-adenosyl methionine
- tAg:
-
tumor antigen
- TAM:
-
Tumor-associated macrophages
- TCR:
-
T-cell receptor
- TF:
-
Transcription factor
- Th1:
-
Type 1 helper T cell
- TIE:
-
Tumor immune escape
- TIGIT:
-
T cell immunoreceptor with immunoglobulin and ITIM domain
- TIM3:
-
T-cell immunoglobulin and mucin-domain containing 3
- TME:
-
Tumor microenvironment
- TNF:
-
Tumor necrosis factor
- Treg:
-
CD4+ regulatory T cell
- tRNA:
-
Transfer RNA
- WTAP:
-
Wilms tumor 1-associated protein
- ZCCHC4:
-
Zinc finger CCHC-type containing 4
References
Dunn G, Bruce A, Ikeda H, Old L, Schreiber R. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8.
Waldman A, Fritz J, Lenardo M. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–68.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–5.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–46.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–72.
Kretschmer J, Rao H, Hackert P, Sloan KE, Höbartner C, Bohnsack MT. The m(6)A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′-3′ exoribonuclease XRN1. Rna. 2018;24:1339–50.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–5.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5′ UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–8.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Satterwhite ER, Mansfield KD. RNA methyltransferase METTL16: targets and function. Wiley Interdiscip Rev RNA. 2022;13:e1681.
Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15:88–94.
van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47:7719–33.
Richard EM, Polla DL, Assir MZ, Contreras M, Shahzad M, Khan AA, et al. Bi-allelic variants in METTL5 cause autosomal-recessive intellectual disability and microcephaly. Am J Hum Genet. 2019;105:869–78.
Akichika S, Hirano S, Shichino Y, Suzuki T, Nishimasu H, Ishitani R, et al. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science. 2019;363:eaav0080.
Boulias K, Toczydłowska-Socha D, Hawley BR, Liberman N, Takashima K, Zaccara S, et al. Identification of the m(6)Am methyltransferase PCIF1 reveals the location and functions of m(6)Am in the transcriptome. Mol Cell. 2019;75:631–643.e638.
Sun H, Zhang M, Li K, Bai D, Yi C. Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res. 2019;29:80–2.
Koh CWQ, Goh YT, Goh WSS. Atlas of quantitative single-base-resolution N(6)-methyl-adenine methylomes. Nat Commun. 2019;10:5636.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K, et al. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. 2017;7:42271.
Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–72.
Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m(6)A(m) in the 5′ cap controls mRNA stability. Nature. 2017;541:371–5.
Sommer S, Lavi U, Darnell JE Jr. The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J Mol Biol. 1978;124:487–99.
Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31:990–1006.
Darnell RB, Ke S, Darnell JE Jr. Pre-mRNA processing includes N(6) methylation of adenosine residues that are retained in mRNA exons and the fallacy of "RNA epigenetics". Rna. 2018;24:262–7.
Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, Vanacova S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3′-end processing. Nucleic Acids Res. 2017;45:11356–70.
Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, et al. Structure of human RNA N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014;42:4741–54.
Feng C, Liu Y, Wang G, Deng Z, Zhang Q, Wu W, et al. Crystal structures of the human RNA demethylase Alkbh5 reveal basis for substrate recognition. J Biol Chem. 2014;289:11571–83.
Xu C, Liu K, Ahmed H, Loppnau P, Schapira M, Min J. Structural basis for the discriminative recognition of N6-Methyladenosine RNA by the human YT521-B homology domain family of proteins. J Biol Chem. 2015;290:24902–13.
Zou S, Toh JD, Wong KH, Gao YG, Hong W, Woon EC. N(6)-Methyladenosine: a conformational marker that regulates the substrate specificity of human demethylases FTO and ALKBH5. Sci Rep. 2016;6:25677.
Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Brönneke HS, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16:1042–8.
Wei C, Gershowitz A, Moss B. N6, O2'-dimethyladenosine a novel methylated ribonucleoside next to the 5′ terminal of animal cell and virus mRNAs. Nature. 1975;257:251–3.
Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol. 2019;15:340–7.
Garcia-Campos MA, Edelheit S, Toth U, Safra M, Shachar R, Viukov S, et al. Deciphering the “m(6)A code” via antibody-independent quantitative profiling. Cell. 2019;178:731–747.e716.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38:79–96.e11.
Zhang Z, Theler D, Kaminska KH, Hiller M, de la Grange P, Pudimat R, et al. The YTH domain is a novel RNA binding domain. J Biol Chem. 2010;285:14701–10.
Theler D, Dominguez C, Blatter M, Boudet J, Allain FH. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014;42:13911–9.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017;6:e31311.
Jain D, Puno MR, Meydan C, Lailler N, Mason CE, Lima CD, et al. ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. Elife. 2018;7:e30919.
Bailey AS, Batista PJ, Gold RS, Chen YG, de Rooij DG, Chang HY, et al. The conserved RNA helicase YTHDC2 regulates the transition from proliferation to differentiation in the germline. Elife. 2017;6:e26116.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–27.
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m(6)A transcripts by the 3′→5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68:374–387.e312.
Saito Y, Hawley BR, Puno MR, Sarathy SN, Lima CD, Jaffrey SR, et al. YTHDC2 control of gametogenesis requires helicase activity but not m(6)A binding. Genes Dev. 2022;36:180–94.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444–7.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Zaccara S, Jaffrey SR. A unified model for the function of YTHDF proteins in regulating m(6)A-modified mRNA. Cell. 2020;181:1582–1595.e1518.
Patil DP, Pickering BF, Jaffrey SR. Reading m(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. 2018;28:113–27.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24.
Birkaya B, Ortt K, Sinha S. Novel in vivo targets of DeltaNp63 in keratinocytes identified by a modified chromatin immunoprecipitation approach. BMC Mol Biol. 2007;8:43.
Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, et al. m(6)A enhances the phase separation potential of mRNA. Nature. 2019;571:424–8.
Merkurjev D, Hong WT, Iida K, Oomoto I, Goldie BJ, Yamaguti H, et al. Synaptic N(6)-methyladenosine (m(6)A) epitranscriptome reveals functional partitioning of localized transcripts. Nat Neurosci. 2018;21:1004–14.
Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera-Castrejon A, Schneir D, et al. Context-dependent functional compensation between Ythdf m(6)A reader proteins. Genes Dev. 2020;34:1373–91.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4.
Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. 2018;9:420.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051–63.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162:1299–308.
Chao JA, Patskovsky Y, Patel V, Levy M, Almo SC, Singer RH. ZBP1 recognition of beta-actin zipcode induces RNA looping. Genes Dev. 2010;24:148–58.
Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, et al. RNA structure maps across mammalian cellular compartments. Nat Struct Mol Biol. 2019;26:322–30.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
Leone P, Shin E, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105:1172–87.
Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 1898;2019:10.
Spranger S, Bao R, Gajewski T. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5.
Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9:1124–41.
Liu X, Su K, Sun X, Jiang Y, Wang L, Hu C, et al. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/β-catenin pathway. J Exp Clin Cancer Res. 2021;40:132.
Xu J, Wan Z, Tang M, Lin Z, Jiang S, Ji L, et al. N(6)-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating β-catenin signaling. Mol Cancer. 2020;19:163.
Liu L, Wang J, Sun G, Wu Q, Ma J, Zhang X, et al. m(6)A mRNA methylation regulates CTNNB1 to promote the proliferation of hepatoblastoma. Mol Cancer. 2019;18:188.
Cui X, Wang Z, Li J, Zhu J, Ren Z, Zhang D, et al. Cross talk between RNA N6-methyladenosine methyltransferase-like 3 and miR-186 regulates hepatoblastoma progression through Wnt/β-catenin signalling pathway. Cell Prolif. 2020;53:e12768.
Wu H, Xu H, Jia D, Li T, Xia L. METTL3-induced UCK2 m(6)A hypermethylation promotes melanoma cancer cell metastasis via the WNT/β-catenin pathway. Ann Transl Med. 2021;9:1155.
Liu ZF, Yang J, Wei SP, Luo XG, Jiang QS, Chen T, et al. Upregulated METTL3 in nasopharyngeal carcinoma enhances the motility of cancer cells. Kaohsiung J Med Sci. 2020;36:895–903.
Wang W, Shao F, Yang X, Wang J, Zhu R, Yang Y, et al. METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N(6)-methyladenosine-dependent YTHDF binding. Nat Commun. 2021;12:3803.
Miao W, Chen J, Jia L, Ma J, Song D. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem Biophys Res Commun. 2019;516:719–25.
Zhang R, Li SW, Liu L, Yang J, Huang G, Sang Y. TRIM11 facilitates chemoresistance in nasopharyngeal carcinoma by activating the β-catenin/ABCC9 axis via p62-selective autophagic degradation of Daple. Oncogenesis. 2020;9:45.
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–7.
Liu B, Zhou J, Wang C, Chi Y, Wei Q, Fu Z, et al. LncRNA SOX2OT promotes temozolomide resistance by elevating SOX2 expression via ALKBH5-mediated epigenetic regulation in glioblastoma. Cell Death Dis. 2020;11:384.
Pi J, Wang W, Ji M, Wang X, Wei X, Jin J, et al. YTHDF1 promotes gastric carcinogenesis by controlling translation of FZD7. Cancer Res. 2021;81:2651–65.
Han B, Yan S, Wei S, Xiang J, Liu K, Chen Z, et al. YTHDF1-mediated translation amplifies Wnt-driven intestinal stemness. EMBO Rep. 2020;21:e49229.
Liu X, Qin J, Gao T, Li C, He B, Pan B, et al. YTHDF1 facilitates the progression of hepatocellular carcinoma by promoting FZD5 mRNA translation in an m6A-dependent manner. Mol Ther Nucleic Acids. 2020;22:750–65.
Chen MH, Fu LS, Zhang F, Yang Y, Wu XZ. LncAY controls BMI1 expression and activates BMI1/Wnt/β-catenin signaling axis in hepatocellular carcinoma. Life Sci. 2021;280:119748.
Li Y, Sheng H, Ma F, Wu Q, Huang J, Chen Q, et al. RNA m(6)A reader YTHDF2 facilitates lung adenocarcinoma cell proliferation and metastasis by targeting the AXIN1/Wnt/β-catenin signaling. Cell Death Dis. 2021;12:479.
Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257–70.
Bonavita E, Bromley CP, Jonsson G, Pelly VS, Sahoo S, Walwyn-Brown K, et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity. 2020;53:1215–1229.e1218.
Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. 2018;24:1178–91.
Xuan Y, Wang J, Ban L, Lu JJ, Yi C, Li Z, et al. hnRNPA2/B1 activates cyclooxygenase-2 and promotes tumor growth in human lung cancers. Mol Oncol. 2016;10:610–24.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270–4.
Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell. 2011;147:1355–68.
Samie M, Cresswell P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat Immunol. 2015;16:729–36.
Shen S, Yan J, Zhang Y, Dong Z, Xing J, He Y. N6-methyladenosine (m6A)-mediated messenger RNA signatures and the tumor immune microenvironment can predict the prognosis of hepatocellular carcinoma. Ann Transl Med. 2021;9:59.
Tang R, Zhang Y, Liang C, Xu J, Meng Q, Hua J, et al. The role of m6A-related genes in the prognosis and immune microenvironment of pancreatic adenocarcinoma. PeerJ. 2020;8:e9602.
Zhao H, Xu Y, Xie Y, Zhang L, Gao M, Li S, et al. m6A regulators is differently expressed and correlated with immune response of esophageal cancer. Front Cell Dev Biol. 2021;9:650023.
He X, Tan L, Ni J, Shen G. Expression pattern of m(6)A regulators is significantly correlated with malignancy and antitumor immune response of breast cancer. Cancer Gene Ther. 2021;28:188–96.
Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307.
Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32.
Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–42.
Tong J, Cao G, Zhang T, Sefik E, Amezcua Vesely MC, Broughton JP, et al. m(6)A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018;28:253–6.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21:485–98.
Grover A, Sanseviero E, Timosenko E, Gabrilovich DI. Myeloid-derived suppressor cells: a propitious road to clinic. Cancer Discov. 2021;11:2693–706.
Ni HH, Zhang L, Huang H, Dai SQ, Li J. Connecting METTL3 and intratumoural CD33(+) MDSCs in predicting clinical outcome in cervical cancer. J Transl Med. 2020;18:393.
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–37.
Gu X, Zhang Y, Li D, Cai H, Cai L, Xu Q. N6-methyladenosine demethylase FTO promotes M1 and M2 macrophage activation. Cell Signal. 2020;69:109553.
Wang X, Ji Y, Feng P, Liu R, Li G, Zheng J, et al. The m6A reader IGF2BP2 regulates macrophage phenotypic activation and inflammatory diseases by stabilizing TSC1 and PPARγ. Adv Sci (Weinh). 2021;8:2100209.
Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X, et al. ALKBH5 facilitates hypoxia-induced paraspeckle assembly and IL8 secretion to generate an immunosuppressive tumor microenvironment. Cancer Res. 2021;81:5876–88.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, et al. The next decade of immune checkpoint therapy. Cancer Discov. 2021;11:838–57.
Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis V. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5:ra46.
Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel). 2020;12:738.
Ai Y, Liu S, Luo H, Wu S, Wei H, Tang Z, et al. METTL3 intensifies the progress of oral squamous cell carcinoma via modulating the m6A amount of PRMT5 and PD-L1. J Immunol Res. 2021;2021:6149558.
Tsuruta N, Tsuchihashi K, Ohmura H, Yamaguchi K, Ito M, Ariyama H, et al. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem Biophys Res Commun. 2020;530:235–9.
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M(6)A demethylase ALKBH5 regulates PD-L1 expression and tumor Immunoenvironment in intrahepatic cholangiocarcinoma. Cancer Res. 2021;81:4778–93.
Wang L, Zhang S, Li H, Xu Y, Wu Q, Shen J, et al. Quantification of m6A RNA methylation modulators pattern was a potential biomarker for prognosis and associated with tumor immune microenvironment of pancreatic adenocarcinoma. BMC Cancer. 2021;21:876.
Lin X, Wang Z, Yang G, Wen G, Zhang H. YTHDF2 correlates with tumor immune infiltrates in lower-grade glioma. Aging (Albany NY). 2020;12:18476–500.
Koppenol W, Bounds P, Dang C. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37.
Doherty J, Cleveland J. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685–92.
Choi SY, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol. 2013;230:350–5.
Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159–70.
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. 2020;19:72.
Chen H, Gao S, Liu W, Wong CC, Wu J, Wu J, et al. RNA N(6)-Methyladenosine methyltransferase METTL3 facilitates colorectal cancer by activating the m(6)A-GLUT1-mTORC1 Axis and is a therapeutic target. Gastroenterology. 2021;160:1284–1300.e1216.
Huang S, Wu Z, Cheng Y, Wei W, Hao L. Insulin-like growth factor 2 mRNA binding protein 2 promotes aerobic glycolysis and cell proliferation in pancreatic ductal adenocarcinoma via stabilizing GLUT1 mRNA. Acta Biochim Biophys Sin Shanghai. 2019;51:743–52.
Brandi J, Cecconi D, Cordani M, Torrens-Mas M, Pacchiana R, Dalla Pozza E, et al. The antioxidant uncoupling protein 2 stimulates hnRNPA2/B1, GLUT1 and PKM2 expression and sensitizes pancreas cancer cells to glycolysis inhibition. Free Radic Biol Med. 2016;101:305–16.
Wang Q, Guo X, Li L, Gao Z, Su X, Ji M, et al. N(6)-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell Death Dis. 2020;11:911.
Han H, Fan G, Song S, Jiang Y, Qian C, Zhang W, et al. piRNA-30473 contributes to tumorigenesis and poor prognosis by regulating m6A RNA methylation in DLBCL. Blood. 2021;137:1603–14.
Yu H, Zhao K, Zeng H, Li Z, Chen K, Zhang Z, et al. N(6)-methyladenosine (m(6)A) methyltransferase WTAP accelerates the Warburg effect of gastric cancer through regulating HK2 stability. Biomed Pharmacother. 2021;133:111075.
Liu H, Qin S, Liu C, Jiang L, Li C, Yang J, et al. m(6)A reader IGF2BP2-stabilized CASC9 accelerates glioblastoma aerobic glycolysis by enhancing HK2 mRNA stability. Cell Death Dis. 2021;7:292.
Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell. 2021;81:922–939.e929.
Li Z, Peng Y, Li J, Chen Z, Chen F, Tu J, et al. N(6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat Commun. 2020;11:2578.
Jia G, Wang Y, Lin C, Lai S, Dai H, Wang Z, et al. LNCAROD enhances hepatocellular carcinoma malignancy by activating glycolysis through induction of pyruvate kinase isoform PKM2. J Exp Clin Cancer Res. 2021;40:299.
Yang N, Wang T, Li Q, Han F, Wang Z, Zhu R, et al. HBXIP drives metabolic reprogramming in hepatocellular carcinoma cells via METTL3-mediated m6A modification of HIF-1α. J Cell Physiol. 2021;236:3863–80.
Tanabe A, Tanikawa K, Tsunetomi M, Takai K, Ikeda H, Konno J, et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett. 2016;376:34–42.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–33.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–34.
Li F, Yi Y, Miao Y, Long W, Long T, Chen S, et al. N(6)-Methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79:5785–98.
Visvanathan A, Patil V, Abdulla S, Hoheisel JD, Somasundaram K. N6-Methyladenosine landscape of glioma stem-like cells: METTL3 is essential for the expression of actively transcribed genes and sustenance of the oncogenic signaling. Genes (Basel). 2019;10:141.
Gao Q, Zheng J, Ni Z, Sun P, Yang C, Cheng M, et al. The m(6)A methylation-regulated AFF4 promotes self-renewal of bladder cancer stem cells. Stem Cells Int. 2020;2020:8849218.
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. 2019;38:3667–80.
Zhou R, Gao Y, Lv D, Wang C, Wang D, Li Q. METTL3 mediated m(6)A modification plays an oncogenic role in cutaneous squamous cell carcinoma by regulating ΔNp63. Biochem Biophys Res Commun. 2019;515:310–7.
Zhang Y, Kang M, Zhang B, Meng F, Song J, Kaneko H, et al. m(6)A modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18:185.
Yin H, Chen L, Piao S, Wang Y, Li Z, Lin Y, et al. M6A RNA methylation-mediated RMRP stability renders proliferation and progression of non-small cell lung cancer through regulating TGFBR1/SMAD2/SMAD3 pathway. Cell Death Differ. 2021.
Shi Y, Dou Y, Zhang J, Qi J, Xin Z, Zhang M, et al. The RNA N6-Methyladenosine methyltransferase METTL3 promotes the progression of kidney cancer via N6-Methyladenosine-dependent translational enhancement of ABCD1. Front Cell Dev Biol. 2021;9:737498.
Xu T, Zhang W, Chai L, Liu C, Zhang S, Xu T. Methyltransferase-like 3-induced N6-methyladenosine upregulation promotes oral squamous cell carcinoma by through p38. Oral Dis. 2021;00:1–10.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22:191–205.e199.
Liu Z, Wu K, Gu S, Wang W, Xie S, Lu T, et al. A methyltransferase-like 14/miR-99a-5p/tribble 2 positive feedback circuit promotes cancer stem cell persistence and radioresistance via histone deacetylase 2-mediated epigenetic modulation in esophageal squamous cell carcinoma. Clin Transl Med. 2021;11:e545.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.e596.
Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527–42.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–56.
Hao L, Wang JM, Liu BQ, Yan J, Li C, Jiang JY, et al. m6A-YTHDF1-mediated TRIM29 upregulation facilitates the stem cell-like phenotype of cisplatin-resistant ovarian cancer cells. Biochim Biophys Acta Mol Cell Res. 2021;1868:118878.
Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discov. 2021;11:480–99.
Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25:137–148.e136.
Zhang C, Huang S, Zhuang H, Ruan S, Zhou Z, Huang K, et al. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene. 2020;39:4507–18.
Zhu P, He F, Hou Y, Tu G, Li Q, Jin T, et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40:1609–27.
Yang Y, Wu J, Liu F, He J, Wu F, Chen J, et al. IGF2BP1 promotes the liver cancer stem cell phenotype by regulating MGAT5 mRNA stability via m6A RNA methylation. Stem Cells Dev. 2021;30:1115–25.
Hu X, Peng WX, Zhou H, Jiang J, Zhou X, Huang D, et al. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ. 2020;27:1782–94.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18:142.
Zhao C, Ling X, Xia Y, Yan B, Guan Q. The m6A methyltransferase METTL3 controls epithelial-mesenchymal transition, migration and invasion of breast cancer through the MALAT1/miR-26b/HMGA2 axis. Cancer Cell Int. 2021;21:441.
Liang X, Zhang Z, Wang L, Zhang S, Ren L, Li S, et al. Mechanism of methyltransferase like 3 in epithelial-mesenchymal transition process, invasion, and metastasis in esophageal cancer. Bioengineered. 2021;12:10023–36.
Hou P, Meng S, Li M, Lin T, Chu S, Li Z, et al. LINC00460/DHX9/IGF2BP2 complex promotes colorectal cancer proliferation and metastasis by mediating HMGA1 mRNA stability depending on m6A modification. J Exp Clin Cancer Res. 2021;40:52.
Chen C, Yuan W, Zhou Q, Shao B, Guo Y, Wang W, et al. N6-methyladenosine-induced circ1662 promotes metastasis of colorectal cancer by accelerating YAP1 nuclear localization. Theranostics. 2021;11:4298–315.
Wang H, Deng Q, Lv Z, Ling Y, Hou X, Chen Z, et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol Cancer. 2019;18:181.
Ni XF, Xie QQ, Zhao JM, Xu YJ, Ji M, Hu WW, et al. The hepatic microenvironment promotes lung adenocarcinoma cell proliferation, metastasis, and epithelial-mesenchymal transition via METTL3-mediated N6-methyladenosine modification of YAP1. Aging (Albany NY). 2021;13:4357–69.
Cheng C, Wu Y, Xiao T, Xue J, Sun J, Xia H, et al. METTL3-mediated m(6)A modification of ZBTB4 mRNA is involved in the smoking-induced EMT in cancer of the lung. Mol Ther Nucleic Acids. 2021;23:487–500.
Rong D, Wu F, Lu C, Sun G, Shi X, Chen X, et al. m6A modification of circHPS5 and hepatocellular carcinoma progression through HMGA2 expression. Mol Ther Nucleic Acids. 2021;26:637–48.
Xu H, Wang H, Zhao W, Fu S, Li Y, Ni W, et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics. 2020;10:5671–86.
Li E, Wei B, Wang X, Kang R. METTL3 enhances cell adhesion through stabilizing integrin β1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am J Cancer Res. 2020;10:1012–25.
Wang D, Qu X, Lu W, Wang Y, Jin Y, Hou K, et al. N(6)-Methyladenosine RNA demethylase FTO promotes gastric cancer metastasis by down-regulating the m6A methylation of ITGB1. Front Oncol. 2021;11:681280.
Hao L, Yin J, Yang H, Li C, Zhu L, Liu L, et al. ALKBH5-mediated m(6)A demethylation of FOXM1 mRNA promotes progression of uveal melanoma. Aging (Albany NY). 2021;13:4045–62.
Tan L, Tang Y, Li H, Li P, Ye Y, Cen J, et al. N6-Methyladenosine modification of LncRNA DUXAP9 promotes renal cancer cells proliferation and motility by activating the PI3K/AKT signaling pathway. Front Oncol. 2021;11:641833.
Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–14.
Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2013;32:1638–50.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22.
Maccalli C, Rasul KI, Elawad M, Ferrone S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol. 2018;53:189–200.
Li Z, Qian P, Shao W, Shi H, He X, Gogol M, et al. Suppression of mA reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018;28:904–17.
Dongre A, Weinberg R. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69–84.
Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195–206.
Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017;77:3982–9.
Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat Commun. 2019;10:2065.
Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m(6)A methylation in cancer. Cancer Res. 2019;79:1285–92.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–71.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46.
Shen Z, Zhu B, Li J, Qin L. Rhein augments antiproliferative effects of atezolizumab based on breast cancer (4T1) regression. Planta Med. 2019;85:1143–9.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84.
Xiao L, Li X, Mu Z, Zhou J, Zhou P, Xie C, et al. FTO inhibition enhances the antitumor effect of temozolomide by targeting MYC-miR-155/23a cluster-MXI1 feedback circuit in glioma. Cancer Res. 2020;80:3945–58.
Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS One. 2016;11:e0159072.
Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME, et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Nerosci. 2014;5:658–65.
Cockova Z, Honc O, Telensky P, Olsen MJ, Novotny J. Streptozotocin-induced astrocyte mitochondrial dysfunction is ameliorated by FTO inhibitor MO-I-500. ACS Chem Nerosci. 2021;12:3818–28.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35:677–691.e610.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90–105.e123.
Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10:818–27.
Müller S, Bley N, Busch B, Glaß M, Lederer M, Misiak C, et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020;48:8576–90.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601.
He ZH, He MF, Ma SC, But PP. Anti-angiogenic effects of rhubarb and its anthraquinone derivatives. J Ethnopharmacol. 2009;121:313–7.
Gao Q, Qin WS, Jia ZH, Zheng JM, Zeng CH, Li LS, et al. Rhein improves renal lesion and ameliorates dyslipidemia in db/db mice with diabetic nephropathy. Planta Med. 2010;76:27–33.
Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394.
Dong L, Chen C, Zhang Y, Guo P, Wang Z, Li J, et al. The loss of RNA N(6)-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8(+) T cell dysfunction and tumor growth. Cancer Cell. 2021;39:945–957.e910.
Acknowledgements
Not applicable.
Funding
This work was supported by funding from the Project Nn10 of Harbin Medical University Cancer Hospital (Grant Number Nn102017-02), the National Natural Science Foundation of China (Grant Number 81972706, 82173235, 82072904, 81872149), Outstanding Youth Project of Heilongjiang Provincial Natural Science Foundation (Grant Number YQ2019H027), Distinguished Young Scholars of Harbin Medical University Cancer Hospital (Grant Number JCQN2018-03), Yong Elite Training Foundation Grant of Harbin Medical University Cancer Hospital (Grant Number JY2016-02) and Heilongjiang Health and Family Planning Commission Foundation (Grant Numbers 2019-056).
Author information
Authors and Affiliations
Contributions
D.P., S.X., and W.L. conceived the idea, design the study; W.L. retrieved and analyzed the data, and drafted the manuscript; W.L., Y.H. and X.Z. revised and polished the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, W., Hao, Y., Zhang, X. et al. Targeting RNA N6-methyladenosine modification: a precise weapon in overcoming tumor immune escape. Mol Cancer 21, 176 (2022). https://doi.org/10.1186/s12943-022-01652-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01652-3