
Abstract
Autophagy is a highly conserved catabolic process seen in eukaryotes and is essentially a lysosome-dependent protein degradation pathway. The dysregulation of autophagy is often associated with the pathogenesis of numerous types of cancers, and can not only promote the survival of cancer but also trigger the tumor cell death. During cancer development, the microbial community might predispose cells to tumorigenesis by promoting mucosal inflammation, causing systemic disorders, and may also regulate the immune response to cancer. The complex relationship between autophagy and microorganisms can protect the body by activating the immune system. In addition, autophagy and microorganisms can crosstalk with each other in multifaceted ways to influence various physiological and pathological responses involved in cancer progression. Various molecular mechanisms, correlating the microbiota disorders and autophagy activation, control the outcomes of protumor or antitumor responses, which depend on the cancer type, tumor microenvironment and disease stage. In this review, we mainly emphasize the leading role of autophagy during the interaction between pathogenic microorganisms and human cancers and investigate the various molecular mechanisms by which autophagy modulates such complicated biological processes. Moreover, we also highlight the possibility of curing cancers with multiple molecular agents targeting the microbiota/autophagy axis. Finally, we summarize the emerging clinical trials investigating the therapeutic potential of targeting either autophagy or microbiota as anticancer strategies, although the crosstalk between them has not been explored thoroughly.
Similar content being viewed by others


Background
Autophagy is an evolutionarily conserved intracellular recycling and cellular self-degradation process that occurs in eukaryotes and plays a critical role in the maintenance of homeostasis in various biological processes [1, 2]. Many studies have indicated that autophagy, as a cell death mechanism, plays an important pathophysiological role in various disease processes, including cell death, infection, heart diseases, neurodegeneration, autoimmune diseases and cancer [3,4,5,6,7,8]. Autophagy serves multiple functions in cancer progression by modulating cell death, and further studies have shown that autophagy plays a dual role in cancer. It can promote malignant transformation in certain tumors and suppress tumor growth in others [1, 2, 9]. Previous reports showed that BECN1, an essential autophagy-related gene (ATG), was deleted in 40 to 75% of breast, ovarian, and prostate cancers, suggesting the role of autophagy in tumor growth suppression [10,11,12]. In addition, autophagy is upregulated in RAS-transformed tumors, hence promoting their survival, growth and tumorigenesis [13,14,15]. Autophagy can be classified based on the mechanism into common (nonselective) or selective types. Common autophagy involves the packaging of cytoplasmic portions into autophagosomes and the delivery of these cargoes to lysosomes for degradation. In contrast, selective autophagy is activated when specific targets, such as protein aggregates, damaged cell organelles, and intracellular pathogens, are recognized [16,17,18].
The correlation between cancer and microbiota is still unclear. It is well known that genetic and environmental factors are critical for the initiation and progression of cancer, but recent studies have shown that microorganisms are also indispensable [19]. Gastric [20], ovarian [21], pancreatic [22], prostate [23], lung [24] and breast cancers [25], in addition to cholangiocarcinoma [26], have all been found associated with microorganism infection. Recently, mucosa-associated microbiota was shown to be essential for the microenvironment of various malignant tumors; moreover, intratumoral organisms can influence tumorigenesis and metastasis [27,28,29,30,31]. Garrett W.S. also indicated that the ways in which microbes and microbiota contribute to carcinogenesis, whether by enhancing or diminishing a host’s risk, fall into three broad categories: (i) altering the balance of host cell proliferation and death, (ii) guiding immune system function, and (iii) influencing the metabolism of host-produced factors, ingested foodstuffs, and pharmaceuticals [32]. Thus, micromolecular drugs targeting microorganisms have become a research hotspot in the context of antitumor treatment, mainly for tumors caused by microbial infections. Moreover, certain microorganisms that can affect the response of tumors to other therapy strategies can be used to treat cancer, referred as “use microorganisms to treat microorganisms” [33, 34].
Aside from their respective functions in pathological and physiological conditions, the crosstalk between autophagy and microbiota is vital in tumorigenesis and resistance to chemotherapy drugs [35, 36]. Autophagy induced by Porphyromonas gingivalis infection controls cell proliferation and G1 arrest in host oral cancer cells [35]. Fusobacterium nucleatum promotes cancer by upregulating ULK1 and ATG7 to induce resistance to oxaliplatin and 5-fluorouracil (5-FU) in colorectal cancer (CRC) [36]. More studies on the crosstalk between microbiota and autophagy are needed to better explore different variables and define the correlation, as well as the mechanisms through which they affect cancer progression. A deeper understanding of the molecular mechanisms of the microbiota/autophagy axis may help to design improved anticancer drugs for clinical use.
In this review, we described the critical roles of microbiota and autophagy in tumors separately and their characteristics in tumor progression. Moreover, this review analyzed the effects and mechanisms of autophagy regulated by different bacteria on the biological behavior of tumors, as well as the impact of autophagy on the carcinogenesis of microbiota, emphasizing the dominant role of autophagy in the interaction between the microbiota and human tumors. In addition, multiple drugs were summarized, and clinical trials or animal experiments were evaluated to assess the therapeutic potential of targeting the microbiota/autophagy axis as anticancer strategies.
Overview of microbiota and autophagy
Microbiota and cancer
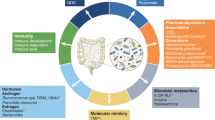
During cancer development, the microbial community may have an indirect carcinogenic function by promoting mucosal inflammation or causing systemic disorders [37], where most studies focused on the oral and intestinal microbiota (Fig. 1).

Overview of the mechanisms by which bacteria regulate tumor progression. Pathogenic bacteria mainly come from oral and gastrointestinal flora. Different kinds of bacteria can promote the occurrence, development and metastasis of tumors by causing host adaptive immune responses, cell cycle arrest, DNA transcription changes and DNA damage, reactive oxygen species accumulation and the activation of various signaling pathways, such as Wnt/β-catenin, NF-κB, and STAT3 signaling
The oral microbiome is very complex, and common pathogenic bacteria include Streptococcus anginosus, Veillonella, F. nucleatum and P. gingivalis [38, 39]. Studies have shown that pathogenic bacteria in the oral cavity are associated with multiple cancers of the digestive system [38]. The development of esophageal carcinoma has been reported to be related to P. gingivalis [40] and F. nucleatum [41]. The detection rate of P. gingivalis in esophageal squamous cell carcinoma (ESCC) is higher than that in adjacent tissue or normal controls [42]. In addition, P. gingivalis might utilize the miR-194/GRHL3/PTEN/AKT axis to promote ESCC proliferation and migration [43]. The F. nucleatum DNA concentration in esophageal cancer tissue is significantly higher than that in normal esophageal tissue [41]. The levels of oral bacteria (such as Streptococcus and Clostridium) in gastric cancer tissue are significantly higher than those in normal tissue, while Lactobacillus brevis is less enriched than that in the non-tumor tissue conversely [ 44]. The proportions of Bacteroides and Firmicutes in patients with non-cardia gastric cancer decrease significantly [ 45]. The abundance of Neisseria elongata and Streptococcus mitis in the salivary microbiome of pancreatic cancer patients are significantly lower than those in the healthy population [46]. Researchers also found a significantly higher ratio of Leptotrichia to Porphyromonas in the saliva of patients with pancreatic cancer than in that of healthy patients or those with other diseases [47]. In a controlled study, P. gingivalis and F. nucleatum were found to be associated with a higher risk of pancreatic cancer [48]. P. gingivalis can promote the proliferation of pancreatic cancer cells without the function of TLR2 [49]. Studies have shown that the total abundance of F. nucleatum in CRC is 415 times higher than that in adjacent normal tissue, and a positive correlation with lymph node metastasis has also been observed [50].
Certain bacteria in the gastrointestinal tract are associated with cell dysplasia and carcinogenic effects. Campylobacter jejuni [51] and F. nucleatum [52] have been shown to be cancer-causing intestinal bacteria in CRC. Helicobacter pylori increases the expression of VCAM1 in cancer-associated fibroblasts (CAFs) via JAK/STAT1 signaling pathway in gastric carcinoma, and the level of VCAM1 in patients with gastric cancer is positively correlated with tumor progression and a poor prognosis. Moreover, the interaction between CAF-derived VCAM1 and integrin αvβ1/5 could promote gastric cancer cell invasion both in vitro and in vivo [53]. In a study of CRC, it was found that chronic inflammation can be promoted via the accumulation of certain bacteria (such as Escherichia coli) and induce carcinogenesis by cell lethal expansion toxin produced by C. jejuni [51]. Furthermore, oncogenic transcriptional changes in CRC cell lines are associated with FadA adhesion complexes of F. nucleatum [27, 52]. F. nucleatum level is found increased in the stool samples of colorectal adenoma and cancer patients, and is also enriched in adenocarcinomas and adenomas compared with normal colonic tissues [54]. F. nucleatum increases the expression of miR-21 through the TLR4/MYD88/ NF-κB pathway and induces CRC cell proliferation and migration [55]. Pks+ E. coli could output DNA adducts and then strengthen the ability of colibactin to generate mutations in tumor suppressor genes or oncogenes, which contribute to cancer initiation and progression in mammalian and mouse cells [56]. Enterotoxigenic Bacteroides fragilis (ETBF), a common commensal bacterium in human intestine, strongly induces colonic tumors in multiple intestinal neoplasia (Min) mice via STAT3- and Th17- dependent pathways [57, 58]. Peptostreptococcus anaerobius is more abundant in the stool samples of CRC patients than in those of normal controls without CRC [59], which could combine with α2/β1 integrin through its surface protein PCWBR2 [60]. The crosstalk between PCWBR2 and α2/β1 integrin could induce focal adhesion kinase phosphorylation and then activate the PI3K/AKT pathway in CRC cells, increase cell proliferation, activate NF-κB signaling, and finally contribute to chronic inflammation and tumor progression [60]. The abovementioned bacteria and other cancer-related microbiota are all summarized in Table 1.
Autophagy and cancer
Autophagy is a degradation pathway by which eukaryotic cells degrade damaged organelles and proteins through lysosomes and is widely found in both normal cells and malignant tumor cells [71, 72]. The process of autophagy mainly includes the formation and extension of isolation membranes or phagocytic bubbles, the formation of autophagosomes, the fusion of autophagosomes and lysosomes to form autophagolysosomes, and the final degradation of intracellular substances [17]. Degradation products, such as amino acids and fatty acids, can be reused by cells [4], which is considered to be a favorable repair and defense mechanism [73]. Gozuacik and other scholars have pointed out that the occurrence of some malignant tumors is accompanied by the inhibition of autophagy [74]. A large number of studies have found that autophagy is closely but complexly related to malignant tumors, particularly affecting the processes of recurrence, metastasis and drug resistance [75] (Fig. 2). TRPM3 is highly expressed in clear cell renal cell carcinoma and induces a high level of autophagy by activating the upstream CAMKK2/ULK1 cascade and inhibiting endogenous miR-214 through the CAMKK2/AMPK pathway, ultimately promoting tumor growth [76]. TRIM59, which contains tripartite motifs, inhibits the NF-κB pathway, downregulates the transcription of BECN1, and affects the ubiquitination level of BECN1 via TRAF6-induced K63 linkage at the same time. As a result, it blocks the establishment of the BECN1/PIK3C3 complex, which triggers the downstream autophagy cascade [77]. POX induces protective autophagy, which can promote HT-29 cell survival in the hypoxic tumor microenvironment through AMPK activation [78]. In non-small-cell lung cancer (NSCLC), casein kinase 1 alpha (CK1α), an autophagy inducer, activates the PTEN/AKT/FOXO3a/ATG7 axis, which increases autophagy and suppresses tumor progression to negatively regulate tumor growth [79]. IFN-γ induces the formation of autophagosomes and the conversion of LC3 through the IRF-1 signaling pathway, which aids to construct the autophagy complex. IFN-γ not only inhibits cell growth, rather it induces non-apoptotic cell death in Huh7 HCC cells [80]. In addition, IFN-γ can also upregulate the expression of Beclin-1, which is vital in autophagy in gastric epithelial cells, and can suppress IL-1β-induced inflammation, H. pylori-induced epithelial apoptosis, cell proliferation, and Dckl1+ cell elevation to aid in inhibiting bacterial infection and gastric mucosa carcinogenesis [81]. Knockdown of lncRNA HOTAIR with siRNA reduces autophagy, inhibits EMT, decreases cell viability, suppresses cell proliferation, induces cell apoptosis, and enhances sensitivity to radiotherapy in radioresistant HeLa cells through downregulating the activity of the Wnt/β-catenin signaling pathway [82]. When overexpressed, lncRNA CTA inhibits autophagy by decreasing the level of LC3-II isoforms and BNIP3/BNIP3L expression. It also promotes cell apoptosis induced by chemotherapy in osteosarcoma [83]. MiR-93 can suppress autophagic activity by downregulating the expression of BECN1, ATG4B, ATG5, and SQSTM1 in glioblastoma stem cells (GSCs), which results in the suppression of tumor cell growth and glioma sphere self-renewal and the enhancement of temozolomide (TMZ) activity to combat tumor progression by inhibiting autophagy [84]. In CRC, miR-18a* and miR-4802, which target ULK1/ATG7, regulate autophagy via the TLR4 and MYD88 signaling pathways. Both of these miRNAs promote CRC chemoresistance to oxaliplatin and 5-FU [36]. The silencing of FXYD6 promotes prosurvival autophagy and inhibits apoptosis by regulating the activity of ATP-α1, whereas the overexpression of FXYD6 increases chemosensitivity in CRC [85]. Knockdown of IRF1, which negatively correlates with ATG7, increases the level of autophagy by blocking IGF1 receptor and BECN1 expansion, thus promoting resistance to antiestrogens in breast cancer [86]. HMGB1 induces autophagy by increasing LC3-II expression, decreasing p62 expression and inhibiting the formation of autophagosomes through the PI3K/MEK/ERK pathway, thus promoting chemotherapy resistance in leukemia cells [87]. Studies have suggested that the absence of autophagy might lead to cancer development, but autophagy itself may also promote tumorigenesis [88, 89]. To a large extent, changes in autophagy levels may increase autophagy induction or inhibit autophagy activity in tumor cells, but in general, there are three main situations in which changes occur: first, the hypoxic “low-nutrient environment” of the tumor leads to increased autophagy [90]; second, some cancer-related genes are closely related to the autophagy process, and their aberrations lead to changes in autophagy activity [91]; third, changes in lysosomal activity and transport capacity in tumor cells may also lead to large variations in autophagy levels [92]. Autophagy levels vary among different types of tumor cells and different stages of tumorigenesis and even in different parts of tumor tissue [93]. Consistent with the complex changes in autophagy activity, the effects of autophagy on cancer also require special analysis. First, autophagy can provide tumor cells with the metabolites needed for growth and maintain the stability of the internal environment, thus promoting cancer [94,95,96]. Second, autophagy can avoid the threats of oxidative stress, persistent inflammation, and DNA damage and thus playing a role in inhibiting cancer [97]. The above molecules and their mechanisms of regulating autophagy in tumor progression are summarized in Table 2.

Different proteins or small molecules regulate autophagy pathways. Autophagy is a key regulator during tumorigenesis. It can not only promote cancer by providing nutrition for tumor cells but can also inhibit cancer progression by increasing apoptosis. Autophagy can be divided into four parts: initiation, elongation, maturation, and the fusion of autophagosomes and lysosomes. ATGs participate in each step and eventually promote or inhibit cancer
Crosstalk between microbiota and autophagy in cancer development
H. pylori is a major risk factor for gastric cancer [98]. Studies have shown that approximately 2% ~ 3% of people infected with H. pylori eventually develop gastric cancer [99]. Studies have also shown that autophagy can be induced by the concerted action of H. pylori and virulence factors in tumor cells [100]. Cytotoxic-related gene A (CagA) and vacuolar toxin A (VacA) are the main pathogenic factors of H. pylori, and both factors are related to autophagy and gastric cancer [101] (Fig. 3). CagA was reported to inhibit autophagy by activating the PI3K/AKT/mTOR pathway [102]. As soon as CagA enters gastric epithelial cells, it may be phosphorylated by tyrosine, which is activated by many signaling factors, therefore changing the cytoskeleton of polyactin, inducing an inflammatory response, initiating apoptosis, suppressing autophagy, and leading to cell scattering [103,104,105,106,107]. VacA is another main factor mediating the involvement of H. pylori in regulating autophagy [108]. When gastric mucosal cells are exposed to VacA for a short period of time, autophagy levels can increase, therefore inhibiting tumor growth [103]. When gastric epithelial cells are exposed to VacA for a long period of time, antiphagocytic pathways are affected [109]. Long-term H. pylori infection decreases the level of autophagy and induces the collection of the autophagic substrate p62, which can subsequently interact with Rad51, a DNA repair marker, directly through its UBA domain, resulting in the promotion of Rad51 ubiquitination and degradation, thus suppressing the capability to repair damaged DNA. In addition, H. pylori might promote gastric tumorigenesis by promoting double-strand breaks (DSBs) and genomic instability [110]. Therefore, eradicating H. pylori and increasing autophagy can help prevent gastric cancer [111, 112]. At present, the mechanism of autophagy in promoting gastric cancer formation by H. pylori is unclear, thus further studies are mandated.

Crosstalk between autophagy and microbiota in cancer. The crosstalk between autophagy and microbiota regulates multiple physiological and pathological responses, including cancer progression. Autophagy can play a role in microbiota-mediated tumorigenesis, metastasis and drug resistance in different cancers. H. pylori regulates miR-543, miR-125b-5p and autophagy through its virulence factor CagA to promote tumor occurrence. F. nucleatum mainly regulates autophagy through TLR/MyD88 and its downstream miRNA-18a*/4802 and then plays a cancer-promoting role in colorectal cancer. P. gingivalis can induce G1 cell cycle arrest. C. jejuni can enhance cell radiosensitivity by producing cytolethal distending toxin (CDT). S. typhimurium inhibits tumor cell survival through autophagy
As mentioned above, F. nucleatum is closely related to the occurrence and development of CRC [50]. With the development of CRC, the abundance of F. nucleatum in tumor tissues increases gradually compared to that in normal tissues [113]; this finding has also been supported by the positive correlation between F. nucleatum and the American Joint Committee on Cancer (AJCC) stage of CRC patients [114]. Fang JY from Shanghai Institute of Digestive Diseases pointed out that F. nucleatum can promote resistance to chemotherapeutic drugs in CRC patients by affecting autophagy-regulating miRNAs [36]. For other cancers, studies have shown that F. nucleatum regulates the expression of endogenous LC3 and ATG7 and the formation of autophagosomes, resulting in chemoresistance to 5-FU, cisplatin (CDDP), and docetaxel [115].
The role of microbiota in autophagy-related cancer
Helicobacter pylori
The detailed mechanism of the occurrence and development of gastric cancer mediated by H. pylori is still unclear and is influenced by many variables, including strain-specific bacterial components, the complex role of the inflammatory response, the diversity of host heredity, environmental impact, and so on. Recent studies have revealed that megakaryocyte autophagy induced by H. pylori is a conserved process by which eukaryotic cells maintain intracellular environmental stability and combat external stress, thus promoting tumor progression [116, 117]. It has also been reported that H. pylori can escape autophagy by downregulating the expression of autophagy proteins and can facilitate gastric carcinogenesis [118]. The mechanism of autophagy caused by H. pylori infection is complex in gastric cancer, and autophagy might have different functions at different infection stages [119]. Terebiznik first reported that H. pylori triggered autophagy in gastric adenocarcinoma epithelial cells and identified LC3 as an autophagy-related marker [104]. Furthermore, Yahiro et al. demonstrated that low-density lipoprotein receptor-associated protein-1 (LRP-1) could mediate autophagy through VacA in gastric cancer epithelial cells, which might be associated with the activation of VacA channels to promote the formation of tyrosine phosphatase-related vacuoles [120]. As another important virulence factor of H. pylori, CagA has been suggested to trigger the overexpression of miR-543 (Fig. 3), which can suppress autophagy by interacting with SIRT1, to increase EMT and promote tumor cell migration and invasion in gastric cancer [121].
In recent years, a growing number of scholars have reported that the occurrence of colon cancer is closely related to H. pylori infection [122]. Pyrogallol treatment significantly reduced the viability rate of H. pylori to 62% and exhibited marked antimetastatic potential via inhibiting the migration of HT-29 cells [123]. IHC staining was applied and demonstrated that Beclin-1 was abnormally expressed in colon cancer tissue infected with H. pylori. Therefore, it was speculated that Beclin-1 mediated the tumor-promoting activity of H. pylori. Moreover, CagA-positive H. pylori might decrease the level of miR-125b-5p (Fig. 3), resulting in high expression of LC3-II/LC3-I and Beclin-1, which are key molecules related to autophagy in colon cancer, thereby inducing the autophagy, proliferation and invasion of colon cancer cells, hence inducing colon cancer [124]. In addition to regulating the autophagy and apoptosis of colon cancer cells, H. pylori can inhibit gastric acid secretion, indirectly causing the excessive release of gastrin, which leads to the abnormal proliferation of intestinal cells, inducing colon polyps and even colon cancer [125].
Fusobacterium nucleatum
Available data show that F. nucleatum infection is mainly related to the development of esophageal and pancreatic cancers [41, 48]. In addition, Gallimidi et al. found that F. nucleatum plays an essential role in the development of oral cancer. F. nucleatum can stimulate the level of IL-6 through the TLR signaling pathway in tumor cells and subsequently activate the STAT3 pathway, thereby promoting the growth of oral squamous cell carcinoma (OSCC) [126]. Fewer studies have investigated the relationship between IL-6 and autophagy in the development of oral cancer. Existing studies revealed that IL-6 can upregulate Beclin-1 and induce autophagy by increasing the level of NS5ATP9 through NF-κB activation; in turn, NS5ATP9 can upregulate IL-6 levels, which subsequently further induced autophagy in liver cancer [127]. Moreover, IL-6 promotes the formation of autophagosomes, strengthens autophagic flux through the IL-6/JAK2/BECN1 pathway and induces chemotherapy resistance in CRC [128]. LC3-II is a key molecule in tumor proliferation and progression, and its expression level can directly reflect the activity of autophagy [129]. Excessive secretion of IL-6 is an adaptive mechanism of autophagy that occurs by means of LC3-II induction; however, there is still no consensus on its effects.
As mentioned above, autophagy plays a critical role in the induction of CRC via F. nucleatum infection [48, 50]. F. nucleatum can resist chemotherapeutic drugs by regulating autophagy. In vivo and in vitro studies demonstrated that autophagy levels increased in intestinal cancer cells infected with F. nucleatum. Western blotting results demonstrated that F. nucleatum infection increases the expression of various autophagy signaling elements, such as pAMPK, ATG7, pULK1, and ULK1, in HCT116 and HT-29 cells [36]. The expression levels of miR-18a* and miR-4802 are decreased in intestinal cancer cells infected with F. nucleatum to induce chemoresistance, while the expression levels of target genes ULK1 and ATG7 are significantly increased in these cells. Overexpression of miR-18a* and miR-4802 inhibited autophagy induced by F. nucleatum in intestinal cancer cells, while inhibition of miR-18a* and miR-4802 had the opposite effect. Therefore, in summary, F. nucleatum plays a vital role in inducing CRC chemoresistance through selectively silencing miR-18a* and miR-4802 and activating autophagy pathways [36]. In addition, the F. nucleatum concentration was found to be positively related to the level of CARD3, which was lower in those without metastasis. Downregulation of CARD3 decreased the expression of proteins related to migration, invasion, metastasis, and autophagy and the formation of autophagosomes induced by F. nucleatum in vitro or in vivo. Thus, F. nucleatum was also demonstrated to activate autophagy by targeting CARD3 to promote CRC metastasis [130].
Porphyromonas gingivalis
P. gingivalis is an important pathogen causing periodontitis, OSCC, and even esophageal cancer [43, 131, 132]. Bélanger et al. found that the survival of P. gingivalis depended upon autophagy activation of endothelial host cells [133]. After P. gingivalis invades the cells, the endogenous transport and autophagy process is aberrantly regulated; thus, it can escape degradation by host cells. At the same time, a microenvironment suitable for bacterial colonization and proliferation is created in modified autophagosomes. In addition, P. gingivalis infection is considered to promote autophagy and inhibit tumor cell proliferation by inducing G1 cell cycle arrest in host oral cancer cells [35]. The detailed autophagy-related mechanism by which microbiota regulate human cancer is summarized in Table 3.
Targeting microbiota/autophagy in cancer therapy
Since autophagy plays an important role in the occurrence and development of tumors, it may promote tumor formation, or inhibit tumor cell growth and metastasis through certain mechanisms. In recent years, breakthrough results have been obtained in clinical trials assessing the relationship between autophagy and cancer. A series of drugs targeting autophagy have also been introduced through animal experiments or clinical trials. In the following section, we systematically summarized the drugs that individually target autophagy and exert anticancer effects and their mechanisms.
Alternatively, the abovementioned microbial community members, especially H. pylori, may indirectly cause cancer by promoting mucositis or causing systemic diseases. Studies have shown that H. pylori is one of the most important risk factors for gastric cancer. Therefore, we considered whether inhibiting the carcinogenic microbial community in the tumor microenvironment can delay the occurrence of tumors and even play an anticancer role. Similarly, we also summarized drugs targeting the microbial community in the tumor microenvironment and their mechanisms, as well as the corresponding clinical trials.
Most importantly, we emphasized the critical role of autophagy in mediating microbial communities and cancer. A variety of bacteria regulate tumor formation, development, invasion and metastasis by inducing or inhibiting autophagy. Therefore, the simultaneous use of drugs targeting autophagy and microorganisms may play a synergistic anticancer role. However, few such studies have been performed, and more clinical trials are urgently needed to better understand the therapeutic potential of drugs targeting the microbiota/autophagy axis, which could help to develop new drugs to prevent or treat human cancer.
Clinical trials of drugs targeting autophagy
Chloroquine (CQ) is a quinoline derivative belonging to the heterocyclic aromatic compound family and has been used as an antimalarial therapy for many years [145]. Moreover, CQ and its derivatives block autophagy by regulating lysosomal function [146]. However, there are few animal experiments or clinical trials on its anticancer activity as a monotherapy drug. Studies have shown that CQ can inhibit autophagy and enhance the efficacy of many anticancer agents in a variety of mouse cancer models, such as colon cancer [147, 148], head and neck cancer [149] and gallbladder cancer [150]. We found clinical trials that attempted to investigate the antitumor activity of CQ in small cell lung cancer (SCLC) and breast cancer [151] patients, as shown in Table 4.
Hydroxychloroquine (HCQ) is a derivative of CQ that has an extra hydroxyl group; however, it has 40% lower toxicity than that of CQ. Moreover, HCQ has stronger anti-inflammatory effects; therefore, it has been used to treat rheumatoid arthritis and systemic lupus erythematosus [152, 153]. Similarly, HCQ can increase the cytotoxicity of various chemotherapies and target therapies by inhibiting autophagy [154]. However, a phase II clinical trial and pharmacodynamic study of HCQ in metastatic pancreatic adenocarcinoma patients showed that HCQ alone could induce severe autophagy inhibition but with negligible therapeutic efficacy [155]. A clinical trial (NCT02013778) was developed to confirm the dose limiting toxicity and maximum tolerated dose of the oral administration of HCQ combined with transarterial chemoembolization (TACE) in the management of hepatocellular carcinoma (HCC). Furthermore, another phase 0 clinical trial, NCT00962845, investigated the role of HCQ in disrupting autophagy, which may promote the survival of cancer cells under chemotherapy in prostate cancer (PCa). Other clinical studies on HCQ are summarized in Table 4. In addition, there are other micromolecular drugs that can play anticancer roles by regulating autophagy. Compared with HCQ, Lys05, a water-soluble salt of Lys01 that has a 10-fold greater ability to inhibit autophagy than HCQ and can improve the degradation of lysosomes more potently, ultimately downregulating autophagy and tumor growth. Contrary to HCQ, Lys05 shows single-agent antitumor activity and less toxicity at lower doses in animal models [136]. By adjusting the structure of Lys05, DQ661 emerged and has been regarded as an anticancer compound that inhibits mTOR and autophagy. DQ661, as the most important lysosomal inhibitor [137], can promote DNA damage independent of apoptosis, decrease autophagic flux and contribute to lysosomal membrane permeability (LMP). DQ661 treatment significantly reduced the tumor volume compared with that of the control mice, and the weight of the mice did not significantly change. Immunoblotting revealed that mTORC1 and autophagy were inhibited in a melanoma mouse model. In addition, DQ661 improved survival in a colon cancer mouse model and showed antitumor activity in a syngeneic pancreatic model that was resistant to immunotherapy [138]. SAR405, a selective PIK3C3/Vps34 inhibitor, can prevent the formation of late endosome and lysosome compartments by inhibiting the activity of PIK3C3 kinase and can suppress autophagy and mTOR signaling synergistically in tumor cells. Vps34i inhibits autophagic flux in multiple cancer mouse models, including melanoma, CRC and renal cell carcinoma models. Systemic treatment (oral gavage) with Vps34i contributed to significant tumor weight reduction, tumor growth suppression and survival improvement in cancer-bearing mouse, which means that SB02024 and SAR405 (Vps34i) do not target one specific cancer type for combating cancer, and both might be used in multiple tumor models [139]. 3-Methyladenine (3-MA) [156], wortmannin [157] and LY294002 [158] are all PI3K inhibitors that inhibit autophagy by disrupting the production of PI3P through inhibiting the class III PI3K complex [159], which plays a vital role in recruiting other ATG proteins for the activation of autophagy at the isolation membrane [160, 161]. The effects of 3-MA and wortmannin in regulating autophagy are slightly different. 3-MA can promote autophagy in complete medium when applied for a long time, but it can also inhibit starvation-induced autophagy, as shown in previous studies. Nevertheless, wortmannin is capable of suppressing autophagy in any nutrient status. 3-MA promotes autophagy by transiently suppressing class III PI3K, while wortmannin persistently inhibits class III PI3K [162]. Many clinical trials have been developed to explore the anticancer effects of 3-MA and wortmannin in combination with other cancer treatments. For example, 3-MA inhibits autophagy, which might be a resistance mechanism of colon cancer cells against 5-FU, and enhances the apoptosis of colon cancer cells treated with 5-FU; thus, inhibition of autophagy could decrease the chemoresistance of colon cancer [163]. Lin J et al. demonstrated that wortmannin could markedly enhance the antitumor effect of Ag nanoparticles (NPs) in a B16 mouse melanoma cell model [164]. However, there are few clinical trials on the single-agent antitumor activity of 3-MA and wortmannin; therefore, more work needs to be done on this topic. Mefloquine hydrochloride suppresses the expression of lysosomal LAMP1/LAMP2, which plays an important role in the formation of autolysosomes and inhibits CD133/CD44v9 colon cancer stem cells (CSCs) to exert its antitumor effect. Notably, mefloquine alone can achieve almost the same effects as other anticancer agents [165]. Spautin-1 was developed for autophagy attenuation and can induce the degradation of PI3K/Vps34 complexes by inhibiting USP10 and USP13, two ubiquitin-specific peptidases targeting the Beclin-1 subunit [141]. Additionally, spautin-1 has also been shown to suppress the phosphorylation of EGFR and the activation of its downstream signaling, contributing to cell cycle arrest and programmed cell death in PCa in a USP10/USP13 independent manner [166]. In melanoma, spautin-1 can decrease tumor growth and enhance the antitumor effect of cisplatin by targeting USP10 and USP13 [142]. Rapamycin (sirolimus), a macrolide immunosuppressant, was first isolated from Streptomyces hygroscopicus and was found to inhibit mTOR protein kinase, which plays an important role in downregulating autophagy [143, 167]. Temsirolimus is an ester of rapamycin that can selectively inhibit mTOR kinase and subsequently disrupt cell cycle regulatory protein translation and then exert an anticancer effect [168]. Both rapamycin and temsirolimus can modulate autophagy by inhibiting the mTOR pathway, and some associated clinical trials have started to study their anticancer effects in various cancer entities. Autophagy-related drugs and related clinical trials are summarized in Table 4.
Clinical trials of drugs targeting microbiota
H. pylori infection is an important risk factor for gastrointestinal diseases, such as gastric inflammation, gastric cancer and gastric mucosa-related lymphoid-tissue lymphoma [169]. Therefore, the eradication of H. pylori may inhibit the occurrence of gastric cancer. Traditionally, the standard scheme for the eradication of H. pylori is the triple therapy of anti-secretory agents, such as proton pump inhibitors, combined with two kinds of antibiotics, such as amoxicillin, levofloxacin, clarithromycin, and metronidazole [169]. In addition, phytomedicines and probiotics have also been used for the treatment of H. pylori infection in recent studies [170]. The purpose of NCT04660123 is to observe the eradication rate of H. pylori infection, the improvement of symptoms, and the incidence of adverse effects in gastric cancer patients treated with bismuth colloidal pectin granule quadruple therapy. Itraconazole is a broad-spectrum antibiotic and is a triazole antifungal agent, which has commendable pharmacodynamic and pharmacokinetic profiles and is broadly applied for preventing or treating systemic fungal infections [171, 172]. Recent studies demonstrated that itraconazole can induce autophagy, downregulate the expression of steroid carrier protein 2; and redistribute intracellular cholesterol to inhibit glioblastoma growth [173]. In addition, itraconazole can exert its anticancer effect through the Hedgehog signaling pathway [174]. Itraconazole promotes apoptosis and autophagy by inhibiting the Hedgehog pathway, resulting in a reduction in tumor cells in vitro, and this phenomenon was also observed in a human xenograft breast cancer mouse model, demonstrating its anticancer effect in breast cancer [175]. Itraconazole induces cell cycle arrest and cell apoptosis and then decreases the proliferation, invasion and migration of OSCC cells by inhibiting the Hedgehog pathway. Similarly, itraconazole disrupted the growth of tumor cells, decreased the expression of Ki67, and induced cell apoptosis in the OSCC patient-derived xenograft (PDX) model [176]. Itraconazole can decrease the proliferation and growth of colon cancer cells by promoting autophagy and apoptosis; at the same time, it can reduce the protein expression levels of shh and Gli1 in a dose-dependent manner [177]. Furthermore, the purpose of NCT02749513 is to demonstrate that orally administered itraconazole can inhibit the Hedgehog pathway in patients with esophageal cancer, including adenocarcinoma and squamous cell carcinoma. Probiotics can regulate the gut microbiota, positively affect the interaction between the immune system and microbiota and are beneficial for preventing inflammation and CRC [178]. The purpose of NCT03072641 is to confirm whether probiotic bacteria have a beneficial effect on the colon cancer-associated microbiota and epigenetic alterations. The clinical trials of the above drugs are summarized in Table 5. Taking Bifidobacterium orally alone enhances the local control rate of tumors to the same degree as PD-L1-specific antibody therapy, and combination therapy almost stops tumor growth. Bifidobacterium-treated mice showed significantly enhanced local tumor control compared with the untreated group, and this effect was accompanied by the accumulation of antigen-specific CD8+ T cells in the tumor microenvironment [179]. In addition, oral administration of the probiotic candidate DTA81 might have beneficial effects for preventing CRC development [180]. The relevant clinical trials of the above drugs are shown in Table 5.
Antitumor effects of antibiotics combined with autophagy inhibitors
At present, there are few antitumor studies evaluating the effect of combined therapy with antibiotics and autophagy inhibitors, and rapamycin is the most commonly used antibiotic. Some scholars attempted to use rapamycin combined with the autophagy inhibitor 3-MA to treat human lung cancer in vitro. Western blotting and MTT results showed that the combination with an autophagy inhibitor could significantly increase the decline in cell survival caused by rapamycin. Liu et al. found that rapamycin and CQ have certain antitumor effects respectively in vitro, and their combined use can enhance the antitumor effect on osteosarcoma [181]. Moreover, rapamycin was further confirmed to promote autophagy by blocking the mTOR pathway in the human osteosarcoma cell line MG63; and CQ was found to enhance apoptosis by blocking autophagy, suggesting that CQ may amplify the effects of rapamycin in inducing apoptosis via autophagy inhibition [182].
Concluding remarks
Autophagy is a conserved degradation mechanism in eukaryotic cells. The response of eukaryotic cells to external antigens and intracellular aging substances triggers autophagy activation. An imbalance in autophagy is often observed in human cancer. Therefore, it is speculated that autophagy can play a regulatory role in tumor cells, as it can promote tumor growth by providing metabolic energy for tumor cells or inhibit tumor cell proliferation and metastasis by activating certain intracellular signaling pathways. Microbiota are leading causes of cancer that act either by inducing mucosal inflammation or causing systemic disorders. More importantly, bacteria have been globally proven to promote or inhibit cancer via autophagy regulation, suggesting a complex interaction between autophagy and bacteria. This review explains how bacteria regulate tumor development, progression, invasion and metastasis by inducing or inhibiting autophagy and summarizes the influence of various bacteria-mediated autophagy mechanisms on the biological behavior of cancer.
Considering that both autophagy and microbiota can play a certain role in tumor progression and that crosstalk between these factors has been discovered recently, the inhibition of autophagy and/or microbiota with drugs may be beneficial for controlling tumor development. This strategy appears more achievable with the emergence of innovatively developed direct and/or nonspecific small molecule inhibitors of the autophagy/microbiota axis.
Availability of data and materials
The materials that support the conclusion of this review have been included within the article.
References
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–42.
Chung C, Seo W, Silwal P, Jo EK. Crosstalks between inflammasome and autophagy in cancer. J Hematol Oncol. 2020;13:100.
Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–41.
Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73.
Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–55.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75.
Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–9.
Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19:12.
Yun CW, Lee SH. The roles of autophagy in cancer. Int J Mol Sci. 2018;19:3466.
Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65.
Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6.
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70.
Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–78.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29.
Seglen PO, Gordon PB, Holen I. Non-selective autophagy. Semin Cell Biol. 1990;1:441–8.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
Zaffagnini G, Martens S. Mechanisms of selective autophagy. J Mol Biol. 2016;428:1714–24.
de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15.
Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC, et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. 2018;67:226–36.
Banerjee S, Tian T, Wei Z, Shih N, Feldman MD, Alwine JC, et al. The ovarian cancer oncobiome. Oncotarget. 2017;8:36225–45.
Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018;8:403–16.
Sfanos KS, Sauvageot J, Fedor HL, Dick JD, De Marzo AM, Isaacs WB. A molecular analysis of prokaryotic and viral DNA sequences in prostate tissue from patients with prostate cancer indicates the presence of multiple and diverse microorganisms. Prostate. 2008;68:306–20.
Mao Q, Jiang F, Yin R, Wang J, Xia W, Dong G, et al. Interplay between the lung microbiome and lung cancer. Cancer Lett. 2018;415:40–8.
Urbaniak C, Gloor GB, Brackstone M, Scott L, Tangney M, Reid G. The microbiota of breast tissue and its association with breast cancer. Appl Environ Microbiol. 2016;82:5039–48.
Aviles-Jimenez F, Guitron A, Segura-Lopez F, Mendez-Tenorio A, Iwai S, Hernandez-Guerrero A, et al. Microbiota studies in the bile duct strongly suggest a role for helicobacter pylori in extrahepatic cholangiocarcinoma. Clin Microbiol Infect. 2016;22:178 e111–22.
Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15:317–28.
Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–12.
Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12:661–72.
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–71.
Debnath J. The multifaceted roles of autophagy in tumors-implications for breast cancer. J Mammary Gland Biol Neoplasia. 2011;16:173–87.
Garrett WS. Cancer and the microbiota. Science. 2015;348:80–6.
Ko JY, Lee EJ, Park JH. Interplay between primary cilia and autophagy and its controversial roles in cancer. Biomol Ther (Seoul). 2019;27:337–41.
Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, Wargo JA. The microbiome, cancer, and cancer therapy. Nat Med. 2019;25:377–88.
Cho TJ, Wee SW, Woo VH, Choi JI, Kim SJ, Shin HI, et al. Porphyromonas gingivalis-induced autophagy suppresses cell proliferation through G1 arrest in oral cancer cells. Arch Oral Biol. 2014;59:370–8.
Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170:548–563 e516.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
Sun J, Tang Q, Yu S, Xie M, Xie Y, Chen G, et al. Role of the oral microbiota in cancer evolution and progression. Cancer Med. 2020;9:6306–21.
Irfan M, Delgado RZR, Frias-Lopez J. The oral microbiome and cancer. Front Immunol. 2020;11:591088.
Lin EW, Karakasheva TA, Hicks PD, Bass AJ, Rustgi AK. The tumor microenvironment in esophageal cancer. Oncogene. 2016;35:5337–49.
Yamamura K, Baba Y, Nakagawa S, Mima K, Miyake K, Nakamura K, et al. Human microbiome fusobacterium nucleatum in esophageal cancer tissue is associated with prognosis. Clin Cancer Res. 2016;22:5574–81.
Yuan X, Liu Y, Kong J, Gu B, Qi Y, Wang X, et al. Different frequencies of porphyromonas gingivalis infection in cancers of the upper digestive tract. Cancer Lett. 2017;404:1–7.
Liang G, Wang H, Shi H, Zhu M, An J, Qi Y, et al. Porphyromonas gingivalis promotes the proliferation and migration of esophageal squamous cell carcinoma through the miR-194/GRHL3/PTEN/Akt Axis. ACS Infect Dis. 2020;6:871–81.
Chen XH, Wang A, Chu AN, Gong YH, Yuan Y. Mucosa-associated microbiota in gastric cancer tissues compared with non-cancer tissues. Front Microbiol. 2019;10:1261.
Wu J, Xu S, Xiang C, Cao Q, Li Q, Huang J, et al. Tongue coating microbiota community and risk effect on gastric cancer. J Cancer. 2018;9:4039–48.
Farrell JJ, Zhang L, Zhou H, Chia D, Elashoff D, Akin D, et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut. 2012;61:582–8.
Torres PJ, Fletcher EM, Gibbons SM, Bouvet M, Doran KS, Kelley ST. Characterization of the salivary microbiome in patients with pancreatic cancer. PeerJ. 2015;3:e1373.
Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2018;67:120–7.
Gnanasekaran J, Binder Gallimidi A, Saba E, Pandi K, Eli Berchoer L, Hermano E, et al. Intracellular porphyromonas gingivalis promotes the tumorigenic behavior of pancreatic carcinoma cells. Cancers (Basel). 2020;12:2331.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306.
He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S, et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68:289–300.
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206.
Shen J, Zhai J, You Q, Zhang G, He M, Yao X, et al. Cancer-associated fibroblasts-derived VCAM1 induced by H. pylori infection facilitates tumor invasion in gastric cancer. Oncogene. 2020;39:2961–74.
Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–15.
Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-kappaB, and up-regulating expression of microRNA-21. Gastroenterology. 2017;152:851–866 e824.
Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, Carra A, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science. 2019;363:eaar7785.
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–22.
Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest. 2014;124:4166–72.
Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, et al. Peptostreptococcus anaerobius induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology. 2017;152:1419–1433 e1415.
Long X, Wong CC, Tong L, Chu ESH, Ho Szeto C, Go MYY, et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat Microbiol. 2019;4:2319–30.
Abdulamir AS, Hafidh RR, Bakar FA. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: inflammation-driven potential of carcinogenesis via IL-1, COX-2, and IL-8. Mol Cancer. 2010;9:249.
Chen D, Jin D, Huang S, Wu J, Xu M, Liu T, et al. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020;469:456–67.
Lee YK, Mehrabian P, Boyajian S, Wu WL, Selicha J, Vonderfecht S, et al. The protective role of bacteroides fragilis in a murine model of colitis-associated colorectal cancer. mSphere. 2018;3:e00587.
Li Q, Hu W, Liu WX, Zhao LY, Huang D, Liu XD, et al. Streptococcus thermophilus inhibits colorectal tumorigenesis through secreting beta-galactosidase. Gastroenterology. 2021;160:1179–1193 e1114.
Fox JG, Feng Y, Theve EJ, Raczynski AR, Fiala JL, Doernte AL, et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut. 2010;59:88–97.
Chang C, Wang H, Liu J, Pan C, Zhang D, Li X, et al. Porphyromonas gingivalis infection promoted the proliferation of oral squamous cell carcinoma cells through the miR-21/PDCD4/AP-1 negative signaling pathway. ACS Infect Dis. 2019;5:1336–47.
Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, et al. Commensal microbiota promote lung cancer development via gammadelta T cells. Cell. 2019;176:998–1013 e1016.
Tsay JJ, Wu BG, Badri MH, Clemente JC, Shen N, Meyn P, et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am J Respir Crit Care Med. 2018;198:1188–98.
Parida S, Wu S, Siddharth S, Wang G, Muniraj N, Nagalingam A, et al. A procarcinogenic colon microbe promotes breast tumorigenesis and metastatic progression and concomitantly activates notch and beta-catenin axes. Cancer Discov. 2021;11:1138–57.
Ma J, Sun L, Liu Y, Ren H, Shen Y, Bi F, et al. Alter between gut bacteria and blood metabolites and the anti-tumor effects of Faecalibacterium prausnitzii in breast cancer. BMC Microbiol. 2020;20:82.
Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32.
Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–73.
Gomes LR, Menck CFM, Leandro GS. Autophagy roles in the modulation of DNA repair pathways. Int J Mol Sci. 2017;18:2351.
Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906.
Ojha R, Bhattacharyya S, Singh SK. Autophagy in cancer stem cells: a potential link between chemoresistance, recurrence, and metastasis. Biores Open Access. 2015;4:97–108.
Hall DP, Cost NG, Hegde S, Kellner E, Mikhaylova O, Stratton Y, et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell. 2014;26:738–53.
Han T, Guo M, Gan M, Yu B, Tian X, Wang JB. TRIM59 regulates autophagy through modulating both the transcription and the ubiquitination of BECN1. Autophagy. 2018;14:2035–48.
Liu W, Glunde K, Bhujwalla ZM, Raman V, Sharma A, Phang JM. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012;72:3677–86.
Cai J, Li R, Xu X, Zhang L, Lian R, Fang L, et al. CK1alpha suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nat Cell Biol. 2018;20:465–78.
Li P, Du Q, Cao Z, Guo Z, Evankovich J, Yan W, et al. Interferon-gamma induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012;314:213–22.
Tu SP, Quante M, Bhagat G, Takaishi S, Cui G, Yang XD, et al. IFN-gamma inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011;71:4247–59.
Guo X, Xiao H, Guo S, Li J, Wang Y, Chen J, et al. Long noncoding RNA HOTAIR knockdown inhibits autophagy and epithelial-mesenchymal transition through the Wnt signaling pathway in radioresistant human cervical cancer HeLa cells. J Cell Physiol. 2019;234:3478–89.
Wang Z, Liu Z, Wu S. Long non-coding RNA CTA sensitizes osteosarcoma cells to doxorubicin through inhibition of autophagy. Oncotarget. 2017;8:31465–77.
Huang T, Wan X, Alvarez AA, James CD, Song X, Yang Y, et al. MIR93 (microRNA −93) regulates tumorigenicity and therapy response of glioblastoma by targeting autophagy. Autophagy. 2019;15:1100–11.
Luo W, Liu Q, Chen X, Liu H, Quan B, Lu J, et al. FXYD6 regulates chemosensitivity by mediating the expression of Na+/K+-ATPase alpha1 and affecting cell autophagy and apoptosis in colorectal cancer. Biomed Res Int. 2021;2021:9986376.
Schwartz-Roberts JL, Cook KL, Chen C, Shajahan-Haq AN, Axelrod M, Warri A, et al. Interferon regulatory factor-1 signaling regulates the switch between autophagy and apoptosis to determine breast cancer cell fate. Cancer Res. 2015;75:1046–55.
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011;25:23–31.
Zhou S, Zhao L, Kuang M, Zhang B, Liang Z, Yi T, et al. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 2012;323:115–27.
Russo M, Russo GL. Autophagy inducers in cancer. Biochem Pharmacol. 2018;153:51–61.
Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–30.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64.
Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy. 2019;15:169–71.
Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–73.
Bialik S, Kimchi A. Autophagy and tumor suppression: recent advances in understanding the link between autophagic cell death pathways and tumor development. Adv Exp Med Biol. 2008;615:177–200.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
Zhi X, Zhong Q. Autophagy in cancer. F1000Prime Rep. 2015;7:18.
Lee DS, Moss SF. Targeting helicobacter pylori in gastric carcinogenesis. Expert Opin Ther Targets. 2007;11:757–69.
El Khadir M, Boukhris Alaoui S, Benajah DA, Ibrahimi SA, Chbani L, El Abkari M, et al. VacA genotypes and cagA-EPIYA-C motifs of helicobacter pylori and gastric histopathological lesions. Int J Cancer. 2020;147:3206–14.
Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, et al. Effect of helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy. 2009;5:370–9.
Nejati S, Karkhah A, Darvish H, Validi M, Ebrahimpour S, Nouri HR. Influence of helicobacter pylori virulence factors CagA and VacA on pathogenesis of gastrointestinal disorders. Microb Pathog. 2018;117:43–8.
Li N, Tang B, Jia YP, Zhu P, Zhuang Y, Fang Y, et al. Helicobacter pylori CagA protein negatively regulates autophagy and promotes inflammatory response via c-met-PI3K/Akt-mTOR signaling pathway. Front Cell Infect Microbiol. 2017;7:417.
Tohidpour A. CagA-mediated pathogenesis of helicobacter pylori. Microb Pathog. 2016;93:44–55.
Selbach M, Moese S, Hurwitz R, Hauck CR, Meyer TF, Backert S. The helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 2003;22:515–28.
Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the helicobacter pylori CagA protein. Proc Natl Acad Sci U S A. 2005;102:9300–5.
Eslami M, Yousefi B, Kokhaei P, Arabkari V, Ghasemian A. Current information on the association of helicobacter pylori with autophagy and gastric cancer. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.28279.
Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of helicobacter pylori CagA protein activities. Mol Cell. 2002;10:745–55.
Ricci V. Relationship between VacA toxin and host cell autophagy in helicobacter pylori infection of the human stomach: a few answers, many questions. Toxins (Basel). 2016;8:203.
Raju D, Hussey S, Ang M, Terebiznik MR, Sibony M, Galindo-Mata E, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote helicobacter pylori infection in humans. Gastroenterology. 2012;142:1160–71.
Xie C, Li N, Wang H, He C, Hu Y, Peng C, et al. Inhibition of autophagy aggravates DNA damage response and gastric tumorigenesis via Rad51 ubiquitination in response to H. pylori infection. Gut Microbes. 2020;11:1567–89.
Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomark Prev. 2014;23:700–13.
Yang JC, Shun CT, Chien CT, Wang TH. Effective prevention and treatment of helicobacter pylori infection using a combination of catechins and sialic acid in AGS cells and BALB/c mice. J Nutr. 2008;138:2084–90.
Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–8.
Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2016;65:1973–80.
Liu Y, Baba Y, Ishimoto T, Tsutsuki H, Zhang T, Nomoto D, et al. Fusobacterium nucleatum confers chemoresistance by modulating autophagy in oesophageal squamous cell carcinoma. Br J Cancer. 2021;124:963–74.
Wang Y, Zhang H. Regulation of autophagy by mTOR signaling pathway. Adv Exp Med Biol. 2019;1206:67–83.
Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9:1167–81.
Muhammad JS, Nanjo S, Ando T, Yamashita S, Maekita T, Ushijima T, et al. Autophagy impairment by helicobacter pylori-induced methylation silencing of MAP1LC3Av1 promotes gastric carcinogenesis. Int J Cancer. 2017;140:2272–83.
Zhang F, Chen C, Hu J, Su R, Zhang J, Han Z, et al. Molecular mechanism of helicobacter pylori-induced autophagy in gastric cancer. Oncol Lett. 2019;18:6221–7.
Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J, et al. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by helicobacter pylori VacA. J Biol Chem. 2012;287:31104–15.
Shi Y, Yang Z, Zhang T, Shen L, Li Y, Ding S. SIRT1-targeted miR-543 autophagy inhibition and epithelial-mesenchymal transition promotion in helicobacter pylori CagA-associated gastric cancer. Cell Death Dis. 2019;10:625.
Teimoorian F, Ranaei M, Hajian Tilaki K, Shokri Shirvani J, Vosough Z. Association of helicobacter pylori infection with colon cancer and adenomatous polyps. Iran J Pathol. 2018;13:325–32.
Revathi S, Hakkim FL, Kumar NR, Bakshi HA, Rashan L, Al-Buloshi M, et al. Induction of HT-29 colon cancer cells apoptosis by pyrogallol with growth inhibiting efficacy against drug-resistant helicobacter pylori. Anti Cancer Agents Med Chem. 2018;18:1875–84.
Zhong X, Chen O, Zhou T, Lu M, Wan J. Cytotoxin-associated gene A-positive helicobacter pylori promotes autophagy in colon cancer cells by inhibiting miR-125b-5p. Can J Infect Dis Med Microbiol. 2021;2021:6622092.
Hartwich A, Konturek SJ, Pierzchalski P, Zuchowicz M, Labza H, Konturek PC, et al. Helicobacter pylori infection, gastrin, cyclooxygenase-2, and apoptosis in colorectal cancer. Int J Color Dis. 2001;16:202–10.
Binder Gallimidi A, Fischman S, Revach B, Bulvik R, Maliutina A, Rubinstein AM, et al. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget. 2015;6:22613–23.
Lu H, Han M, Yuan X, Tursun K, Zhang Y, Li Y, et al. Role of IL-6-mediated expression of NS5ATP9 in autophagy of liver cancer cells. J Cell Physiol. 2018;233:9312–9.
Hu F, Song D, Yan Y, Huang C, Shen C, Lan J, et al. IL-6 regulates autophagy and chemotherapy resistance by promoting BECN1 phosphorylation. Nat Commun. 2021;12:3651.
Huang R, Liu W. Identifying an essential role of nuclear LC3 for autophagy. Autophagy. 2015;11:852–3.
Chen Y, Chen Y, Zhang J, Cao P, Su W, Deng Y, et al. Fusobacterium nucleatum promotes metastasis in colorectal cancer by activating autophagy signaling via the upregulation of CARD3 expression. Theranostics. 2020;10:323–39.
Mysak J, Podzimek S, Sommerova P, Lyuya-Mi Y, Bartova J, Janatova T, et al. Porphyromonas gingivalis: major periodontopathic pathogen overview. J Immunol Res. 2014;2014:476068.
Olsen I, Yilmaz O. Possible role of Porphyromonas gingivalis in orodigestive cancers. J Oral Microbiol. 2019;11:1563410.
Belanger M, Rodrigues PH, Dunn WA Jr, Progulske-Fox A. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy. 2006;2:165–70.
Liu B, Jiang Y, Dong T, Zhao M, Wu J, Li L, et al. Blockage of autophagy pathway enhances Salmonella tumor-targeting. Oncotarget. 2016;7:22873–82.
Lin HJ, Liu HH, Lin CD, Kao MC, Chen YA, Chiang-Ni C, et al. Cytolethal distending toxin enhances radiosensitivity in prostate cancer cells by regulating autophagy. Front Cell Infect Microbiol. 2017;7:223.
McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8.
Allemailem KS, Almatroudi A, Alrumaihi F, Almatroodi SA, Alkurbi MO, Basfar GT, et al. Novel approaches of dysregulating lysosome functions in cancer cells by specific drugs and its nanoformulations: a smart approach of modern therapeutics. Int J Nanomedicine. 2021;16:5065–98.
Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A, et al. A unified approach to targeting the Lysosome’s degradative and growth signaling roles. Cancer Discov. 2017;7:1266–83.
Pasquier B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy. 2015;11:725–6.
Jiang H, Courau T, Borison J, Ritchie AJ, Mayer AT, Krummel MF, et al. Activating immune recognition in pancreatic ductal adenocarcinoma via autophagy inhibition, MEK blockade and CD40 Agonism. Gastroenterology. 2021. https://doi.org/10.1053/j.gastro.2021.09.066.
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–34.
Guo J, Zhang J, Liang L, Liu N, Qi M, Zhao S, et al. Potent USP10/13 antagonist spautin-1 suppresses melanoma growth via ROS-mediated DNA damage and exhibits synergy with cisplatin. J Cell Mol Med. 2020;24:4324–40.
Liu X, Meng L, Li X, Li D, Liu Q, Chen Y, et al. Regulation of FN1 degradation by the p62/SQSTM1-dependent autophagy-lysosome pathway in HNSCC. Int J Oral Sci. 2020;12:34.
Bi Y, Jiang Y, Li X, Hou G, Li K. Rapamycin inhibits lung squamous cell carcinoma growth by downregulating glypican-3/Wnt/beta-catenin signaling and autophagy. J Cancer Res Clin Oncol. 2021;147:499–505.
Martinez GP, Zabaleta ME, Di Giulio C, Charris JE, Mijares MR. The role of chloroquine and hydroxychloroquine in immune regulation and diseases. Curr Pharm Des. 2020;26:4467–85.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36.
Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O'Dwyer PJ. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res. 2013;19:2995–3007.
Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10:370.
Cerniglia GJ, Karar J, Tyagi S, Christofidou-Solomidou M, Rengan R, Koumenis C, et al. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol Pharmacol. 2012;82:1230–40.
Wang FT, Wang H, Wang QW, Pan MS, Li XP, Sun W, et al. Inhibition of autophagy by chloroquine enhances the antitumor activity of gemcitabine for gallbladder cancer. Cancer Chemother Pharmacol. 2020;86:221–32.
Arnaout A, Robertson SJ, Pond GR, Lee H, Jeong A, Ianni L, et al. A randomized, double-blind, window of opportunity trial evaluating the effects of chloroquine in breast cancer patients. Breast Cancer Res Treat. 2019;178:327–35.
Lebin JA, LeSaint KT. Brief review of chloroquine and hydroxychloroquine toxicity and management. West J Emerg Med. 2020;21:760–3.
Ponticelli C, Moroni G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin Drug Saf. 2017;16:411–9.
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–66.
Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19:637–8.
Ito S, Koshikawa N, Mochizuki S, Takenaga K. 3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition. Int J Oncol. 2007;31:261–8.
Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994;54:2419–23.
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–6.
Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–8.
Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90:313–23.
Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–70.
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850–61.
Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol. 2009;16:761–71.
Lin J, Huang Z, Wu H, Zhou W, Jin P, Wei P, et al. Inhibition of autophagy enhances the anticancer activity of silver nanoparticles. Autophagy. 2014;10:2006–20.
Takeda M, Koseki J, Takahashi H, Miyoshi N, Nishida N, Nishimura J, et al. Disruption of endolysosomal RAB5/7 efficiently eliminates colorectal cancer stem cells. Cancer Res. 2019;79:1426–37.
Liao Y, Guo Z, Xia X, Liu Y, Huang C, Jiang L, et al. Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer. J Exp Clin Cancer Res. 2019;38:157.
Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32.
Schulze M, Stock C, Zaccagnini M, Teber D, Rassweiler JJ. Temsirolimus. Recent Results Cancer Res. 2014;201:393–403.
Yang JC, Lu CW, Lin CJ. Treatment of helicobacter pylori infection: current status and future concepts. World J Gastroenterol. 2014;20:5283–93.
Vitor JM, Vale FF. Alternative therapies for helicobacter pylori: probiotics and phytomedicine. FEMS Immunol Med Microbiol. 2011;63:153–64.
Pierard GE, Arrese JE, Pierard-Franchimont C. Itraconazole. Expert Opin Pharmacother. 2000;1:287–304.
Odds FC, Oris M, Van Dorsselaer P, Van Gerven F. Activities of an intravenous formulation of itraconazole in experimental disseminated Aspergillus, Candida, and Cryptococcus infections. Antimicrob Agents Chemother. 2000;44:3180–3.
Liu R, Li J, Zhang T, Zou L, Chen Y, Wang K, et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking. Autophagy. 2014;10:1241–55.
Wei X, Liu W, Wang JQ, Tang Z. “Hedgehog pathway”: a potential target of itraconazole in the treatment of cancer. J Cancer Res Clin Oncol. 2020;146:297–304.
Wang X, Wei S, Zhao Y, Shi C, Liu P, Zhang C, et al. Anti-proliferation of breast cancer cells with itraconazole: hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Lett. 2017;385:128–36.
Ban L, Mei T, Su Q, Li W, Huang Z, Liu L, et al. Anti-fungal drug itraconazole exerts anti-cancer effects in oral squamous cell carcinoma via suppressing hedgehog pathway. Life Sci. 2020;254:117695.
Deng H, Huang L, Liao Z, Liu M, Li Q, Xu R. Itraconazole inhibits the hedgehog signaling pathway thereby inducing autophagy-mediated apoptosis of colon cancer cells. Cell Death Dis. 2020;11:539.
Ambalam P, Raman M, Purama RK, Doble M. Probiotics, prebiotics and colorectal cancer prevention. Best Pract Res Clin Gastroenterol. 2016;30:119–31.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–9.
Vinícius da Silva Duarte V, Dos Santos Cruz BC, Tarrah A, Sousa Dias R, de Paula Dias Moreira L, Lemos Junior WJF, et al. Chemoprevention of DMH-induced early colon carcinogenesis in male BALB/c mice by administration of lactobacillus paracasei DTA81. Microorganisms. 2020;8:1994.
Liu WD, Sun W, Hua YQ, Wang SG, Cai ZD. Effect of rapamycin and chloroquine on osteosarcoma. Zhonghua Yi Xue Za Zhi. 2017;97:1510–4.
Ishibashi Y, Nakamura O, Yamagami Y, Nishimura H, Fukuoka N, Yamamoto T. Chloroquine enhances rapamycin-induced apoptosis in MG63 cells. Anticancer Res. 2019;39:649–54.
Acknowledgements
Not applicable.
Funding
This work was supported by the China National Natural Scientific Fund No. 82002892 (to W.Y.), 82073010 (to Z.X.), 82172764 (to R.Y.); the Tianjin Education Commission Funded Projects No.2019KJ188 (to W.Y.); the Fundamental Research Funds for the Central Universities No.3332020079 (to W.SN.)
Author information
Authors and Affiliations
Contributions
WY, JD and XMW collected the related papers and drafted the manuscript. CL, A. Abdelrehem and YR participated in the design of the review, and SNS, XZ and YW initiated the study and revised and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Du, J., Wu, X. et al. Crosstalk between autophagy and microbiota in cancer progression. Mol Cancer 20, 163 (2021). https://doi.org/10.1186/s12943-021-01461-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-021-01461-0