
Abstract
N6-methyladenosine (m6A) is the most abundant epigenetic modification of RNA, and its dysregulation drives aberrant transcription and translation programs that promote cancer occurrence and progression. Although defective gene regulation resulting from m6A often affects oncogenic and tumor-suppressing networks, m6A can also modulate tumor immunogenicity and immune cells involved in anti-tumor responses. Understanding this counterintuitive concept can aid the design of new drugs that target m6A to potentially improve the outcomes of cancer immunotherapies. Here, we provide an up-to-date and comprehensive overview of how m6A modifications intrinsically affect immune cells and how alterations in tumor cell m6A modifications extrinsically affect immune cell responses in the tumor microenvironment (TME). We also review strategies for modulating endogenous anti-tumor immunity and discuss the challenge of reshaping the TME. Strategies include: combining specific and efficient inhibitors against m6A regulators with immune checkpoint blockers; generating an effective programmable m6A gene-editing system that enables efficient manipulation of individual m6A sites; establishing an effective m6A modification system to enhance anti-tumor immune responses in T cells or natural killer cells; and using nanoparticles that specifically target tumor-associated macrophages (TAMs) to deliver messenger RNA or small interfering RNA of m6A-related molecules that repolarize TAMs, enabling them to remodel the TME. The goal of this review is to help the field understand how m6A modifications intrinsically and extrinsically shape immune responses in the TME so that better cancer immunotherapy can be designed and developed.
Similar content being viewed by others

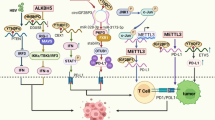
Background
Recent studies of RNA modifications have revealed that RNA is not simply an intermediary between DNA and protein or an effector molecule [like ribosomal RNA (rRNA) and transfer RNA (tRNA)] but that it also plays significant roles in post-transcriptional gene regulation [1]. To date, more than 100 types of chemical modification have been identified in cellular RNAs, including N1-methyladenosine (m1A), N6-methyladenosine (m6A), 5-methylcytosine (m5C), N7-methylguanosine (m7G), RNA cap methylations, pseudouridine, and uridylation [2]. Among these, m6A, which forms when adenosine is methylated at the nitrogen-6 position, is the most abundant and conserved internal RNA modification [1]. More recently, the identification of methyltransferase, demethylase, and binding proteins that install, remove or recognize m6A has revealed unappreciated roles of m6A in almost every aspect of RNA metabolism as well as in various physiological and pathological processes [3, 4].
Recent studies have revealed that the m6A modification modulated immune cell activation and infiltration into the tumor microenvironment (TME) and thus may affect the efficacy of immunotherapy. Therefore, the m6A modification is a potential target for cancer immunotherapy that could perhaps complement immune checkpoint inhibitor therapies and chimeric antigen receptor (CAR) T cell therapy, which have dramatically improved the survival and quality of life for cancer patients [5, 6]. Here, we provide an up-to-date and comprehensive overview of the m6A modification in immune cells and associated anti-tumor immune responses in the TME. Additionally, we discuss the potential therapeutic value of targeting m6A regulators for cancer immunotherapy.
Regulation of the m6A modification
The RNA m6A modification process is dynamically and reversibly regulated by three types of enzymes: m6A methyltransferases (“writers”), m6A demethylases (“erasers”), and m6A binding proteins (“readers”) [7]. Together, these enzymes ensure normal expression and translation of RNAs [8, 9]. The regulation and functions of the m6A regulators are summarized in Fig. 1.

Overview of the m6A modification in the mRNA life cycle. ①The m6A methylation complex, which consists of the core methyltransferase-like protein 3 (METTL3) and its adaptors (writer), adds m6A onto target RNAs in the nucleus. ②The two main demethylases (eraser), fat mass and obesity-associated protein (FTO) and alkB homolog 5, RNA demethylase (ALKBH5), erase the methylation modification in the nucleus. ③ m6A is recognized by diverse readers, such as YTHDC1, HNRNPC/G, and HNRNPA2B1, that mediate various posttranscriptional processes including RNA splicing and miRNA processing in the nucleus. ④ In the cytoplasm, m6A binds to different specific reader proteins, such as IGF2BPs, PRRC2A, YTHDF1/2/3, and YTHDC2, that mediate the stability, translation, and decay of the mRNA. Figure created with BioRender.com
Installation of m6A is catalyzed by a big writer complex, which consists of methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), RNA binding motif protein 15 (RBM15), wlms tumor 1 associated protein (WTAP), vir-like m6A methyltransferase associated (VIRMA) subunits, and zinc finger CCCH domain-containing protein 13 (ZC3H13) [10]. METTL3 is the predominant catalytic subunit of the writer complex, but its catalytic activity is enhanced by METTL14, an allosteric activator [11, 12]. WTAP, a regulatory component of the complex, recruits the METTL3/METTL14 complex to form a catalytic core on a target RNA [13]. RBM15, an interacting partner of WTAP, is an adapter protein that recruits the m6A writer complex to U-rich regions of mRNA [14]. VIRMA, another important subunit of the writer complex, can situate the m6A modification in the 3′UTR near stop codon [15]. ZC3H13 controls nuclear m6A methylation by combining with other cofactors, such as WTAP and RBM15 [16]. This writer complex usually installs the m6A modification on a specific and consensus RNA sequence—RRACH (R = G or A; H = U, A or C)—that is often found in 3′ untranslated regions (UTRs), long exons, and near stop codons [17, 18].
Erasers remove the m6A decoration from RNA by removing the adenosine [19]. The two main erasers are fat mass and obesity-associated protein (FTO) and alpha-ketoglutarate-dependent dioxygenase alkB homologue 5 (ALKBH5) [20, 21]. It is now clear that erasers can have different biological effects depending on which specific target RNAs they demethylate. However, what leads to this selectivity is still unknown and needs to be addressed.
A number of different readers recognize the m6A decorations of target genes. According to their location, readers can be further divided into nuclear readers and cytoplasmic readers. The nuclear readers, which consist of YTH domain containing 1–2 (YTHDC1, YTHDC2), members of the heterogeneous nuclear ribonuclease (HNRNP) family (HNRNPA2B1, HNRNPC, HNRNPG), and fragile x mental retardation protein (FMRP), play multiple roles in modulating mRNA splicing, epigenetic silencing, nuclear export of mRNA, regulation of non-coding RNA, and RNA structure switching [22,23,24,25]. The cytoplasmic readers affect mRNA stability, translation, and degradation, and they include YTHDC2, YTH domain family 1–3 (YTHDF1, YTHDF2, YTHDF3), insulin-like factor-2 mRNA-binding proteins (IGF2BPs), and proline-rich coiled-coil 2A (PRRC2A) [26,27,28].
Role of the m6A modification in immune cells
It is widely known that intrinsic m6A modification regulates tumor cell fate by targeting specific genes in different cancers [29, 30]. However, few studies have focused on how m6A modification regulates anti-tumor functions of immune cells. Here, we summarize recent findings.
Natural killer cells
Natural killer (NK) cells are innate lymphoid immune cells with important roles in cancer immune surveillance due to their ability to directly recognize and kill cancer cells [31, 32]. Our group first reported the multifaceted roles of YTHDF2-mediated m6A methylation in NK cell immunity [33]. This important m6A reader is critical for maintaining NK cell homeostasis, maturation, interleukin (IL)-15-mediated survival, and antitumor and antiviral activity because it regulates several downstream target genes, including signal transducer and activator of transcription 5 (STAT5), Eemesodermin (Eomes), and Tardbp [33]. Subsequently, Chen et al. observed that mRNA levels of the m6A writer METTL3 were decreased in tumor-infiltrating NK cells of certain cancer patients. Using mouse models, they also showed that deleting METTL3 reduced NK cell hyporesponsiveness to IL-15, promoted tumor progression and metastasis by targeting SH2 domain-containing protein tyrosine phosphatase-2 (SHP-2) [34]. As both the m6A writer METTL3 and the reader YTHDF2 positively regulate the anti-tumor immunity of NK cells, METTL3- and YTHDF2-mediated m6A methylation might be important regulators of anti-tumor immunity and homeostasis of NK cells (Fig. 2A) [34]. However, the effector functions and regulatory mechanisms of other m6A regulators on NK cells remain to be determined.

Mechanisms that regulate the m6A modification in immune cells. A METTL3 (a writer) and YTHDF2 (a reader) positively regulate the survival and anti-tumor immunity of NK cells by respectively targeting Ptpn11, STAT5, or Tardbp. B METTL3 in macrophages promotes the production of proinflammatory cytokines such as TNF-α and IL-6 by targeting Irakm (an inhibitor of TLR4) or Spred2 (an inhibitor of the ERK pathway), thereby reshaping the TME and inhibiting tumor progression. METTL14-mediated m6A methylation boosts the degradation of Ebi3 in macrophages, which promotes CD8+ T cell activation and inhibits tumor growth. C YTHDF1 in dendritic cells (DCs) enhances the translation of mRNAs encoding proteases that can degrade antigens inside lysosomes. Without YTHDF1, the translation of lysosomal proteases wanes, favoring antigen cross-presentation and promoting more CD8+ T cell responses against tumors. D In CD4+ T cells, METTL3 inhibits the expression of the SOCS family proteins (SOCS1, SOCS3, and CISH), which inhibit JAK, thereby enhancing the activation of IL-7-mediated JAK/STAT5 to ultimately promote the homeostasis and differentiation of CD4+ T cells. METTL3 also reduces the stability of Tcf7 mRNA, promotes the expression of T follicular helper (Tfh) cell regulators, and subsequently enhances the functional maturation of Tfh cells. ALKBH5 decreases m6A modification on interferon-γ and C-X-C motif chemokine ligand 2 mRNA, increasing the stability of their mRNAs and boosting the expression of their proteins in CD4+ T cells. E METTL3 in regulatory T (Treg) cells reduces the stability of Socs mRNA by m6A modification. This activates IL-2/STAT5 signaling and inhibits the secretion of T cell effector cytokines, diminishing the anti-tumor response of effector T cells, such as CD8+ T cells, in the TME. Figure created with BioRender.com
Macrophages
Macrophages are phagocytic cells of the innate immune system and mainly involved in the recognition, phagocytosis, and degradation of pathogens and tumor cells [35]. Macrophages are highly involved in tumor initiation and progression. Specifically, tumor-associated macrophages (TAMs) in the TME are very plastic, being able to switch their functions to inhibit or promote tumor progression in response to different environmental stimuli [36]. Activation of classical anti-tumor TAMs (M1 type) or polarization of pro-tumor TAMs (M2 type) into M1 TAMs are perceived to be inhibiting forces against various cancers [36, 37]. Recently, Yin et al. found that METTL3 in macrophages helps regulate tumor progression. Ablating macrophage METTL3 promoted tumor growth and lung metastasis [38]. It also reshaped the TME by enhancing infiltration of M1- and M2-like TAMs, upsetting their homeostasis, and recruiting regulatory T (Treg) cells into tumor sites [38]. METTL3 depletion in macrophages also reduced the efficacy of programmed cell death protein 1 (PD-1) blockade therapy, suggesting an immune-relevant function for macrophage METTL3 [38]. This finding was supported by the work from Tong et al. who found that METTL3-deficient macrophages produced subnormal levels of tumor necrosis factor (TNF)-α when stimulated with lipopolysaccharide (LPS) in vitro [39]. Ablating METTL3 in macrophages also increased susceptibility to bacterial infection and tumor growth [39]. When Dong et al. characterized TAMs by single-cell RNA sequencing (scRNA-seq), they found that C1q+ macrophages expressed a set of immunomodulatory ligands and that their functions were regulated by the METTL14. Also, METTL14 deficiency in macrophages inhibited the anti-tumor function of CD8+ T cells and promoted tumor growth [40]. In contrast to m6A writers’ positive roles in macrophages, knocking down the m6A reader YTHDF2 promotes macrophages to express LPS-induced inflammatory cytokines, such as IL-6, TNF-α, IL-1β, and IL-12, suggesting that YTHDF2 plays a negative regulatory role in LPS-induced inflammatory responses of macrophages [41]. Taken together, these findings highlight the complicated roles of m6A regulators in macrophages and suggest that METTL3/14 and YTHDF2 may be potential targets for cancer immunotherapy (Fig. 2B).
Dendritic cells
Dendritic cells (DCs) are important antigen-presenting cells (APCs) that act as a bridge between innate and adaptive immune responses [42, 43]. DCs can either take up tumor antigens into the phagosome for presentation on major histocompatibility complex (MHC) class II ( MHC-II) molecules or transport the antigens to the cytosol for MHC class I ( MHC-I) presentation [44]. Further cross-presentation of antigens from DCs to CD8+ cytotoxic T lymphocytes (CTL) enables more accurate recognition and efficient killing of tumor cells by CD8+ T cells [45]. Recently, METTL3-mediated m6A modification was found to promote DC activation and maturation, causing them to present new antigens to and thereby activate T cells [46]. The underlying mechanism was the enhancement of the translation efficiency of CD40, CD80, and Toll/interleukin-1 receptor (TIR) domain-containing adaptor protein (TIRAP) by METTL3, leading to the secretion of proinflammatory cytokines [46]. METTL3-silenced DCs have reduced expression levels of MHC-II, costimulatory molecules (CD80, CD86), and inflammatory cytokines (IFN-γ, IL-12), thereby inducing immune tolerance and prolonging allograft survival in mouse heart transplantation [47]. This suggests that METTL3 helps maintain the mature properties of DCs. In contrast, Han et al. reported that the m6A reader YTHDF1 negatively regulates the anti-tumor immune responses of DCs [48]. Specifically, YTHDF1 enhanced the translation of mRNAs that encode lysosomal proteases, which can degrade antigens in lysosomes. Without YTHDF1, translation of lysosomal proteases was diminished, favoring antigen cross-presentation and promoting more CTL responses against tumors (Fig. 2C) [48].
T cells
T cells are developed in the thymus, and when T cells mature, they migrate to peripheral organs and constitute the foundation of the adaptive immune system [49]. T cells offer important protection against viral infection and tumor cells [50]. They are generally classified into two groups depending on whether their cell surface receptor is CD4 or CD8 [51]. Depletion of METTL3 in CD4+ T cells disrupts T cell homeostasis and differentiation by downregulating the activation of IL-7-mediated STAT5/suppressor of cytokine signaling (SOCS) (Fig. 2D) [52]. Interestingly, depletion of METTL3 suppresses the function and stability of Treg cells by inhibiting IL-2/STAT5 signaling and promoting the cytokine secretion of T effector cells, resulting in enhancement of the anti-tumor immune responses in the TME (Fig. 2E) [53]. T follicular helper (Tfh) cells are a specialized CD4+ T cell subset critical to humoral immunity [54]. Yao et al. reported that conditional ablation of METTL3 in CD4+ T cells dampened Tfh differentiation and functional maturation, further inhibiting the antibody response of B cells by impairing the stability of m6A-modified Tcf7 mRNA (Fig. 2D) [55]. A very recent study showed that ALKBH5 deficiency decreased the mRNA stability and protein secretion of IFN-γ and C-X-C motif chemokine ligand 2 (CXCL2) in CD4+ T cells, thereby alleviating experimental autoimmune encephalomyelitis (Fig. 2D) [56]. As IFN-γ is an important anti-tumor cytokine and ALKBH5 has a positive effect on the expression of IFN-γ in autoimmunity, ALKBH5 may have the potential to positively regulate the anti-tumor effect of CD4+ T cells. Therefore, the different functions of the m6A modification in T cells may depend on cell types and cellular contexts and this merits further investigation.
Role of m6A modification in remodeling the tumor microenvironment
The TME plays an important role in cancer progression and significantly affects responsiveness to immunotherapy [57, 58]. In recent years, compelling evidence indicates that m6A regulators in tumor cells are associated with immune cell responses and immune checkpoint blockers (ICBs) efficacy. Alteration of the m6A modification in tumor cells influences the infiltration, activation, and effector functions of infiltrated immune cells in the TME [59], making targeting m6A—and especially m6A regulators in tumor cells—a promising strategy for improving cancer immunotherapy.
m6A writers
METTL3 plays a dual role as either an oncogene or a tumor suppressor gene in various types of cancers, including hepatocellular carcinoma (HCC) [60, 61], hepatoblastoma [62], gastric cancer (GC) [63], colorectal cancer (CRC) [64], non-small cell lung cancer (NSCLC) [65, 66], and bladder cancer (BLC) [67, 68]. Abnormal expression of METTL3 in tumor cells affects the infiltration of immune cells. In testicular germ cell tumors (TGCT), METTL3 expression was significantly downregulated in TGCT tissues, and its level correlated positively with patient survival rates and levels of tumor-infiltrating CD8+ T cells, CD4+ T cells, and NK cells [69]. However, METTL3 was highly expressed in CRC tumor cells. Depletion of METTL3 or METTL14 in tumor cells increased cytotoxic tumor-infiltrating CD8+ T cells and elevated secretion of IFN-γ, CXCL9, and CXCL10 in the TME, thereby enhancing the response to anti-PD-1 treatment in mismatch-repair-proficient or microsatellite instability-low CRC tumors [70]. In cervical cancer (CC), METTL3 was expressed much more highly in tumor tissues than in tumor-adjacent tissues, and its level was positively related to the density of CD33+ myeloid-derived suppressor cells (MDSCs), which in turn was linked to poor patient survival rate [71]. Expression levels of METTL3 in the tumor also negatively correlated with breast cancer (BC) patient survival rate and tumor-infiltrating CD8+ T cells, helper T cells, and activated NK cells. In contrast, they positively correlated with M2 TAMs in BC [72]. For HCC, Shen et al. analyzed 433 samples from The Cancer Genome Atlas (TCGA) database, finding that METTL3 expression was negatively related to infiltration of DCs into tumors [73]. In head and neck squamous cell carcinoma (HNSCC), METTL3 and HNRNPC were highly expressed in tumor tissue than normal tissue, high expression of METTL3 and HNRNPC were positively associated with the infiltration of CD4 naive T cells, CD4 memory-activated T cells, and eosinophils [74].
As a critical component of the multicomponent methyltransferase complex, METTL14 is abnormally expressed and plays key regulatory roles in various cancers [75, 76]. In recent years, the effects of tumor cells’ METTL14 on the immune cells of the TME have only begun to be revealed. Low levels of METTL14 predicted an unfavorable prognosis in BC, in which METTL14 expression levels significantly and positively correlated with infiltrating levels of CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and DCs. In contrast, they correlated negatively with Treg cells in BC [77]. In clear cell renal cell carcinoma (ccRCC), METTL14 possessed a good diagnostic and prognostic value. Also, METTL14 expression correlated negatively with Treg cells in BC, and the METTL14/CCL5/Tregs axis was a potential signaling pathway for regulating anti-tumor immunity in ccRCC [78].
In GC, WTAP was highly expressed in tumor cells, and its expression level correlated negatively with T cell infiltration and T cell-related immune responses, which was linked to poor prognosis [79]. In esophageal cancer (EC), however, high expression levels of WTAP were negatively associated with patient survival times and positively correlated with both immuno-inhibitors and immuno-stimulators such as cancer-associated fibroblasts, myeloid DCs, T cells, neutrophils, Treg cells, and macrophages (Table 1) [80].
m6A erasers
Upregulated expression of FTO intrinsically modulates genes in tumor cells that relate to malignant potential, promoting the progression of various types of cancer such as acute myeloid leukemia (AML) [81], glioblastoma (GBM) [82], CRC [83], and GC [84]. Genetic depletion or pharmacological inhibition of FTO dramatically attenuates leukemia stem/initiating cell self-renewal and reprograms immune responses by suppressing the expression of immune checkpoint genes such as leukocyte immunoglobulin-like receptor B4 (LILRB4) [85]. FTO inhibition sensitizes leukemia cells to T cell cytotoxicity and overcomes immune evasion induced by hypomethylating agents. As well as regulating immune checkpoint genes, FTO functions as an oncogene by reprogramming the glycolytic metabolism of tumor cells; it also further inhibits CD8+ T cell activity [86]. Targeting FTO with a small-molecule inhibitor blocks FTO-mediated immune evasion and synergizes with the PD-1/PD-L1 checkpoint blockade [86, 87]. Another eraser, ALKBH5, utilizes different mechanisms but performs a similar function in regulating anti-tumor responses in melanoma, and its expression in tumor cells correlates positively with Treg cell infiltration. ALKBH5 deletion enhances the efficacy of anti–PD-1 therapy in melanoma patients [88]. In addition, in intrahepatic cholangiocarcinoma (ICC), ALKBH5 positively regulates PD-L1 expression in tumor cells and inhibits the expansion and cytotoxicity of T cells through PD-1/PD-L1 signaling (Table 1) [89].
m6A readers
Readers mediate the effects of the m6A modification by controlling the fate of modified RNA. Previous studies showed that upregulated expression of readers such as YTHDF1 promotes the progression of some cancers, such as NSCLC [90] and ovarian cancer [91]. The latest research also found that readers, mainly YTHDF1, YTHDF2, and YTHDC2, correlated positively with expression levels of several immune checkpoint receptors (including PD-1, T cell immunoglobulin and mucin-domain containing-3, and cytotoxic T-lymphocyte-associated antigen 4) as well as with the abundance of tumor-infiltrating lymphocytes (such as CD8+ T cells, CD4+ T cells, macrophages, and DCs) in several cancers, such as lower-grade glioma (LGG), NSCLC, KIRC and BC (Table 1) [92,93,94,95,96,97,98,99].
The above studies show that most of the m6A regulatory factors are abnormally expressed in tumors, where they create an immunosuppressive microenvironment. It would therefore seem beneficial to reshape the TME by enhancing or reducing m6A modification or by targeting m6A regulators to prevent that immunosuppression. However, current knowledge is still in its infancy. Further identification of other m6A-related modulators may greatly improve our understanding of tumor immune regulation and provide effective anti-tumor therapeutics for cancer patients.
The potential role of the m6A modification in gut microbiota-mediated tumor immunity
Gut microbiota is a group of microorganisms that lives in the intestines of humans or other animals and is mutually beneficial and symbiotic with the body. Preclinical mouse models and clinical trials indicate that the gut microbiome modulates tumor response to ICBs [100,101,102,103]. Recent studies have revealed the interaction between host microbiota and RNA modifications. Transcriptome-wide profiling showed that the microbiome had a strong effect on host m6A modifications [104]. Lack of microbiome exposure resulted in higher m6A modifications in the brain and intestine in germ-free (GF) mice than specific pathogen-free (SPF) mice [104]. Another study reported that GF mice and SPF mice had differentially methylated peaks in the cecum, which were mainly associated with metabolic and inflammatory pathways [105]. Some specific microbial species, such as Akkermansia muciniphila and Lactobacillus plantarum, could directly change specific m6A modifications in mono-associated mice (animals colonized by only one microbial species) [105]. Currently, the relationship between m6A and gut microbiota in cancer development has not been explored. It is reported that virus infection, such as rotavirus and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), induces global m6A modifications on mRNA transcripts of host cells and affects host immune defense against virus infections [106,107,108,109]. Therefore, a better understating of the potential functional interaction of gut microbes with the host m6A machinery may offer new strategies for the development of innovative therapies against cancer.
Strategies and challenges of targeting m6A modification for cancer immunotherapy
In the above discussion, we concluded that m6A regulators, such as METTL3, YTHDF1, and YTHDF2, directly contribute to the regulation of anti-tumor immunity in different ways. Meanwhile, abnormal expression of m6A regulators in tumor cells leads to an immunosuppressive TME, further accelerating cancer progression. Therefore, targeting m6A modification should 1) directly inhibit tumor cell growth, 2) enhance the anti-tumor ability of immune cells, as by boosting the cytotoxicity of CD8+ T cells and NK cells, 3) remodel the TME by reprogramming M2 TAMs into M1 TAMs. Here, we discuss strategies and challenges inherent in targeting m6A modification for cancer immunotherapy.
Developing specific inhibitors against m6A regulators to boost endogenous anti-tumor immunity
It is recognized that most of the m6A regulators are abnormally high-expressed in tumor cells. Therefore, targeted inhibition of those regulators should be an effective way to directly inhibit tumor cell proliferation and further activate endogenous anti-tumor responses that kill cancer cells or cancer stem cells. In recent years, a series of small-molecule inhibitors targeting m6A regulators such as FTO, ALKBH5, and METTL3 have been developed, but among them, FTO is the most attractive target. Between 2012 and 2019, researchers developed and identified a set of FTO inhibitors, such as rhein, MO-I-500, meclofenamic acid (MA), fluorescein, 2-hydroxylglutarate (R-2HG), FB23, and FB23-2, that showed marked anti-tumor effects in vitro and in vivo (Table 2) [110,111,112,113,114,115]. Between 2020 and the present, several upgraded FTO inhibitors have been developed, including CS1/CS2 [85] and Dac51 [86], which not only suppress cancer cell proliferation and cancer stem cell self-renewal but also improve anti-tumor immunity (Fig. 3). Most importantly, they promote the infiltration and cytotoxicity of CD8+ T cells [85, 86].

Inhibitors of m6A regulators in tumor cells indirectly augment T cell trafficking and decrease immunosuppression. A High expression in tumor cells of m6A regulators, such as FTO, ALKBH5, and others, leads to an immune-suppressed TME characterized by high expression of immune checkpoints [PD-1 and leukocyte immunoglobulin-like receptor B4 (LILRB4)], reduced infiltration, decreased cytotoxic function of CD8+ T cells, and enhanced infiltration of Treg cells and myeloid-derived suppressor cells (MDSCs). B Targeting FTO or ALKBH5 with specific inhibitors, such as CS1/2, Dac51, or ALK-04, or combining with ICBs, reverses the immunosuppressive TME by increasing the infiltration and cytotoxicity of CD8+ T cells and inhibiting the infiltration of Treg cells and MDSCs, thereby creating an immune-activated TME. Figure created with BioRender.com
With continuous technological breakthroughs, screening of ALKBH5 inhibitors is also proceeding steadily. Two ALKBH5 inhibitors—2-{[1-hydroxy-2-oxo-2-phenylethyl]sulfanyl} acetic acid and 4-{[furan-2-yl]methyl}amino-1,2-diazinane-3,6-dione—suppress the proliferation of leukemia cell lines (HL-60, CCRF-CEM, and K562) when used in micromolar amounts (Table 2) [116]. In 2020, Li et al. identified a specific inhibitor of ALKBH5, named ALK-04, which decreases the infiltration of Treg cells and MDSCs and inhibits tumor growth by enhancing the efficacy of anti-PD-1 therapy (Fig. 3) [88]. Recently, through high-throughput screening of 250,000 drug-like compounds, Eliza et al. discovered a highly potent and selective first-in-class inhibitor of METTL3 and METTL14, named STM2457 (Table 2). This inhibitor showed significant antileukemic effects in preclinical AML models, providing proof of concept that targeting m6A writers is a promising strategy for cancer therapy [117].
Although several inhibitors that target m6A regulators have been developed, there are still the following challenges: 1) Inhibitors that target readers, which decode the m6A mark to mediate downstream effects of m6A-modified mRNAs, have not been discovered. The possible reason is that the binding sites of a reader might be overlapping with other readers with opposed functions, which could have both beneficial and detrimental effects on cancer growth. A recent screening of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) identified YTHDF2 as a therapeutic target for triple-negative breast cancer [118]. In this study, the authors used an alternative approach to inhibit YTHDF2’s effects by targeting its downstream effectors to directly stabilize the target mRNAs or overexpress the proteins that they encode. 2) The established m6A small-molecule inhibitors are being used mainly in hematological malignancies. The main reason is that the role and associated mechanism of the m6A modification in solid tumors still have not yet been fully understood. It is worth noting that m6A plays either tumorigenesis or tumor suppressor roles in the development of solid tumors, such as glioblastoma [119, 120] and colorectal cancer [121, 122], while in general speaking, the m6A modification plays a promoting role in hematological malignancies. Another reason is the complexity of the TME of solid tumors, which is tightly associated with the clinical outcome of cancer patients [123]. In a couple of solid tumors, the other group and we have uncovered beneficial roles for regulators, such as METTL3 and YTHDF2, in the immune response to tumor cells by NK cells [33, 34]. Therefore, m6A inhibitors that target tumor cells may impair the anti-tumor immune response. A comprehensive understanding of the different roles of m6A in regulating immune cells and tumor cells within the TME is needed to develop m6A-based targeted drugs with the specificity to distinguish tumor cells from immune cells during immunotherapy. 3) Preventing the exhaustion of anti-tumor immune cells and activating endogenous anti-tumor immunity are two effective approaches to treat tumors. However, most current inhibitors affect mainly tumor cells, though a few, such as CS1/2, Dac51, and ALK-04, can activate endogenous anti-tumor immune cells or decrease the infiltration of immunosuppressive cells. 4) Most inhibitors are still at the preclinical stage, and therefore, their therapeutic value needs to be explored in the clinic. Some pharmaceutical or biotech companies (e.g., STORM Therapeutics, Accent Therapeutics, Gotham Therapeutics, and Genovel Biotech Corp.) have started to develop highly potent and selective small-molecule inhibitors that directly target m6A regulators such as METTL3, METTL14, FTO, and ALKBH5 [30]. In addition, PROTAC (proteolysis targeting chimera)-based or molecular glue-based inhibitors could be developed to selectively degrade dysregulated m6A regulatory proteins for cancer therapy [30, 124]. Also, gene silencing using oligonucleotide-based therapeutics (ONTs), such as small interfering RNA (siRNA), antisense oligonucleotides, or decoy oligodeoxynucleotides, is a promising strategy for targeting m6A regulators in tumor cells and immune cells.
Developing an effective programmable m6A editing system
Most investigations of m6A change overall RNA methylation by interfering with m6A regulators, or they explore regional functions of m6A by mutating methylation sites [125]. However, mutating a methylation site changes mRNA nucleotide sequence and introduces other unknown features, complicating the interpretation of the phenotype. Moreover, the distribution of m6A on mRNA is not uniform, as most of these modifications cluster near the stop codon [126]. Recently, several groups have attempted to manipulate m6A in a site-specific manner by using CRISPR/Cas9 technology, which enables precise genome editing, including targeted DNA cutting/repair, direct base editing, and site-specific epigenome editing [127, 128]. In 2019, Liu et al. created the first programmable RNA m6A editing machinery by fusing CRISPR-dead Cas9 (dCas9), which lacks the ability to cut DNA, with a single-chain m6A methyltransferase or demethylase [129]. These engineered m6A writers or erasers can install or remove m6A at specific sites without changing the primary sequence. Subsequently, other groups have developed and optimized precise m6A editors by replacing dCas9 with smaller dCas13 [130,131,132]. This new platform enables efficient manipulation of individual m6A sites within endo-transcripts with minimal off-target alterations in both normal mammalian cells and cancer cells. Thus, m6A editors offer a powerful approach to fine-tuning the mRNA modification of specific genes and their related biological functions. Further investigations are warranted to develop smaller m6A editors that allowed for easier cell delivery with higher specificity and more adjustable features for immunotherapy that targets the m6A modification.
Developing an effective m6A modification system of adoptive cell therapy
NK cells account for 10%–20% of human peripheral blood leukocytes, and they are an attractive source of immunotherapy based on genetically modified immune cells [31, 133, 134]. Adoptive NK cell-based immunotherapy, such as chimeric antigen receptor (CAR) NK cells and induced pluripotent stem cell (iPSC)-derived NK or CAR NK cells, are effective for anti-tumor therapy [31, 135,136,137]. However, it seems to be difficult to obtain a large number of NK cells in a short time in vitro due to the limited supply of initial primary or initial iPSC-derived NK cells [138, 139]. Current studies have found that adding cytokines (IL-2 + IL-15) or co-cultivating with K562 feeder cells greatly improves the expansion efficiency of NK cells in vitro [136, 137, 140]. As cell culture time increases, however, the proliferation efficiency and effector functions of NK cells may decrease. Therefore, increasing the expression of genes that promote the proliferation of NK cells and enhance their effector functions may be an effective way to increase the efficiency of NK expansion in vitro.
Our lab and another group found that in mice the m6A regulators METTL3 and YTHDF2 and their corresponding target genes, Ptpn11 (encoding SHP-2), and Tardbp (encoding TDP-43), associated with a gain of effector function and proliferation in NK cells [33, 34]. With lentivirus- or retrovirus-mediated overexpression of METTL3, SHP-2, or YTHDF2, knockdown of Tardbp, or dCasRx-conjugated METTL3 to manipulate methylation events at target mRNA m6A sites of Ptpn11, it might be possible to improve the effector function and proliferation ability of NK cells (Fig. 4). As far as we know, there have been no attempts to modulate m6A regulators to enhance the proliferation and cytotoxicity of human NK cells in vitro. As the m6A regulatory site is highly conserved between humans and mice [18], such investigation might help inform future protocols for the optimal generation of more CAR NK cells and iPSC-derived NK cells.

m6A modification strategies for NK cell-based immunotherapy. During the production of CAR NK and iPSC-derived NK cells, several approaches targeting the m6A modification can be used to increase expansion in vitro. They include lentivirus- or retrovirus-mediated gene delivery of METTL3, YTHDF2, and SHP-2; short hairpin RNA interference targeting Tardbp; and m6A editing machinery that manipulates the m6A site in Ptpn11 mRNA. These m6A-based strategies may improve the functionality and proliferation of NK cells. Figure created with BioRender.com
Besides CAR NK cells and iPSC-derived NK cells therapy, CAR T cell therapy is one of the most successful cancer immunotherapies in the clinic [6]. As discussed above, m6A regulators, such as ALKBH5 and METTL3, and their targeting genes affect T cell function (Fig. 2D). Therefore, modulating m6A modifications in CAR T cells may also represent a potentially promising strategy to enhance anti-tumor immune responses.
Encapsulating m6A modification molecules into nanoparticles to specifically target TAMs
Nanoparticles (NPs) platforms have emerged as promising carriers in cancer therapy [141]. In recent years, scientists have developed NPs that can precisely and efficiently deliver mRNAs, siRNAs, and protein-based drugs into tumor cells [142, 143]. Recent research in HCC found that METTL3 can stabilize the RNA transcript of a long non-coding RNA-LINC00958 via m6A modification, and aberrant overexpression of LINC00958 is an important cause of accelerated HCC. Moreover, specifically delivering NP-encapsulated siRNA of LINC00958 to tumor cells in the TME reduced m6A modification in LINC00958 and inhibited the progression of HCC [144].
Similar to being used for targeted delivery of drugs into tumors cells, many kinds of NPs, such as lipid-based NPs [145], polymer-based NPs [146], and inorganic NPs [ 147], can be recognized by TAMs and then deliver drugs or RNA into TAMs of the TME [148]. For example, C-C motif chemokine receptor 2 siRNA-loaded lipid NPs prevent the recruitment of TAMs into the tumors [145]. Polymeric NPs can be loaded with siRNAs to target vascular endothelial growth factor and placental growth factor signaling in both tumor cells and M2 TAMs, skewing the immunosuppressive M2 TAMs to the M1 type and thereby inhibiting tumor growth [146]. Recent studies found that METTL14, METTL3, and their target genes, Spred2 and Irakm, in macrophages are associated with tumor progression [38, 39]. Therefore, targeted delivery of NP-encapsul ated M ettl3 , Mettl14, or Spred2 mRNA or of Irakm siRNA into TAMs might promote TAM polarization, reduce Treg cell infiltration, promote the cytotoxic function of CD8+ T cells, and reverse immunosuppression in the TME (Fig. 5).

Nanoparticles (NPs) encapsulating m6A modification molecules can specifically target TAMs. NPs that deliver Mettl3, Mettl14, or Sp red2 mRNA or Irakm siRNA specifically into TAMs can reprogram the macrophages from the M2-type to the M1-type. This switch reshapes the TME and inhibits tumor progression. Figure created with BioRender.com
Conclusions and perspectives
Although the biological sequelae of m6A modification in tumors have been extensively investigated in recent y ears, strategies for developing m6A-based targeted drugs for cancer immunotherapy are still in their infancy. In this review, we summarized current progress in understanding the roles and mechanisms of m6A regulators in immune cells and their effects on immune responses in the TME. It is important to acknowledge that the ways in which the RNA m6A machinery affects immune cells and tumor cells are complicated and still not well understood. For example, METTL3 regulates DC functions positively, whereas YTHDF1 regulates them negatively. Similar discrepancies are found in macrophages, where METTL3/14 exert positive control on polarization and immune functions, whereas YTHDF2 exerts negative control. Moreover, both METTL3 and YTHDF2 are positive regulators of NK cell effector functions. Thus, additional studies of the roles of other potential regulators, such as YTHDF3, FTO, and ALKBH5, in immune cells are warranted. In addition, as most m6A regulators play oncogenic roles in tumor development, m6A-based inhibitors that target tumor cells may have opposite effects to those intended as inhibition of m6A and impair host anti-tumor immune responses. Therefore, a more comprehensive understanding of how m6A modulators regulate immune cells and tumor cells will expand our knowledge of the biological roles of m6A in anti-tumor immunity and help with the development of m6A-based targeted drugs with the specificity to distinguish tumor cells from immune cells during immunotherapy.
We also proposed four reasonable strategies for improving m6A-based cancer immunotherapy, including developing: 1) inhibitors against m6A regulators to modulate anti-tumor immunity, 2) a programmable m6A gene-editing system to manipulate individual m6A sites, 3) m6A-modified system for adoptive cell therapy, 4) nanoparticles that can specifically deliver m6A modification molecules into TAMs to remodel the TME. Of note, ICBs therapy has achieved remarkable success in the treatment of cancer. As discussed above, m6A modifications not only modify the immune checkpoint expression pattern in many types of cancers [89, 92,93,94,95,96,97,98,99] but also regulate the sensitivity and efficacy of ICBs therapy in several preclinical animal models [38, 86,87,88,89]. Although there are no clinical trials using m6A inhibitors for the treatment of cancer yet, it is reasonable to speculate a synergistic anticancer effect through the combination of m6A inhibitors and ICBs. Future clinical trials are needed to prove this in humans.
Overall, m6A modification is dynamic and reversible, and many of its regulators are yet to be discovered. Targeting m6A modification for cancer immunotherapy is challenging because the expected “on-target, off-tumor” activity may potentially lead to significant toxicity. Therefore, a better understanding of this process and the ability to control it accurately are still significant challenges that warrant further study.
Availability of data and materials
Not applicable.
Abbreviations
- m6A:
-
N6-Methylation of adenosine
- TME:
-
Tumor microenvironment
- TAMs:
-
Tumor-associated macrophages
- rRNA:
-
Ribosomal RNA
- tRNA:
-
Transfer RNA
- m1A:
-
N1-Methyladenosine
- m5C:
-
5-Methylcytosine
- m7G:
-
N7-Methylguanosine
- CAR:
-
Chimeric antigen receptor
- METTL3:
-
Methyltransferase-like 3
- METTL14:
-
Methyltransferase-like 14
- RBM15:
-
RNA binding motif protein 15
- WTAP:
-
Wlms tumor 1 associated protein
- VIRMA:
-
Vir-like m6A methyltransferase associated subunits
- ZC3H13:
-
Zinc finger CCCH domain-containing protein 13
- UTRs:
-
Untranslated regions
- FTO:
-
Fat mass and obesity-associated protein
- ALKBH5:
-
Alpha-ketoglutarate-dependent dioxygenase alkB homologue 5
- YTHDC1:
-
YTH domain containing 1
- YTHDC2:
-
YTH domain containing 2
- HNRNP:
-
Heterogeneous nuclear ribonuclease
- FMRP:
-
Fragile x mental retardation protein
- YTHDF1:
-
YTH domain family 1
- YTHDF2:
-
YTH domain family 2
- YTHDF3:
-
YTH domain family 3
- IGF2BPs:
-
Insulin-like factor-2 mRNA binding proteins
- PRRC2A:
-
Proline rich coiled-coil 2A
- NK cells:
-
Natural killer cells
- IL-15:
-
Interleukin 15
- STAT5:
-
Signal transducer and activator of transcription 5
- Eomes:
-
Eomesodermin
- SHP-2:
-
SH2 domain-containing protein tyrosine phosphatase-2
- Treg cells:
-
Regulatory T cells
- PD-1:
-
Programmed cell death protein 1
- LPS:
-
Lipopolysaccharide
- scRNA-seq:
-
Single-cell RNA sequencing
- DCs:
-
Dendritic cells
- APCs:
-
Antigen-presenting cells
- MHC-II:
-
MHC class II
- MHC-I:
-
MHC class I
- CTL:
-
Cytotoxic T lymphocytes
- TIRAP:
-
Toll/interleukin-1 receptor domain-containing adaptor protein
- Tfh cells:
-
T follicular helper cells
- CXCL2:
-
C-X-C motif chemokine ligand 2
- ICBs:
-
Immune checkpoint blockers
- HCC:
-
Hepatocellular carcinoma
- GC:
-
Gastric cancer
- CRC:
-
Colorectal cancer
- NSCLC:
-
Non-small cell lung cancer
- BLC:
-
Bladder cancer
- TGCT:
-
Testicular germ cell tumors
- CC:
-
Cervical cancer
- MDSCs:
-
Myeloid-derived suppressor cells
- BC:
-
Breast cancer
- TCGA:
-
The Cancer Genome Atlas
- HNSCC:
-
Head and neck squamous cell carcinoma
- ccRCC:
-
Clear cell renal cell carcinoma
- EC:
-
Esophageal cancer
- AML:
-
Acute myeloid leukemia
- GBM:
-
Glioblastoma
- LILRB4:
-
Leukocyte immunoglobulin like receptor B4
- ICC:
-
Intrahepatic cholangiocarcinoma
- LGG:
-
Lower-grade glioma
- GF:
-
Germ-free
- SPF:
-
Pathogen-free
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- MA:
-
Meclofenamic acid
- R-2HG:
-
2-Hydroxylglutarate
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas9:
-
CRISPR-associated protein 9
- PROTAC:
-
Proteolysis targeting chimera
- ONTs:
-
Oligonucleotide-based therapeutics
- siRNA:
-
Small interfering RNA
- dCas9:
-
CRISPR-dead Cas9
- iPSC:
-
Induced pluripotent stem cell
- NPs:
-
Nanoparticles
References
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–200.
Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–22.
He PC, He C. m6A RNA methylation: from mechanisms to therapeutic potential. EMBO J. 2021;40:e105977.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640–50.
Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev of Pathol. 2021;16:223–49.
June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73.
Zhao W, Qi X, Liu L, Ma S, Liu J, Wu J. Epigenetic regulation of m6A modifications in human cancer. Mo Ther Nucleic Acids. 2020;19:405–12.
Yang C, Hu Y, Zhou B, Bao Y, Li Z, Gong C, Yang H, Wang S, Xiao Y. The role of m6A modification in physiology and disease. Cell Death Dis. 2020;11:960.
Lee Y, Choe J, Park OH, Kim YK. Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet. 2020;36:177–88.
Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, Feederle R, Bruckmann A, Meister G. Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. RNA. 2018;24:499–512.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–47.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Huang W, Chen TQ, Fang K, Zeng ZC, Ye H, Chen YQ. N6-methyladenosine methyltransferases: functions, regulation, and clinical potential. J Hematol Oncol. 2021;14:117.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. VIRMA mediates preferential m6A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villaseñor R, Hess D. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl (2) d. Genes Dev. 2018;32:415–29.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–46.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, Yang C, Chen Y. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Zhao Y, Shi Y, Shen H, Xie W. m6A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. 2020;13:35.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162:1299–308.
Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, Song H, Wu H, Shu Q, Jin P. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. 2018;27:3936–50.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, Min J. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10:927–9.
Li F, Zhao D, Wu J, Shi Y. Structure of the YTH domain of human YTHDF2 in complex with an m(6)A mononucleotide reveals an aromatic cage for m(6)A recognition. Cell Res. 2014;24:1490–2.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Wu R, Li A, Sun B, Sun JG, Zhang J, Zhang T, Chen Y, Xiao Y, Gao Y, Zhang Q. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019;29:23–41.
Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m6A methylation in cancer. Cancer Res. 2019;79:1285–92.
Huang H, Weng H, Chen J. m6A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37:270–88.
Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13:168.
Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19.
Ma S, Yan J, Barr T, Zhang J, Chen Z, Wang LS, Sun JC, Chen J, Caligiuri MA, Yu J. The RNA m6A reader YTHDF2 controls NK cell antitumor and antiviral immunity. J Ex Med. 2021;218:e20210279.
Song H, Song J, Cheng M, Zheng M, Wang T, Tian S, Flavell RA, Zhu S, Li HB, Ding C. METTL3-mediated m6A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat Commun. 2021;12:5522.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
Caux C, Ramos RN, Prendergast GC, Bendriss-Vermare N, Ménétrier-Caux C. A milestone review on how macrophages affect tumor growth. Cancer Res. 2016;76:6439–42.
Shrivastava R, Asif M, Singh V, Dubey P, Malik SA, Tewari BN, Baghel KS, Pal S, Nagar GK, Chattopadhyay N. M2 polarization of macrophages by Oncostatin M in hypoxic tumor microenvironment is mediated by mTORC2 and promotes tumor growth and metastasis. Cytokine. 2019;118:130–43.
Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, Yu Y, Wu Y, Wang Y, Zhang J. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394.
Tong J, Wang X, Liu Y, Ren X, Wang A, Chen Z, Yao J, Mao K, Liu T, Meng FL. Pooled CRISPR screening identifies m6A as a positive regulator of macrophage activation. Sci Adv. 2021;7:eabd4742.
Dong L, Chen C, Zhang Y, Guo P, Wang Z, Li J, Liu Y, Liu J, Chang R, Li Y. The loss of RNA N6-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8+ T cell dysfunction and tumor growth. Cancer Cell. 2021;39:945–57.
Yu R, Li Q, Feng Z, Cai L, Xu Q. m6A reader YTHDF2 regulates LPS-induced inflammatory response. Int J Mol Sci. 2019;20:1323.
Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–8.
Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26.
Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–69.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, Zhou Q, Cao X. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat Commun. 2019;10:1898.
Wu H, Xu Z, Wang Z, Ren Z, Li L, Ruan Y. Dendritic cells with METTL3 gene knockdown exhibit immature properties and prolong allograft survival. Genes Immun. 2020;21:193–202.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, Huang X, Liu Y, Wang J, Dougherty U. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270–4.
Takaba H, Takayanagi H. The mechanisms of T cell selection in the thymus. Trends Immunol. 2017;38:805–16.
Park SL, Gebhardt T, Mackay LK. Tissue-resident memory T cells in cancer immunosurveillance. Trends Immunol. 2019;40:735–47.
Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2:309–22.
Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–42.
Tong J, Cao G, Zhang T, Sefik E, Vesely MCA, Broughton JP, Zhu S, Li H, Li B, Chen L. m6A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018;28:253–6.
Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity. 2019;50:1132–48.
Yao Y, Yang Y, Guo W, Xu L, You M, Zhang YC, Sun Z, Cui X, Yu G, Qi Z, et al. METTL3-dependent m6A modification programs T follicular helper cell differentiation. Nat Commun. 2021;12:1333.
Zhou J, Zhang X, Hu J, Qu R, Yu Z, Xu H, Chen H, Yan L, Ding C, Zou Q. m6A demethylase ALKBH5 controls CD4+ T cell pathogenicity and promotes autoimmunity. Sci Adv. 2021;7:eabg0470.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50.
Tang H, Qiao J, Fu YX. Immunotherapy and tumor microenvironment. Cancer Letts. 2016;370:85–90.
Gu Y, Wu X, Zhang J, Fang Y, Pan Y, Shu Y, Ma P. The evolving landscape of N6-methyladenosine modification in the tumor microenvironment. Mol Ther. 2021;29:1703–15.
Liu X, Qin J, Gao T, Li C, Chen X, Zeng K, Xu M, He B, Pan B, Xu X. Analysis of METTL3 and METTL14 in hepatocellular carcinoma. Aging (Albany NY). 2020;12:21638–59.
Pan F, Lin X, Hao L, Chu X, Wan H, Wang R. The role of RNA methyltransferase METTL3 in hepatocellular carcinoma: results and perspectives. Front Cell Dev Biol. 2021;9:674919.
Chen H, Duan F, Wang M, Zhu J, Zhang J, Cheng J, Li L, Li S, Li Y, Yang Z. Polymorphisms in METTL3 gene and hepatoblastoma risk in Chinese children: A seven-center case-control study. Gene. 2021;800:145834.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, Zhao G. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18:142.
Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, Ji D, Wang Q, Zhang Z, Tang J. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res. 2019;38:393.
Xue L, Li J, Lin Y, Liu D, Yang Q, Jian J, Peng J. m6A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J Cell Physiol. 2021;236:2649–58.
Zhang Y, Liu S, Zhao T, Dang C. METTL3-mediated m6A modification of Bcl-2 mRNA promotes non-small cell lung cancer progression. Oncol Rep. 2021;46:163.
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18:110.
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, Liang Y, Zhu F, Zhang Y, Zhang X. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. 2019;38:3667–80.
Luo Y, Sun Y, Li L, Mao Y. METTL3 may regulate testicular germ cell tumors through EMT and immune pathways. Cell Transplant. 2020;29:963689720946653.
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, Yuan J, Rana TM. m6A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39:e104514.
Ni HH, Zhang L, Huang H, Dai SQ, Li J. Connecting METTL3 and intratumoural CD33+ MDSCs in predicting clinical outcome in cervical cancer. J Transl Med. 2020;18:393.
He X, Tan L, Ni J, Shen G. Expression pattern of m6A regulators is significantly correlated with malignancy and antitumor immune response of breast cancer. Cancer Gene Ther. 2021;28:188–96.
Shen S, Yan J, Zhang Y, Dong Z, Xing J, He Y. N6-methyladenosine (m6A)-mediated messenger RNA signatures and the tumor immune microenvironment can predict the prognosis of hepatocellular carcinoma. Ann Transl Med. 2021;9:59.
Yi L, Wu G, Guo L, Zou X, Huang P. Comprehensive analysis of the PD-L1 and immune infiltrates of m6A RNA methylation regulators in head and neck squamous cell carcinoma. Mol Ther Nucleic Acids. 2020;21:299–314.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63:306–17.
Zhou H, Yin K, Zhang Y, Tian J, Wang S. The RNA m6A writer METTL14 in cancers: Roles, structures, and applications. Biochim Biophys Acta Rev Cancer. 2021;1876:188609.
Gong PJ, Shao YC, Yang Y, Song WJ, He X, Zeng YF, Huang SR, Wei L, Zhang JW. Analysis of N6-methyladenosine methyltransferase reveals METTL14 and ZC3H13 as tumor suppressor genes in breast cancer. Front Oncol. 2020;10:578963.
Xu T, Gao S, Ruan H, Liu J, Liu Y, Liu D, Tong J, Shi J, Yang H, Chen K. METTL14 acts as a potential regulator of tumor immune and progression in clear cell renal cell carcinoma. Front Genet. 2021;12:609174.
Li H, Su Q, Li B, Lan L, Wang C, Li W, Wang G, Chen W, He Y, Zhang C. High expression of WTAP leads to poor prognosis of gastric cancer by influencing tumour-associated T lymphocyte infiltration. J Cell Mol Med. 2020;24:4452–65.
Zhao H, Xu Y, Xie Y, Zhang L, Gao M, Li S, Wang F. m6A Regulators Is Differently Expressed and Correlated With Immune Response of Esophageal Cancer. Front Cell Dev Biol. 2021;9:650023.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–41.
Xiao L, Li X, Mu Z, Zhou J, Zhou P, Xie C, Jiang S. FTO inhibition enhances the antitumor effect of temozolomide by targeting MYC-miR-155/23a cluster-MXI1 feedback circuit in glioma. Cancer Res. 2020;80:3945–58.
Tsuruta N, Tsuchihashi K, Ohmura H, Yamaguchi K, Ito M, Ariyama H, Kusaba H, Akashi K, Baba E. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem Biophys Res Commun. 2020;530:235–9.
Wang D, Qu X, Lu W, Wang Y, Jin Y, Hou K, Yang B, Li C, Qi J, Xiao J. N6-Methyladenosine RNA Demethylase FTO Promotes Gastric Cancer Metastasis by Down-Regulating the m6A Methylation of ITGB1. Front Oncol. 2021;11:681280.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, Deng X, Li H, Huang Y, Gao L. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38:79–96.
Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z, Zhang Z, Li F, Huang Y, Li Y. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021;33:1221–33.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, Aplin AE, Lu Z, Hwang S, He C, He YY. m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, Agrawal K, Gonzalez GM, Wang Y, Patel SP. ALKBH5 regulates anti–PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159–70.
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, Shen S, Jeong S, Li Z, Zhu Y. M6A Demethylase ALKBH5 Regulates PD-L1 Expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res. 2021;81:4778–93.
Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L, Shen Q, Xu P, Zeng L, Zhou Y. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. 2019;10:4892.
Liu T, Wei Q, Jin J, Luo Q, Liu Y, Yang Y, Cheng C, Li L, Pi J, Si Y. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020;48:3816–31.
Lin X, Wang Z, Yang G, Wen G, Zhang H. YTHDF2 correlates with tumor immune infiltrates in lower-grade glioma. Aging (Albany NY). 2020;12:18476–500.
Tsuchiya K, Yoshimura K, Inoue Y, Iwashita Y, Yamada H, Kawase A, Watanabe T, Tanahashi M, Ogawa H, Funai K. YTHDF1 and YTHDF2 are associated with better patient survival and an inflamed tumor-immune microenvironment in non-small-cell lung cancer. Oncoimmunology. 2021;10:1962656.
Su G, Liu T, Han X, Sun H, Che W, Hu K, Xiao J, Li Y, Liu Y, Li W. YTHDF2 is a potential biomarker and associated with immune infiltration in kidney renal clear cell carcinoma. Fronts Pharmacol. 2021;12:709548.
Guo J, Wang L, Xu H, Zhu C, Wang J, Zheng Q. Gene Risk Signature of m6A Regulators, KIAA1429, YTHDF1, YTHDF2, METTL3 and Its Relationship with Immune Infiltration in Hepatocellular Carcinoma. 2020. https://doi.org/10.21203/rs.3.rs-47948/v1.
Bai X. The Role of YTHDF1 in Tumor Immune Microenvironment of Gastric Cancer. Hong Kong: Hong Kong University of Science and Technology; 2020.
Hu Y, Pan Q, Wang M, Ai X, Yan Y, Tian Y, Jing Y, Tang P, Jiang J. m6A RNA methylation regulator YTHDF1 correlated with immune microenvironment predicts clinical outcomes and therapeutic efficacy in breast cancer. Front Med (Lausanne). 2021;8:667543.
Hu J, Qiu D, Yu A, Hu J, Deng H, Li H, Yi Z, Chen J, Zu X. YTHDF1 Is a Potential Pan-Cancer Biomarker for Prognosis and Immunotherapy. Front Oncol. 2021;11:607224.
Zhang C, Guo C, Li Y, Ouyang L, Zhao Q, Liu K. The role of YTH domain containing 2 in epigenetic modification and immune infiltration of pan-cancer. J Cell Mol Med. 2021;25:8615–27.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Man Lei Y, Jabri B, Alegre ML. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science. 2015;350:1084–9.
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–8.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews M, Karpinets T, Prieto P, Vicente D, Hoffman K, Wei S. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
Routy B, Le Chatelier E, Derosa L, Duong CP, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Wang X, Li Y, Chen W, Shi H, Eren AM, Morozov A, He C, Luo GZ, Pan T. Transcriptome-wide reprogramming of N6-methyladenosine modification by the mouse microbiome. Cell Res. 2019;29:167–70.
Jabs S, Biton A, Bécavin C, Nahori MA, Ghozlane A, Pagliuso A, Spanò G, Guérineau V, Touboul D, Giai GQ. Impact of the gut microbiota on the m6A epitranscriptome of mouse cecum and liver. Nat Commun. 2020;11:1344.
Wang A, Tao W, Tong J, Gao J, Wang J, Hou G, Qian C, Zhang G, Li R, Wang D. m6A modifications regulate intestinal immunity and rotavirus infection. Elife. 2022;11:e73628.
Zhang X, Hao H, Ma L, Zhang Y, Hu X, Chen Z, Liu D, Yuan J, Hu Z, Guan W. Methyltransferase-like 3 modulates severe acute respiratory syndrome coronavirus-2 RNA N6-methyladenosine modification and replication. MBio. 2021;12:e0106721.
Liu JE, Xu YP, Li K, Ye Q, Zhou HY, Sun H, Li X, Yu L, Deng YQ, Li RT. The m6A methylome of SARS-CoV-2 in host cells. Cell Res. 2021;31:404–14.
Qiu X, Hua X, Li Q, Zhou Q, Chen J. m6A Regulator-Mediated Methylation Modification Patterns and Characteristics of Immunity in Blood Leukocytes of COVID-19 Patients. Front Immunol. 2021;12:774776.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, Peng S, Chen K, Wang M, Gong S. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–71.
Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME, Gabriel GJ, Zhou L, Bae N, Rowles J. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. 2014;5:658–65.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, Gan J, Jiang H, Jia GF, Luo C. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84.
Wang T, Hong T, Huang Y, Su H, Wu F, Chen Y, Wei L, Huang W, Hua X, Xia Y. Fluorescein derivatives as bifunctional molecules for the simultaneous inhibiting and labeling of FTO protein. J Am Chem Soc. 2015;137:13736–9.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172:90–105.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35:677–91.
Selberg S, Seli N, Kankuri E, Karelson M. Rational Design of Novel Anticancer Small-Molecule RNA m6A Demethylase ALKBH5 Inhibitors. ACS Omega. 2021;6:13310–20.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D, Hendrick AG. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601.
Einstein JM, Perelis M, Chaim IA, Meena JK, Nussbacher JK, Tankka AT, Yee BA, Li H, Madrigal AA, Neill NJ. Inhibition of YTHDF2 triggers proteotoxic cell death in MYC-driven breast cancer. Mol Cell. 2021;81:3048–64.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–34.
Visvanathan A, Patil V, Arora A, Hegde A, Arivazhagan A, Santosh V, Somasundaram K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–33.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112.
Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, Liu H, Deng Q, Wu X, Lan P. m6A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Onco Targets Ther. 2019;12:4391.
Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F, Validire P, Ingels A, Cathelineau X, Fridman WH. The clinical role of the TME in solid cancer. Br J Cancer. 2019;120:45–53.
He M, Lv W, Rao Y. Opportunities and Challenges of Small Molecule Induced Targeted Protein Degradation. Front Cell Dev Biol. 2021;9:685106.
Li M, Zha X, Wang S. The role of N6-methyladenosine mRNA in the tumor microenvironment. Biochim Biophys Acta Rev Cancer. 2021;1875:188522.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46.
Qomi SB, Asghari A, Mojarrad M. An overview of the CRISPR-Based genomic-and epigenome-editing system: function, applications, and challenges. Adv Biomed Res. 2019;8:49.
Laufer BI, Singh SM. Strategies for precision modulation of gene expression by epigenome editing: an overview. Epigenetics Chromatin. 2015;8:34.
Liu XM, Zhou J, Mao Y, Ji Q, Qian SB. Programmable RNA N6-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol. 2019;15:865–71.
Xia Z, Tang M, Ma J, Zhang H, Gimple RC, Prager BC, Tang H, Sun C, Liu F, Lin P, et al. Epitranscriptomic editing of the RNA N6-methyladenosine modification by dCasRx conjugated methyltransferase and demethylase. Nucleic Acids Res. 2021;49:7361–74.
Wilson C, Chen PJ, Miao Z, Liu DR. Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol. 2020;38:1431–40.
Li J, Chen Z, Chen F, Xie G, Ling Y, Peng Y, Lin Y, Luo N, Chiang CM, Wang H. Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res. 2020;48:5684–94.
Li X, Dong W, Nalin AP, Wang Y, Pan P, Xu B, Zhang Y, Tun S, Zhang J, Wang LS. The natural product chitosan enhances the anti-tumor activity of natural killer cells by activating dendritic cells. Oncoimmunology. 2018;7:e1431085.
Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47:820–33.
Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, Meng Z, Wu C, Guan KL, Malmberg KJ. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27:224–37.
Teng KY, Mansour AG, Zhu Z, Li Z, Tian L, Ma S, Xu B, Lu T, Chen H, Hou D. Off-the-shelf PSCA-directed chimeric antigen receptor natural killer cell therapy to treat pancreatic cancer. Gastroenterology. 2022. https://doi.org/10.1053/j.gastro.2021.12.281.
Ma R, Lu T, Li Z, Teng KY, Mansour AG, Yu M, Tian L, Xu B, Ma S, Zhang J. An oncolytic virus expressing IL-15/IL-15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 2021;81:3635–48.
Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Front Immunol. 2019;10:1205.
Karagiannis P, Kim SI. iPSC-Derived Natural Killer Cells for Cancer Immunotherapy. Mol Cells. 2021;44:541–8.
Lu T, Ma R, Li Z, Mansour AG, Teng KY, Chen L, Zhang J, Barr T, Caligiuri MA, Yu J. Hijacking TYRO3 from tumor cells via trogocytosis enhances NK-cell effector functions and proliferation. Cancer Immunol Res. 2021;9:1229–41.
Wang Y, Sun S, Zhang Z, Shi D. Nanomaterials for cancer precision medicine. Adv Mater. 2018;30:e1705660.
Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60.
Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. 2019;27:710–28.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, Cao M, Cai J, Wu J, Wang X. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. Hematol Oncol. 2020;13:5.
Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–10.
Song Y, Tang C, Yin C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials. 2018;185:117–32.
Li K, Lu L, Xue C, Liu J, He Y, Zhou J, Xia Z, Dai L, Luo Z, Mao Y. Polarization of tumor-associated macrophage phenotype via porous hollow iron nanoparticles for tumor immunotherapy in vivo. Nanoscale. 2020;12:130–44.
Ovais M, Guo M, Chen C. Tailoring nanomaterials for targeting tumor‐associated macrophages. Adv Mater. 2019;31:e1808303.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
L.X., M.S., and Y. J. conceived the review. L. X. and M. S. wrote the manuscript. D.Y., Y. P., and Y. J. revised the manuscript. All the authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors give consent for the publication of the manuscript in Molecular Cancer.
Competing interests
The authors declare that there is no potential competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, X., Ma, S., Deng, Y. et al. Targeting the RNA m6A modification for cancer immunotherapy. Mol Cancer 21, 76 (2022). https://doi.org/10.1186/s12943-022-01558-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01558-0