
Abstract
Circular RNAs (circRNAs) are a new class of non-coding RNAs formed by covalently closed loops through backsplicing. Recent methodologies have enabled in-depth characterization of circRNAs for identification and potential functions. CircRNAs play important roles in various biological functions as microRNA sponges, transcriptional regulators and combining with RNA binding proteins. Recent studies indicated that some cytoplasmic circRNAs can be effectively translated into detectable peptides, which enlightened us on the importance of circRNAs in cellular physiology function. Internal Ribosome Entry site (IRES)- and N6-methyladenosines (m6A)-mediated cap-independent translation initiation have been suggested to be potential mechanism for circRNA translation. To date, several translated circRNAs have been uncovered to play pivotal roles in human cancers. In this review, we introduced the properties and functions of circRNAs, and characterized the possible mechanism of translation initiation and complexity of the translation ability of circRNAs. We summarized the emerging functions of circRNA-encoded proteins in human cancer. The works on circRNA translation will open a hidden human proteome, and enhance us to understand the importance of circRNAs in human cancer, which has been poorly explored so far.
Similar content being viewed by others

Background
Circular RNAs (circRNAs) are a new type of endogenous RNAs produced by non-canonical back-splicing events [1, 2]. In circRNA, the downstream splice-donor site can be covalently linked to an upstream splice-acceptor site. The first circRNA molecule was discovered in an RNA virus in 1976 [3, 4], and circRNAs were observed in eukaryotic cell lines by electron microscopy in 1991 [5]. Thereafter, circRNAs have long been considered as aberrant splicing events [6]. Genomic and transcriptomic data generated by next generation sequencing projects and bioinformatics algorithms have identified significant amount of circRNAs in eukaryotes [7,8,9,10,11,12,13,14], clearly demonstrating that they are not merely the accidental byproducts or ‘splicing noise’.
High-throughput technologies have enabled in-depth characterization of circRNAs for identification and potential functions [15,16,17], such as ribosomal RNA depleted RNA sequencing, long-read sequencing and more importantly, improved circRNA mining bioinformatics algorithms. CircRNAs have been demonstrated as an abundant and conserved class of RNAs, and are widely expressed in a complex tissue-, cell type- or stage-specific manner [10, 18,19,20,21,22]. Recent studies confirmed the significant biological functions of circRNAs, especially in human cancers [23, 24]. CircRNAs can function as transcriptional regulators to control the expression of host genes [9, 25, 26]. They can also act as microRNA sponge to fine-tune the miRNA-mRNA regulatory axis [27,28,29,30,31]. It has been indicated that circRNA can serve as prognostic biomarkers because of their stable characteristics [32]. Moreover, several works have demonstrated hidden peptides encoded by circRNAs, which will largely broaden our understanding on the cellular physiology functions [33,34,35]. circRNAs have been considered as ‘non-coding’ elements, and the circRNA translation will provide a new perspective and new horizon for cancer treatment and diagnosis. We herein characterized the possible mechanism of translation initiation and complexity of the translation ability of circRNAs, and summarized the emerging functions of circRNA-encoded proteins in human cancers.
Characterization of circRNAs
Properties of circRNAs
CircRNAs are produced from precursor mRNA and are derived from canonical splice site. CircRNA biogenesis can compete with the maturation of its linear counterpart by linking 3′ splice site to a downstream 5′ splice site [25]. CircRNAs can be classified into three different types based on the type of sequence they contain: exonic circRNAs (EcRNA), intronic circRNAs (CiRNA) and exon-intron circRNAs (EIcRNA) [26, 36]. It has been documented that some intron-containing circRNAs are sequestered in the nucleus, while exonic circRNAs are exported to the cytoplasm. The currently discovered circRNAs are predominantly exonic and can be widely detected in the cytoplasma [28, 37].
The structures of circRNAs lack 5′ cap and 3′ end, which makes them resistant to the digestion of ribonucleases, such as RNase R, and confers a long half-life reaching up to 10 times that of linear RNAs [38]. Most circRNA sequences are highly conserved. Recent work demonstrated that approximately 15,000 human circRNA sequences can be detected in mouse or rat genomes [39, 40]. CircRNAs are generally expressed in vast majority of human tissues and are especially highly expressed in human brain [18, 40]. Furthermore, the expression of circRNAs always has a tissue- or cell- specific manner, which makes them a suitable candidate for biomarker studies in human cancer [10, 19, 20, 41].
Genome-wide profiling of circRNAs
Ribosome RNA-depleted RNA-seq method has been widely employed for the discovery of novel circRNAs [2, 9, 16, 41,42,43]. This method can simultaneously provide the expression data for both coding and non-coding RNAs. Reliable quantification of circRNAs requires a substantial sequencing depth to ensure the identification of circRNAs. Salzman et al. identified “scrambled” exons and potential circRNA in human cancer using rRNA-depleted RNA-seq method [17]. In a recent work, Memczak et al. systematically explored circRNAs in human, mouse and nematode, and found that circRNAs are expressed in a tissue- and developmental stage-specific manner [29]. Subsequently, a genome-wide RNA exonuclease enrichment strategy was applied before sequencing. This method led to the enrichment of circRNAs in the sample, and a significantly higher number of circRNAs can be identified [9]. A recent work detected and characterized circRNAs across > 2000 cancer samples using exome capture RNA-seq, which achieved significantly better enrichment for circRNAs [44].
Bioinformatics methods for identifying novel circRNAs are constantly developed. They mainly identify circRNAs based on the presence of backsplice junction-spanning sequencing reads [16, 45, 46]. At first, sequencing reads that contiguously align to the genome and/or the transcriptome were filtered out, and those unaligned reads were used to align to the backsplice junctions. Sequencing errors and alignment mismatch can lead to false-positive identification [28, 46, 47]. These algorithms were developed to reduce false positives based on reads counts, number of samples having been detected, RNase R resistance, which inevitably results in the inability to detect some specific circRNA isoforms. In the absence of a clear gold-standard data, a combination of more than one circRNA prediction algorithms can largely minimize the false positives [48].
Function of circRNAs
To date, the biological implications have only been investigated for a minor fraction of identified circRNAs, and most of them have been proposed to function as miRNA sponges in the cytoplasm [27,28,29,30, 49]. The most representative circRNA is ciRS-7, which contains more than 70 conserved binding sites for miR-7 [50,51,52]. In addition, circHIPK3 acts as a sponge for a variety of miRNAs to fine-tune transcriptional activity [53, 54]. Another important function of circRNAs is acting as protein sponge [25, 55]. Through binding of specific proteins to circRNAs, a molecular reservoir of proteins were created and act as decoys to facilitate a prompt response to extracellular stimuli.
Translation of circRNAs
Based on our conventional opinions, 5′ and 3′ untranslated regions (UTR) are essential elements for the translation initiation in eukaryotic cells. Due to the absence of 5′ and 3′ ends, circRNAs were once considered as non-coding RNAs. Recently, mounting evidence have shown that circRNAs can be associated with polysomes and some of them comprise the initiation codon AUG and putative Open Reading Frames (ORF) with favorable length, which suggests an unexpected protein-coding potential for circRNAs [33, 56,57,58]. CircRNAs can actually encode regulatory peptides, and a hidden proteome encoded by circRNAs might exist.
In a first study, Chen and Sarnow suggested that engineered circRNAs in artificial constructs could recruit the 40s ribosomal subunit and initiate translation of detectable peptides in vitro [59]. However, this assay does not support the notion that circRNAs can also be translatable molecules in vivo. In 2015, Abe et al. demonstrated strong evidence concerning endogenous circRNAs serving as translation templates [56]. CircRNAs containing an infinite ORF can be efficiently translated in a rolling cycle amplification mechanism to produce a large protein concatemer. In 2017, Legnini et al. reported that circ-ZNF609 is the backsplicing product of ZNF609 exon 2, and can be translated into a protein in a splicing dependent and cap-independent manner in Myogenesis [60]. These works strongly supported the coding potential of endogenous circRNAs and raising the question how are circRNAs translated by non-canonical initiation mechanisms?
Cap-independent translation initiation mechanisms of circRNAs
Eukaryotic mRNAs are always translated through the canonical cap-dependent translation. During the RNA synthesis process, a 7-methylguanosine cap is added to the 5′ end of mRNA for translation initiation. This structure can be organized by the eIF4E translation initiation factor, a protein complex including eIF4E, eIF4G and eIF4A components [61,62,63]. This well-known cap-dependent translation is the primary mode of translation initiation in eukaryotic cells. Under certain conditions, such as cellular stress or viral infection, an alternative mechanism known as cap-independent translation is employed to initiate mRNA translation through internal ribosome entry site (IRES) [64, 65]. IRESs are sequences located in the 5′ UTR of mRNAs that can directly recruit ribosomes to initiate translation. IRES-mediated translation was firstly identified in RNA and DNA viruses [66]. Subsequently, some works have reported that eukaryotic mRNAs can be also translated by IRES-mediated manner under stressful conditions [65, 67, 68], when canonical translation is altered, suggesting an alternative translation mechanism in eukaryotes to compensate for the defective cap-dependent translation. Furthermore, IRES-mediated translation has recently been indicated to play a key regulatory role during mammalian development. High-throughput screening for IRES elements in human genomes suggested that ~ 10% of the human mRNAs contain IRES elements [69, 70], however, the biological implications of IRES-mediated translation remain to be discovered. The translation of circRNAs can only be initiated by cap-independent fashion because of lacking 5′ cap and 3′ end. IRES-mediated fashion is one of the widely accepted mechanisms of circRNA translation initiation [35, 71, 72]. Recent works suggested that in vitro synthesized circRNAs in artificial constructs can be successfully translated, with an engineered IRES and split GFP sequences that can be joined together by precise back-splicing [73]. If the resulting circRNA can be translated from an IRES, a functional GFP protein can be successfully generated. To date, the number of IRESs in circRNAs is still unknown. A recent unbiased work using a cell-based reporter system identified a large number of AU-rich motifs (~ 10 nt) with IRES-like activity to initiate circRNA translation [74, 75]. Moreover, any circRNAs with the fragment longer than 50 nt is expected to contain an IRES-like hexamer by chance. The requirement of IRESs for circRNAs can be easily satisfied, suggesting the importance and prevalence of IRES-mediated cap independent translation mechanism of circRNAs in cytoplasm.
Another important cap-independent translation mechanism is through methylated adenosine residues in the form of N6-methyladenosines (m6A) in the 5′ UTR [76]. It has been shown that the m6A in the 5′ UTR can directly bind eukaryotic initiation factor 3 (eIF3) [76]. It is sufficient to recruit the 43S preinitiation complex and initiate translation bypass the m7G cap requirement, enabling a cap-independent mode of translation initiation. A recent work showed that 5′ URT m6A residues control ribosome scanning and subsequent start codon selection during integrated stress response [77]. The reinitiation of ATF4 mRNA is regulated by both the eIF2α signaling pathway and m6A modification in response to amino acid starvation. Interfering with the m6A demethylases and methyltransferases can influence ATF4 translation. So, 5′ UTR methylation in the form of m6A acts as a dynamic regulator in alternative translation [78]. The concept has been enlarged using m6A-seq method, and revealed that m6A in the 5′ UTR of other transcripts can also modulate start codon selection, thereby controlling alternative translation [77]. Using circRNA report genes, it has been reported that some short RNA elements containing m6A have IRES-like activity to initiate circRNA translation. Yang et al. showed that short RNA elements for the most abundant m6A were enriched in the circRNA sequences [79]. A single m6A modification is sufficient to initiate circRNA translation with the initiation factor eIF4G2 and the m6A reader protein YTHDF3. While depleting m6A demethylases FTO suppress circRNAs translation, knocking down m6A methyltransferases METTL3/14 promotes circRNA translation under the stress condition. High-throughput m6A-seq was performed with the RNase R treatment, indicating that at least 13% of circRNAs carry m6A modification. The authors identified 250 circRNAs to be associated with polysomes, which may have actively translation potential [79].
Both IRES- and m6A-mediated translation initiation are important mechanisms for circRNA translation (Fig. 1). It is still unclear whether there is other cap-independent mechanism to initiate circRNA translation in eukaryotic cells.

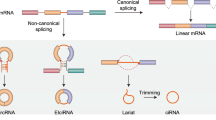
Cap-independent translation of circRNAs in eukaryotic cell. The circRNA can be divided into three different types: exonic (EcRNA), intronic (CiRNA) and exon-intron (EIcRNA) . CiRNAs and EIcRNAs are likely sequestered in the nucleus, while EcRNAs are mostly exported into cytoplasm. Internal Ribosome Entry site (IRES)- and N6-methyladenosines (m6A)-mediated cap-independent translation initiation are potential mechanisms for circRNA translation. These structures allow the internal recruitment of the 40S ribosomal subunit
Bioinformatics tools for identifying circRNA coding potential
Systematic identification for circRNAs with coding potential using bioinformatics ways is necessary, and could provide insights into the function of circRNA-derived proteins. We herein summarized those bioinformatics tools for predicting circRNA coding potential (Table 1). 1) Accurate and quantitative assessment of coding potential of circRNAs. ORF Finder is an easy-to-use graphical analysis tool to find all possible ORFs in the sequence provided by user [80]; CPC (Coding-Potential Calculator) is a widely used algorithm to assess the protein-coding potential of a transcript based on six biologically meaningful sequence features. The prediction of coding potential relied on sequence alignment using pairwise homology search for protein evidence [81]; PhyloCSF (Phylogenetic Codon Substitution Frequencies) determines whether the sequence is likely to represent a conserved protein-coding region using multiple alignments to calculate the phylogenetic conservation score [82]; CPAT (Coding-Potential Assessment Tool) is an alignment-free algorithm to distinguish between coding and noncoding transcripts on the basis of four sequence features using logistic regression [83]. A combination of these tools for the coding potential prediction can largely reduce the false positives. 2) IRES element identification. Sequences and structures of many IRES are well-known. IRESite is a database that can be used to examine the cellular internal ribosome entry sites [80]; CircInteractome database allow the investigation of potential circRNA translation through IRES sequences [84]; IRES finder is an improved computation method which can be used to perform a comprehensive search of IRES [85]. 3) feature analysis of circRNAs with coding potential. Pfam is a tool for the homology search of a putative product sequence [86]. The identification of a domain provides biological insights into its function. N-Glycosylation sites, Mucin-type O-glycosylation sites and phosphorylation sites can be predicted using NetNGlyc 1.0, NetOGlyc 3.1 and NetPhos 3.1 tools [87, 90], respectively. Some works built integrated bioinformatics tools for the identification of circRNAs with protein-coding potential, such as CircPro [88] and CircCode [89].
Experimental approaches for circRNA translation evaluation
After computational assessments, it is desirable to experimentally validate whether the circRNAs are indeed translatable. Ribosome profiling and polysomal fractionation enable translational global analysis. Ribosome profiling can capture footprints of actively translating ribosomes [91, 92]. From these footprints, we can infer the codon-by-codon movements of ribosomes. Recently, genome-wide translatomes were characterized using ribosome profiling in human heart [93]. A total of 40 ribosome bound circRNAs, produced from 39 genes, were identified across the backspliced junctions present in the ribosome footprints. Polysomal fractionation by sucrose density gradient centrifugation allows direct determination of translation efficiencies of circRNAs at a genome-wide scale [91, 94]. These two methods are complementary that enable genome-wide translational analysis. For those potential peptides encoded by circRNAs, mass spectrometry is a widely used platform for peptide detection [95].
CircRNA translation in human cancer
To date, several translated circRNAs have been identified, which play pivotal roles in human cancers [96,97,98,99,100,101]. Here, we present each of them and their functions in human cancers (Table 2).
Translation of circSHPRH in glioma
A new circRNA produced by the SNF2 histone linker PHD RING helicase (SHPRH) gene was identified [96]. Mature circSHPRH is formed by backsplicing of exons 26 to 29 and the total length of circSHPRH is 400 nucleotides. The motif of ‘UGAUGA’ contains the overlapping initiation and termination codon (initiation codon is AUG, and termination codon is UGA). The circSHPRH is the first circRNA having overlapping initiation and termination codon during protein translation in eukaryotic cells. circSHPRH encodes a novel 146 amino acids protein (SHPRH-146aa), and displayed a tumor suppressor activity during tumorigenesis. Further analysis suggests SHPRH-146aa protects SHPRH protein from degradation by the ubiquitin proteasome. Another recent studies indicated that circSHPRH was identified as a biomarker in hepatocellular carcinoma [104, 105]. It is therefore reasonable to speculate that circSHPRH has the potential to be translated in other human cancers.
Translation of circLINC-PINT in gliobalstoma
The circLINC-PINT is originated from the long intergenic non-coding RNA p53-induced transcript (LINC-PINT), and encodes a peptide with 87 amino acids (PINT87aa) [98]. This 1084 nt circRNA is formed by the circularization of exon 2. PINT87aa is concentrated in nucleus, and plays a tumor-suppressive role in the control of cell proliferation and tumorigenesis. PINT87aa can interact with PAF1 complex inhibiting the transcriptional elongation of multiple oncogenes. As the binding partner of PAF1 complex, PINT87aa plays important role in deciding PAF1 complex proper localization.
Translation of circβ-catenin in hepatocellular carcinoma (HCC)
The Wnt/β-catenin pathway has been extensively examined in HCC [99]. GSK3β-induced β-catenin phosphorylation and degradation can lead to the activation of Wnt pathway, which correlates with tumorigenesis and poor prognosis in HCC [102]. The circβ-catenin was generated by the backsplicing of exon 2 to 7 of the CTNNB1 gene, forming a 1129 nt circRNA molecule. In circBase [106], a total of 24 circRNAs can be derived from CTNNB1 gene, and circβ-catenin is the only isoform expressed in HCC. The circβ-catenin produces a 370 amino acid peptide (β-catenin-370aa), and mainly located in the cytoplasm. Further in vitro and in vivo assays indicated that circβ-catenin can promote HCC cell growth through activation of Wnt pathway. Acting as a decoy for GSK3β, β-catenin-370aa can lead to escape from GSK3β-induced β-catenin degradation.
Translation of circFBXW7 in glioma and breast cancer
A circRNA, named circFBXW7, was produced by the tumor suppressor E3 ligase FBXW7 [97]. The circFBXW7 is derived from the backsplicing of exon3 to exon4 of the FBXW7 gene with a length of 620 nt. The circFBXW7 encodes a 185 amino acid peptide (FBXW7-185aa). FBXW7-185aa is thought to function as a tumor suppressor in glioma. It interacts competitively with the deubiquitinating enzyme USP28, which antagonizing USP28-induced c-Myc stabilization. Another recent work demonstrated that FBXW7-185aa can inhibit the proliferation and migration of triple-negative breast cancer cells by increasing the abundance of FBXW7 and inducing c-Myc degradation [103].
Translation of circPPP1R12A in colon cancer
Zheng et al. identified a new circRNA, named circPPP1R12A, produced by the PPP1R12A gene [100]. The circPPP1R12A have a 216 nt ORF to encode a 73 amino acid peptide (circPPP1R12A-73aa). Similar to circSHPRH, the circularization contains overlapping initiation and termination codons during protein translation. The expression of circPPP1R12A is significantly up-regulated in tumors, which displays a tumor suppressor activity in colon cancer. Functional analysis showed that CircPPP1R12A-73aa can promote the proliferation and metastasis abilities through activating Hippo-YAP signaling pathway in colon cancer.
Translation of circAKT3 in glioblastoma
Recently, Xia et al. characterized a circRNA, named circAKT3, produced by AKT3 gene [101]. The circAKT3 is generated from exon 3 to exon 7 of the AKT3 gene with a full length of 524 nt, and encodes a 174 amino acid novel protein (AKT3-174aa). The analysis of circAKT3 expression showed the potential tumor suppressor activity of AKT3-17aa. AKT3-174aa can interact with phosphorylated PDK1 and limits AKT3-thr308 phosphorylation as a molecular decoy, and plays a negative regulatory role in modulating the PI3K/AKT signal intensity.
Future perspective
Although circRNAs have been discovered to be key regulators that mediate many fundamental cellular processes, they were once considered as non-coding because of the arbitrary site restriction. Despite being at an early stage, recent works have shown that circular RNAs play key roles in human cancers. The translation of circRNAs will open a hidden human proteome, and enhance us to understand the importance of circRNAs in human cancer, which has been poorly explored so far. It has been documented that some circRNAs exert their biological functions both through encoded peptides and by RNA-based regulatory mechanism. For example, circ-SHPRH can encode SHPRH-146aa peptide to suppress tumorigenesis in glioma [96], and it can function as miRNA sponge to inhibit HCC progression [104, 105]; circZNF609 can act as miRNA sponge to promote breast cancer progression [25], and it can encode protein in Myogenesis [60]. Some circRNAs are bi-functional in human cancer, and the bi-functional ability of circRNAs remains to be explored.
These reported circRNA encoded peptides have significant anti-tumor functions by interfering cancer metabolic reporgramming or metastasis. Therefore, circRNA-encoded peptides have great potential to become therapeutic target and tumor biomarkers. Given their high tumor specificity, circRNA encoded peptides will become a new resource for anti-tumor protein drug screen. To date, tumor biomarkers play important roles in early detection, precise treatment and prognosis prediction. Those circRNA encoded peptides have great potential to be useful tumor biomarkers. Taken together, the researches on circRNA encoded proteins will provide a new way for cancer diagnosis and therapy.
Conclusion
New insights have been gained bout the translation of circRNAs and their functional roles in human cancer. In this review, we summarized the possible scenarios by which circRNAs can serve as translational template. A new perspective is opening for the research on circRNAs, and there’s no doubt that more and more new circRNA encoded proteins will be discovered in the near future. A hidden human proteome will enlighten us on the importance of circRNA in human cancer.
Availability of data and materials
Not applicable.
Abbreviations
- circRNAs:
-
Circular RNAs
- CiRNA:
-
Intronic circRNAs
- CPAT:
-
Coding-potential assessment tool
- CPC:
-
Coding-potential calculator
- EcRNA:
-
Exonic circRNAs
- EIcRNA:
-
exon
Intron circRNAs
- HCC:
-
Hepatocellular carcinoma
- IRES:
-
Internal ribosome entry site
- m6A:
-
N6-methyladenosines
- ORF:
-
Open reading frames
- PhyloCSF:
-
Phylogenetic codon substitution frequencies
- UTR:
-
Untranslated regions
References
Qu S, Zhong Y, Shang R, Zhang X, Song W, Kjems J, Li H. The emerging landscape of circular RNA in life processes. RNA Biol. 2017;14:992–9.
Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32:453–61.
Kolakofsky D. Isolation and characterization of Sendai virus DI-RNAs. Cell. 1976;8:547–55.
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73:3852–6.
Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nat. 1979;280:339–40.
Cocquerelle C, Mascrez B, Hetuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB J. 1993;7:155–60.
Wang PL, Bao Y, Yee MC, Barrett SP, Hogan GJ, Olsen MN, Dinneny JR, Brown PO, Salzman J. Circular RNA is expressed across the eukaryotic tree of life. PLoS One. 2014;9:e90859.
Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, Rajewsky N. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10:170–7.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–57.
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9:e1003777.
Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker SE, Graveley BR, Lai EC. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014;9:1966–80.
Maass PG, Glazar P, Memczak S, Dittmar G, Hollfinger I, Schreyer L, Sauer AV, Toka O, Aiuti A, Luft FC, Rajewsky N. A map of human circular RNAs in clinically relevant tissues. J Mol Med (Berl). 2017;95:1179–89.
Xia S, Feng J, Lei L, Hu J, Xia L, Wang J, Xiang Y, Liu L, Zhong S, Han L, He C. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief Bioinform. 2017;18:984–92.
Wang Y, Wu N, Liu J, Wu Z, Dong D. FusionCancer: a database of cancer fusion genes derived from RNA-seq data. Diagn Pathol. 2015;10:131.
Derks KW, Misovic B, van den Hout MC, Kockx CE, Gomez CP, Brouwer RW, Vrieling H, Hoeijmakers JH, van IWF PJ. deciphering the RNA landscape by RNAome sequencing. RNA Biol. 2015;12:30–42.
Szabo L, Salzman J. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet. 2016;17:679–92.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733.
Hanan M, Soreq H, Kadener S. CircRNAs in the brain. RNA Biol. 2017;14:1028–34.
Veno MT, Hansen TB, Veno ST, Clausen BH, Grebing M, Finsen B, Holm IE, Kjems J. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015;16:245.
Yang Q, Wu J, Zhao J, Xu T, Zhao Z, Song X, Han P. Circular RNA expression profiles during the differentiation of mouse neural stem cells. BMC Syst Biol. 2018;12:128.
You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015;18:603–10.
Nicolet BP, Engels S, Aglialoro F, van den Akker E, von Lindern M, Wolkers MC. Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res. 2018;46:8168–80.
Qu S, Liu Z, Yang X, Zhou J, Yu H, Zhang R, Li H. The emerging functions and roles of circular RNAs in cancer. Cancer Lett. 2018;414:301–9.
Wang Y, Mo Y, Gong Z, Yang X, Yang M, Zhang S, Xiong F, Xiang B, Zhou M, Liao Q, et al. Circular RNAs in human cancer. Mol Cancer. 2017;16:25.
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56:55–66.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22:256–64.
Karreth FA, Pandolfi PP. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov. 2013;3:1113–21.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8.
Han D, Li J, Wang H, Su X, Hou J, Gu Y, Qian C, Lin Y, Liu X, Huang M, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatol. 2017;66:1151–64.
Kleaveland B, Shi CY, Stefano J, Bartel DP. A network of noncoding regulatory RNAs acts in the mammalian brain. Cell. 2018;174:350–62 e317.
Lei B, Tian Z, Fan W, Ni B. Circular RNA: a novel biomarker and therapeutic target for human cancers. Int J Med Sci. 2019;16:292–301.
Granados-Riveron JT, Aquino-Jarquin G. The complexity of the translation ability of circRNAs. Biochim Biophys Acta. 1859;2016:1245–51.
Dhamija S, Menon MB. Non-coding transcript variants of protein-coding genes - what are they good for? RNA Biol. 2018;15:1025–31.
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, et al. Translation of CircRNAs. Mol Cell. 2017;66:9–21 e27.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18:5–18.
Huang C, Liang D, Tatomer DC, Wilusz JE. A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev. 2018;32:639–44.
Harland R, Misher L. Stability of RNA in developing Xenopus embryos and identification of a destabilizing sequence in TFIIIA messenger RNA. Development. 1988;102:837–52.
Dong R, Ma XK, Chen LL, Yang L. Increased complexity of circRNA expression during species evolution. RNA Biol. 2017;14:1064–74.
Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58:870–85.
Smid M, Wilting SM, Uhr K, Rodriguez-Gonzalez FG, de Weerd V, der Smissen WJC P-V, van der Vlugt-Daane M, van Galen A, Nik-Zainal S, Butler A, et al. the circular RNome of primary breast cancer. Genome Res. 2019;29:356–66.
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–47.
Chen S, Huang V, Xu X, Livingstone J, Soares F, Jeon J, Zeng Y, Hua JT, Petricca J, Guo H, et al. Widespread and functional RNA circularization in localized prostate Cancer. Cell. 2019;176:831–43 e822.
Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Wu YM, Dhanasekaran SM, Engelke CG, Cao X, Robinson DR, et al. The landscape of circular RNA in Cancer. Cell. 2019;176:869–81 e813.
Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt LM, Teupser D, Hackermuller J, Stadler PF. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014;15:R34.
Hansen TB, Veno MT, Damgaard CK, Kjems J. Comparison of circular RNA prediction tools. Nucleic Acids Res. 2016;44:e58.
Zeng X, Lin W, Guo M, Zou Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol. 2017;13:e1005420.
Hansen TB. Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol. 2018;6:20.
Panda AC. Circular RNAs act as miRNA sponges. Adv Exp Med Biol. 2018;1087:67–79.
Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in cancer. Cancer Res. 2013;73:5609–12.
Xu H, Guo S, Li W, Yu P. The circular RNA Cdr1as, via miR-7 and its targets, regulates insulin transcription and secretion in islet cells. Sci Rep. 2015;5:12453.
Zheng XB, Zhang M, Xu MQ. Detection and characterization of ciRS-7: a potential promoter of the development of cancer. Neoplasma. 2017;64:321–8.
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215.
Li Y, Zheng F, Xiao X, Xie F, Tao D, Huang C, Liu D, Wang M, Wang L, Zeng F, Jiang G. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. EMBO Rep. 2017;18:1646–59.
Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S, Kim J, Noh JH, Kim KM, Martindale JL, Gorospe M. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017;14:361–9.
Abe N, Matsumoto K, Nishihara M, Nakano Y, Shibata A, Maruyama H, Shuto S, Matsuda A, Yoshida M, Ito Y, Abe H. Rolling circle translation of circular RNA in living human cells. Sci Rep. 2015;5:16435.
Chen X, Han P, Zhou T, Guo X, Song X, Li Y. circRNADb: a comprehensive database for human circular RNAs with protein-coding annotations. Sci Rep. 2016;6:34985.
Wilusz JE. Circular RNAs: unexpected outputs of many protein-coding genes. RNA Biol. 2017;14:1007–17.
Chen CY, Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Sci. 1995;268:415–7.
Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in Myogenesis. Mol Cell. 2017;66:22–37 e29.
Gross JD, Moerke NJ, von der Haar T, Lugovskoy AA, Sachs AB, McCarthy JE, Wagner G. Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell. 2003;115:739–50.
Schutz P, Bumann M, Oberholzer AE, Bieniossek C, Trachsel H, Altmann M, Baumann U. Crystal structure of the yeast eIF4A-eIF4G complex: an RNA-helicase controlled by protein-protein interactions. Proc Natl Acad Sci U S A. 2008;105:9564–9.
Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, Herdy B, Sonenberg N, Wagner G. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell. 2009;136:447–60.
Godet AC, David F, Hantelys F, Tatin F, Lacazette E, Garmy-Susini B, Prats AC. IRES trans-acting factors, key actors of the stress response. Int J Mol Sci. 2019;20.
Fitzgerald KD, Semler BL. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta. 1789;2009:518–28.
Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol. 1988;62:2636–43.
Macejak DG, Sarnow P. Internal initiation of translation mediated by the 5′ leader of a cellular mRNA. Nature. 1991;353:90–4.
Shatsky IN, Dmitriev SE, Terenin IM, Andreev DE. Cap- and IRES-independent scanning mechanism of translation initiation as an alternative to the concept of cellular IRESs. Mol Cells. 2010;30:285–93.
Spriggs KA, Stoneley M, Bushell M, Willis AE. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol Cell. 2008;100:27–38.
Weingarten-Gabbay S, Elias-Kirma S, Nir R, Gritsenko AA, Stern-Ginossar N, Yakhini Z, Weinberger A, Segal E. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Sci. 2016:351.
Petkovic S, Muller S. RNA circularization strategies in vivo and in vitro. Nucleic Acids Res. 2015;43:2454–65.
Wang Y, Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015;21:172–9.
Perriman R, Ares M, Jr.: Circular mRNA can direct translation of extremely long repeating-sequence proteins in vivo. RNA 1998, 4:1047–1054.
Yang Y, Wang Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J Mol Cell Biol. 2019.
Wang Z. Diverse roles of regulatory non-coding RNAs. J Mol Cell Biol. 2018;10:91–2.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6) a promotes cap-independent translation. Cell. 2015;163:999–1010.
Zhou J, Wan J, Shu XE, Mao Y, Liu XM, Yuan X, Zhang X, Hess ME, Bruning JC, Qian SB. N(6)-Methyladenosine guides mRNA alternative translation during integrated stress response. Mol Cell. 2018;69:636–47 e637.
Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Sci. 2016;352:1413–6.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Chen LL, Wang Y, Wong CC, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27:626–41.
Mokrejs M, Masek T, Vopalensky V, Hlubucek P, Delbos P, Pospisek M. IRESite--a tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 2010;38:D131–6.
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–9.
Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–82.
Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74.
Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M. CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13:34–42.
Chen X, Wang LQ, Huang Y, Qiu P, Murgolo NJ, Greene JR, Wu CH, Jiang Y. IRE_FINDER-computational search of iron response element in human and mouse UTRs. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai). 2002;34:743–7.
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–32.
Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–62.
Meng X, Chen Q, Zhang P, Chen M. CircPro: an integrated tool for the identification of circRNAs with protein-coding potential. Bioinformatics. 2017;33:3314–6.
Sun P, Li G. CircCode: a powerful tool for identifying circRNA coding ability. Front Genet. 2019;10:981.
Julenius K, Molgaard A, Gupta R, Brunak S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology. 2005;15:153–64.
Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23.
Andreev DE, O'Connor PB, Loughran G, Dmitriev SE, Baranov PV, Shatsky IN. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 2017;45:513–26.
van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, Kirchner M, Maatz H, Blachut S, Sandmann CL, et al. The translational landscape of the human heart. Cell. 2019;178:242–60 e229.
Chasse H, Boulben S, Costache V, Cormier P, Morales J. Analysis of translation using polysome profiling. Nucleic Acids Res. 2017;45:e15.
Housman G, Ulitsky I. Methods for distinguishing between protein-coding and long noncoding RNAs and the elusive biological purpose of translation of long noncoding RNAs. Biochim Biophys Acta. 1859;2016:31–40.
Zhang M, Huang N, Yang X, Luo J, Yan S, Xiao F, Chen W, Gao X, Zhao K, Zhou H, et al. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene. 2018;37:1805–14.
Yang Y, Gao X, Zhang M, Yan S, Sun C, Xiao F, Huang N, Yang X, Zhao K, Zhou H, et al. Novel role of FBXW7 circular RNA in repressing Glioma tumorigenesis. J Natl Cancer Inst. 2018;110.
Zhang M, Zhao K, Xu X, Yang Y, Yan S, Wei P, Liu H, Xu J, Xiao F, Zhou H, et al. A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat Commun. 2018;9:4475.
Liang WC, Wong CW, Liang PP, Shi M, Cao Y, Rao ST, Tsui SK, Waye MM, Zhang Q, Fu WM, Zhang JF. Translation of the circular RNA circbeta-catenin promotes liver cancer cell growth through activation of the Wnt pathway. Genome Biol. 2019;20:84.
Zheng X, Chen L, Zhou Y, Wang Q, Zheng Z, Xu B, Wu C, Zhou Q, Hu W, Jiang J. A novel protein encoded by a circular RNA circPPP1R12A promotes tumor pathogenesis and metastasis of colon cancer via hippo-YAP signaling. Mol Cancer. 2019;18:47.
Huang X, Li Z, Zhang Q, Wang W, Li B, Wang L, Xu Z, Zeng A, Zhang X, He Z, et al. Circular RNA AKT3 upregulates PIK3R1 to enhance cisplatin resistance in gastric cancer via miR-198 suppression. Mol Cancer. 2019;18:71.
Nejak-Bowen KN, Monga SP. Beta-catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin Cancer Biol. 2011;21:44–58.
Ye F, Gao G, Zou Y, Zheng S, Zhang L, Ou X, Xie X, Tang H. circFBXW7 inhibits malignant progression by sponging miR-197-3p and encoding a 185-aa protein in triple-negative breast Cancer. Mol Ther Nucleic Acids. 2019;18:88–98.
Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z, Yang J, Fan J, Liu L, Qin W. Hsa_circ_0001649: a circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark. 2016;16:161–9.
Zhang X, Qiu S, Luo P, Zhou H, Jing W, Liang C, Tu J. Down-regulation of hsa_circ_0001649 in hepatocellular carcinoma predicts a poor prognosis. Cancer Biomark. 2018;22:135–42.
Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–70.
Acknowledgements
We thank Prof. Ming Shi for helpful discussion.
Funding
This work is supported by Jiangsu Provincial Key Medical Discipline, The Project of Invigorating Health Care through Science, Technology and Education (NO.ZDXKA2016014).
Author information
Authors and Affiliations
Contributions
DD and JZ designed the study, and DD, ML, GZ, QN and JZ carried out the data analysis. DD wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lei, M., Zheng, G., Ning, Q. et al. Translation and functional roles of circular RNAs in human cancer. Mol Cancer 19, 30 (2020). https://doi.org/10.1186/s12943-020-1135-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-1135-7