
Abstract
Cancer cells are frequently confronted with metabolic stress in tumor microenvironments due to their rapid growth and limited nutrient supply. Metabolic stress induces cell death through ROS-induced apoptosis. However, cancer cells can adapt to it by altering the metabolic pathways. AMPK and AKT are two primary effectors in response to metabolic stress: AMPK acts as an energy-sensing factor which rewires metabolism and maintains redox balance. AKT broadly promotes energy production in the nutrient abundance milieu, but the role of AKT under metabolic stress is in dispute. Recent studies show that AMPK and AKT display antagonistic roles under metabolic stress. Metabolic stress-induced ROS signaling lies in the hub between metabolic reprogramming and redox homeostasis. Here, we highlight the cross-talk between AMPK and AKT and their regulation on ROS production and elimination, which summarizes the mechanism of cancer cell adaptability under ROS stress and suggests potential options for cancer therapeutics.
Similar content being viewed by others


Background
Metabolic stress, prevalently existing in tumor microenvironments and characterized with nutrient, oxygen and growth factor deprivation, is the consequence of aberrant proliferation and relative inadequate angiogenesis and vascularization [1–4]. Glucose deprivation is one of the main patterns of metabolic stress due to the dramatic reliance on glucose for energy production in cancer cells [5, 6]. It is estimated that glucose concentration in tumors may be 3–10 folds lower than noncancerous tissues [7]. This nutrient deficiency directly reduces ATP production and leads to reactive oxygen species (ROS) overproduction [8].
The ROS accumulation activates multiple pathways and exerts discrepant impacts on cancer cell survival [9]. AMP-activated protein kinase (AMPK), the energy sensor in cells, is activated in response to this stress and promotes metabolic reprograming. AKT (also known as protein kinase B, PKB), a proto-oncogene activated in multiple cancers, acts as anti-apoptotic factor to a variety of stimuli such as radiation, hypoxia and chemotherapy [10]. However, growing number of studies indicate the activation of AKT does not inhibit cell death, but renders cells more sensitive to metabolic stress instead [11–14]. It is anticipated that anti-apoptotic ability of AKT is coupled with glucose metabolism. Glucose deprivation could induce ROS overload, causing AKT hyperactivation and accelerating cell death. This implies dual roles of AKT in tumor growth and stress resistance [11].
From the view of ROS production and elimination in combination with these two regulators under metabolic stress, we review the metabolic reprogramming and redox homeostasis of cancer cells, highlighting the cross-talk between AMPK and AKT and their influence on cancer progression and treatment.
ROS-mediated cross-talk between energy and redox homeostasis
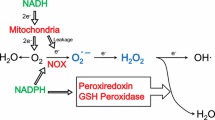
ROS are byproducts of biological reactions of energy generation, and are mainly produced in the mitochondria through the oxidative metabolism [15, 16]. It is estimated that ROS produced by mitochondria are about 1–2% of the total rate of oxygen consumption in normal cells [17]. Cancer cells prevalently exhibit much higher ROS levels than normal cells due to dysfunctional mitochondria, oncogene activation and antioxidant imbalance [8, 18]. ROS are a double-edged sword for oncogenesis. Moderate ROS inactivate the protein tyrosine phosphatases (PTP) such as phosphatase and tensin homolog (PTEN), facilitating phosphoinositide 3-kinase (PI3K) and tyrosine kinase receptor (TKR) signaling, which ultimately leads to tumor progression [19]. However, excess ROS damage cellular structures, such as the lipid membrane, protein and nucleic acid. Specifically for more proliferating cancer cells, excess ROS induce DNA mutations and compromise genome integrity, leading to cell senescence and death [18]. Cancer cells develop an antioxidant system comprised with ROS scavenging enzymes such as superoxide dismutases (SODs), catalase (CAT), glutathione peroxidases (GPX) as well as antioxidant agents like nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH). The antioxidant capacities of cancer cells increase with the rising ROS levels, potentially as a survival adaptation [20–23]. The balance of ROS production and elimination maintains the cellular redox homeostasis, which is vital to cell survival (Fig. 1a).

The cellular redox state is determined by ROS production and elimination: a Under normal condition, cancer cells maintain redox homeostasis by balancing ROS production and elimination. b Under metabolic stress, the redox homeostasis is damaged with enhanced ROS production and decreased ROS elimination
Redox homeostasis is closely linked to glucose metabolism. Cancer cells make prevalent use of glycolysis to produce energy even in aerobic environments, a phenomenon known as the Warburg effect [24]. Although this mode of energy production is less efficient than mitochondrial respiration per unit of glucose, the rate of glycolysis is 10–100 times faster [25]. Glycolysis is an uncomplete energy release process and produces less ROS than mitochondria oxidation. The excess carbons from glycolytic intermediates are ingredients for biosynthesis of lipids, nucleic acids and proteins, which are necessary for increased de novo synthesis of cellular building blocks [26]. Increased glucose absorption is diverted directly or indirectly to pentose phosphate pathway (PPP) to produce NADPH [27], which further facilitates the conversion of oxidized glutathione (GSSG) to reduced GSH by acting as co-substrate of glutathione reductase (GR) [28]. The increased NADPH and GSH not only facilitate biosynthesis, but also create reductive milieu to resist oxidative stress [29]. In different phases of the cell cycle, the main energy-production methods may fluctuate between glycolysis and oxidative phosphorylation (OXPHOS). Glycolysis is a primitive method to produce energy in proliferating cells such as early embryonic cells, stem cells and cancer cells when compared to those rest cells. Enhanced glycolysis contributes to alleviating ROS stress and diminishing the chance of spontaneous mutation during DNA replication [30–32]. Therefore, glycolysis is a protective strategy for rapid ATP synthesis with less ROS stress in cancer cells [33].
The conflicts of redox and metabolic homeostasis under metabolic stress
Under metabolic stress, the redox balance is damaged. Limited glucose sources impair glycolysis, and glycolysis-based NADPH production is depleted by reduced utilization of the PPP [34]. Additionally, glucose limitation leads to overburdening of mitochondria energy production. As a result, the metabolism rewires from glycolysis to OXPHOS and subsequent pro-oxidant production, primarily superoxide and hydrogen peroxide, leads to ROS overload [35, 36]. The disequilibrium of ROS production over ROS scavenging leads to ROS stress (Fig. 1b), which further activates apoptotic pathways. A study by NA Graham et al. based on phospho-tyrosine proteomics showed that metabolic stress provoked a supra-physiological level of TKR signaling, causing a positive feedback loop between ROS, PTPs and TKR signaling, and ultimately leading to cell death [37]. Meanwhile, by using ROS scavenger N-acetylcysteine (NAC), the kinase activation could be reversed under glucose deprivation [38, 39]. In addition, it is reported in M Gao’s study that Reverse Phase Protein Arrays (RPPA) analysis detected the signal change after glucose deprivation. A variety of kinases were activated, which was consistent with NA Graham’s study [37]. Interestingly, though the study of M Gao exhibited a dramatic difference in the spectrum of activated kinases between transient and prolonged glucose deprivation [40]. It was hypothesized that the kinase activation after glucose deprivation and its role in cell survival might be time-dependent and specific to cell lines. These studies underline the importance of the kinase activation loop in mediating ROS-induced cell death.
Although Otto Warburg, who proposed the theory of the Warburg effect, declares mitochondrial dysfunction leads to prevalent use of glycolysis in cancer cells, there are studies suggesting that many cancer cells prioritize OXPHOS to generate ATP [41]. It is generally agreed that enhanced glycolysis in cancer cells does not necessarily correspond to impaired OXPHOS [32]; particularly under metabolic stress, mitochondrial-based energy production is essential for viability. Under metabolic stress, other intermediates like glutamine [42, 43], lactate [44, 45], fatty acids and others [7, 46] are alternatively consumed to produce ATP. J Yun et al. reported that colorectal cancer cells demonstrated higher expression of KRAS and GLUT1 under glucose deprivation [47]. Additionally, glucose deprivation stimulated the tricarboxylic acid (TCA) cycle through mitochondrial glutamine metabolism, which was a source of ATP and generated a-ketoglutarate (α-KG) for biosynthesis [43, 48]. However, enhanced mitochondrial burden under metabolic stress increases the ROS production. The conflicts of producing energy versus maintaining ROS homeostasis dramatically impacts the fate of cancer cells.
ROS regulation by AMPK and AKT under glucose deprivation
Glucose deprivation leads to ATP depletion and ROS accumulation, which in turn activates AMPK. AMPK is a heterotrimer complex, including a catalytic subunit (α) and two regulatory subunits (β and γ). It is phosphorylated on the Thr-172 in the presence of high AMP/ATP ratios due to ATP depletion, allowing it to act as an energy sensor for the cell. It is also regulated by its upstream LKB1, CaMMK or other factors like ADP or Ca2+ [49]. In addition, ROS directly activate AMPK through S-glutathionylation of cysteines on the AMPKα and β subunit [31].
AMPK mediates metabolic reprogramming to survive glucose deprivation by promoting catabolism (glucose uptake, glycolysis, fatty acid oxidation, autophagy, etc.) and suppressing anabolism (protein, fatty acid, glycogen synthesis) [49–52] (Fig. 2). AMPK also regulates the redox state by alleviating the glucose deprivation-induced NADPH depletion via decreased fatty acid synthesis and increased fatty acid oxidation [46]. Further, B Chaube et al. found that AMPK could enhance mitochondrial biogenesis and OXPHOS by activating p38/PGC1α pathway [53]. This indicates AMPK is not only involved in glycolytic regulation, but also participates in ATP generation from OXPHOS.

Cross effects of AMPK and AKT on the cellular metabolism and redox state: The targeted proteins regulated by AMPK and AKT and their regulatory effects are depicted, AMPK is a key player in response to metabolic stress by regulating the metabolism of glucose, lipid and protein. AMPK promotes glucose uptake and glycolysis, facilitating antioxidant production. AMPK also stimulates fatty acid oxidation and limits the fatty acid synthesis. mTOR and FOXO are two main downstream effectors of AMPK. AMPK inhibits mTOR activity, which induces protein synthesis inhibition and autophagy activation. AMPK also promotes FOXO activity to maintain the redox balance through enhanced antioxidant production and glucose metabolism. On the other side, AKT exerts antagonistic effect to regulate mTOR and FOXO activity. AKT stimulates mTOR signaling to promote glucose metabolism and protein synthesis, leading to increased ROS production. Meanwhile, it inhibits FOXO activity and renders cells susceptible to ROS toxicity
The serine/threonine kinase AKT is widely acknowledged as a proto-oncogene. It is activated by extracellular signals (mostly growth factors through PI3K signaling) and is downregulated by PTEN. AKT mediates carcinogenesis and tumor progression mainly through promoting cell survival and inhibiting apoptosis [54] (Fig. 2). In addition, AKT is evolutionarily conserved and regulates glucose metabolism [14]. AKT promotes glycolysis through increasing GLUT1 trafficking to the cell surface, and through phosphofructokinase (PFK) and hexokinase (HK) activation [12]. Moreover, by stimulating oxidative metabolism, AKT promotes mitochondria oxygen consumption and contributes to ROS accumulation [55]. AKT is downstream to multiple growth factors such as EGF, IGF and HGF, etc. These growth factors are increased via autocrine or paracrine signals in nutrient-abundant conditions [56], indicating the role of AKT in proliferation is closely related to a well suitable growth milieu.
Recently, some studies indicate that the ability of AKT to inhibit cell death is dependent on glucose metabolism [13, 57]. JL Coloff et al. found that AKT suppressed Bim-induced cell death only when glucose was present [12]. Additionally, AKT activation rendered glioblastoma cells more sensitive to glucose withdrawal-induced cell death [13], and overexpression of PTEN dramatically reversed this process [37]. Further, V Nogueira et al. found that AKT activation rendered cells more susceptible to ROS-mediated premature senescence and cell death by increasing oxygen consumption and suppressing FOXO activity [14]. These studies imply that AKT acts as a pro-apoptotic factor under ROS stress, which is at odds with the established cognition of AKT as a tumor protective gene. Moreover, AKT is one of the factors involved in the aforementioned glucose deprivation-induced cell death via strengthening the kinase activation loop [37].
The cross-talk between AMPK and AKT under metabolic stress
It is interesting that under glucose deprivation, AKT plays antagonistic roles from AMPK in ROS-mediated cell apoptosis. mTOR and FOXO are two main downstream effectors regulated by both AMPK and AKT, which exert antagonistic effects on ROS homeostasis. In addition, AMPK and AKT also regulate mutual phosphorylation directly or indirectly.
mTOR signaling
mTOR is a nutrient and growth factor sensing complex, which lies the intersection between glucose and amino acid metabolism and contributes to biosynthesis and autophagy [58]. Active mTOR1 phosphorylates ribosomal protein S6 kinase (S6K) and eukaryotic translation initiation factor 4E (eIF4E) binding proteins (4E-BPs), controlling the activity of eukaryotic initiation factors (eIFs) and eukariotic elongation factors (eEFs). This promotes the protein synthesis and ribosome biosynthesis [31, 58]. In addition, mTOR1 inactivates UNC51like kinase 1 (ULK1) at Ser-757 to inhibit autophagy. Interestingly, AMPK phosphorylates ULK1 at Ser-317 and Ser-777 to initiate Beclin1-mediated autophagy [59]. AMPK phosphorylates the regulatory-associated protein of mTOR (Raptor) at Ser-792/Ser-722 and tuberous sclerosis complex 2 (TSC2) at Ser-1387, indirectly leading to inhibition of mTOR1 (Fig. 3). mTOR1 activity is positively correlated with mitochondrial activity and promotes oxidative metabolism [60, 61]. Under glucose deprivation, AMPK inhibits mTOR1 activity, thereby decreasing protein synthesis and increasing autophagy. Decreased anabolism reduces ROS production, while enhanced autophagy and glycolysis increases the resilience of cells to ROS.

Signaling of AMPK and AKT on the ROS homeostasis via mTOR and FOXO regulation: Under metabolic stress, AMPK inhibits mTOR mainly via two ways:phosphorylates TSC2 at Ser-1387 which stimulates the TSC1-TSC2 complex to inhibit Rheb’s ability to activate mTOR; phosphorylates Raptor at Ser-792/Ser-722 to inhibit mTOR1. AKT activates mTOR reversely: AKT phosphorylates TSC2 at another site and activates mTOR via Rheb; AKT phosphorylates PRAS40 to inhibit its ability to suppress mTOR. Activated mTOR in turn promotes protein synthesis through S6K1 and 4E-BP1. AMPK phosphorylates FOXO, promoting the translocation to nucleus. AMPK also facilitates FOXO acetylation and enhances its transcriptional activity of antioxidant genes: SOD, Catalase, Sestrin. Additionally, AMPK promotes NADPH production via the PPP. On the other hand, AKT phosphorylates FOXO and leads to the translocation from the nucleus to the cytoplasm. By ubiquitination of FOXO, AKT leads to its degradation in cytoplasm
However, opposed to AMPK, AKT activates mTOR by inhibiting TSC2 and subsequently allowing Rheb-GAP to phosphorylate mTOR1. Additionally, AKT inactivates PRAS40, which alleviates the PRAS40-mediated inhibition of mTORC1 (Fig. 3). By activating mTORC1, AKT promotes oxygen consumption and increases ROS production under glucose deprivation [54, 62], rendering cancer cells closer to the death threshold of ROS.
FOXO signaling
FOXO acts in response to starvation and oxidative stress. FOXO normally exists in cytoplasm in an inactive form and translocate to nucleus to initiate transcriptional activity once activated [63]. FOXO activation increases resistance to oxidative stress by targeting the expression of SOD, catalase and sestrin, which are the antioxidant enzymes that maintain redox homeostasis [64]. In addition, FOXO participates in glucose metabolism by regulating phosphoenolpyruvate carboxykinase (PEPCK), Glucose-6-phosphatase (G6Pase) and peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α). PEPCK is the key enzyme in gluconeogenesis which is enhanced in energy deprivation and promotes glucose and glutamine metabolism [65, 66]; PGC1α is a major transcription coactivator closely related to mitochondria biogenesis and OXPHOS [67], PGC1α positive cells exhibit increased ROS detoxification capacities in some cancers such as melanoma [68]. In addition, FOXO induces expression of autophagy-related genes (ATG6, ATG7, ATG12, etc.) to elevate autophagic flux and increases the production of mainly fatty acid and amino acids consumed by mitochondria OXPHOS [64, 69].
AMPK can directly regulate FOXO. AMPK enhances FOXO3-mediated transcriptional activity by recruiting CREB-binding protein (CBP) and p300 [63, 70, 71]. It also regulates S-phase kinase-associated protein 2/coactivator-associated arginine methyltransferase 1 (SKP2/CARM1) signaling by FOXO3 phosphorylation to induce autophagy under glucose starvation [72]. In EL Greer’s study, AMPK-mediated FOXO3 phosphorylation did not affect the nuclear localization of FOXO3 [73], indicating AMPK influences FOXO3 activity only when it is in the nucleus. Conversely, there are also conflicting studies that suggest that AMPK may in fact facilitate FOXO3 nuclear localization [64, 74]. The exact effect of FOXO phosphorylation remains unclear. In addition, AMPK may increase nuclear FOXO acetylation and mediate localization to nuclear promyelocytic leukemia (PML) bodies, which act as transcriptional co-activators [75]. AMPK indirectly promotes FOXO acetylation by retaining class II histone deacetylase (HDAC) in the cytosol [63, 76]. Taken together, FOXO post-translational modifications greatly influence its function and AMPK is crucial in this process (Fig. 3).
In contrast to AMPK, FOXO is negatively regulated by AKT signaling. Glucose deprivation-derived ROS production induces the nucleus translocation of FOXO and thereby promotes transcriptional activity of antioxidant-related genes [63, 77]. However, AKT inhibits this process by phosphorylating the FOXO at three conserved residues and inversely translocates FOXO from nucleus to cytoplasm [78]. Besides, AKT also promotes the ubiquitination of FOXO and leads to its degradation [79] (Fig. 3).
Mutual phosphorylation regulation of AMPK and AKT
AKT blunts AMPK activation. In a rat model of ischemia perfusion, AKT phosphorylates AMPKα1/α2 at Ser-485/491 (equivalent to Ser-487/491 in human), while the phosphorylation of Thr-172 is reduced [80]. In another rat model of hypoxia, AKT activation also prevents the phosphorylation of AMPKα at Thr-172 [81]. This phenomenon is consistently found in human normal tissues as well as in tumor such as breast and liver cancer cell [82–85]. Ser-487 of AMPKα is located in the serine/threonine-rich loop (ST loop, residues 472–525 in human) within the C-terminal domain of AMPKα1, which interacts with residues within the kinase domain. Ser-487 phosphorylation hinders the ability of upstream kinase LKB1 or CaMMK to access Thr-172 [83]. Moreover, in specific glioblastoma and breast cancer cells characterized by hyperactivity of AKT due to loss of PTEN, AMPK is resistant to activation by AMPK activator A769662, though this effect is reversed by addition of MK2206 as an AKT inhibitor [83, 86].
AMPK reversely inhibits AKT phosphorylation. AMPK activated by AICAR or phenformin dephosphorylates Ser-473 and Thr-308 of AKT, thereby inhibiting its activity. This blockade of AKT by AMPK agonists consequently lowers the inhibition of AKT’s downstream effector, glycogen synthase kinase-3α/β (GSK3α/β) [87]. In addition, AMPK affects AKT signaling by regulating insulin receptor substrate 1 (IRS1), which is phosphorylated by the insulin receptor and mediates PI3K activation. AMPK phosphorylates IRS1 at Ser-794 (in human, equivalent to Ser-789 in rats) and inhibits AKT signaling [88–90]. However, there are studies indicating AMPK sensitizes AKT phosphorylation through IRS signaling [91]. It is reported that activated AMPK by AICAR stimulates AKT through the same phosphorylation site of Ser-789 [86, 92, 93]. It seems that IRS1 phosphorylation at Ser-794 in humans (Ser-789 in rats) has dual roles in AMPK-mediated AKT signaling, and its function needs to be further determined [93–95]. Taken together, AMPK and AKT have mutual complicated antagonism, which may partially explain their roles in metabolism and redox maintenance (Fig. 4).

Interaction between AMPK and AKT on the phosphorylation: Growth factor activates TKR and promotes the activation of AKT via IRS1/PI3K/PDK1 signaling. Activated AKT phosphorylates AMPKα on Ser-485/491, preventing the active site Thr-172 to get access to LKB1 or CaMMK. AMPK phosphorylates IRS1 at Ser-794 and inhibits AKT signaling. AMPK activated by AICAR or phenformin dephosphorylates Ser-473 and Thr-308 of AKT, inhibiting AKT activity
In general, mTOR inhibition and FOXO activation is essential in AMPK-mediated metabolic reprogramming and ROS scavenging to maintain the redox hemostasis under metabolic stress. On the other hand, AKT activation renders cells more sensitive to ROS-mediated cell death by impairing redox homeostasis through opposite regulation of mTOR and FOXO from AMPK. Additionally, AMPK and AKT have mutual regulation on phosphorylation. These antagonistic regulations are significant in maintaining redox homeostasis under glucose deprivation.
The regulation of AMPK and AKT in response to glucose supply
When glucose is abundant, AMPK activity remains limited and AKT is relatively activated, promoting cancer cell growth, division and metastasis. In addition, growth factor autocrine or paracrine signaling under suitable milieu forms positive feedback loops to activate AKT. Activated AKT also promotes the production of ROS, which stimulates oncogenic pathway, leading to uncontrolled proliferation. However, higher ROS levels render cells closer to the threshold of ROS lethality, which is regarded as the Achilles’ heel of AKT [14].
Under glucose deficiency when AMPK predominates, it resists against glucose deprivation-derived ROS accumulation by increasing glycolysis and the PPP. Meanwhile, autophagy and OXPHOS are enhanced to balance the input and output of energy, relieving the ROS load. This is achieved by the downstream effectors of AMPK, of which FOXO activation and mTOC1 inhibition play key roles (Fig. 5a).

AMPK and AKT mediate ROS regulation in tumor progression and treatment: a-b Under metabolic stress, AMPK and AKT manipulate ROS regulation and influence cell survival. The basal ROS indicate the ROS level under normal condition. c-d ROS load is increased by inducing ROS production or suppressing antioxidants. The effects are magnified when combined with chemo/radiotherapy. e ROS and antioxidant capacities are increased with tumor progression. f Compared to normal tissues, cancer cells are more susceptible to ROS targeting therapy, chemo/radiotherapy or their combined application
Under glucose deprivation when AKT predominates, the anti-apoptotic role of AKT is reversed since glucose is lacking. AKT inhibits FOXO translocation to the nucleus and decreases antioxidant production. Unlike AMPK, which is ubiquitously activated under glucose deprivation, AKT seems to be different among cancer cells. AKT is activated in glioblastoma and Hela cells, for example [13, 14], but inhibited in ovarian cancer and leukemic T cells [12, 96]. In addition, PTEN significantly influences AKT activity under glucose deprivation. When PTEN is present in lung cancer cells, AKT phosphorylation is increased after glucose deprivation. When PTEN is mutated or knocked down, AKT phosphorylation is inhibited instead [97]. This suggests that AKT activation is context-dependent among cell lines and decided by multiple factors. There are reports suggesting that AKT activation can protect cells under glucose deprivation. It is found that site-specific phosphorylation of AKT at site Thr-308 decreased cell death [40]. This shows that under glucose deprivation, AKT function is sophisticated and specific to different cancer cells and backgrounds (Fig. 5b).
The role of ROS regulation through AMPK and AKT in tumor progression
Anoikis resistance
Glucose deficiency is one of the main factors leading to ROS stress in tumors, either in the stage of tumor initiation or progression [18]. At the initial stage of metastasis, cancer cells must leave its original niche to enter the vascular or lymphatic circulation. This entails detaching from the extracellular matrix (ECM) and becoming anchorage independent, which leads to ATP deficiency owing to reduction of glucose transport [22]. In normal human cells, this transition leads to a strong induction of ROS, which leads to anoikis. Tumor cells, on the other hand, may become anoikis resistant and successfully leave their primary niche to become circulation tumor cells (CTC) and facilitate metastatic colonization [98].
Under ECM-detached condition, AMPK promotes survival by autophagy induction and global inhibition of protein synthesis, mitigating the ATP reduction [99, 100]. AMPK also enhances the PPP and increases the NADPH production. Addition of antioxidants like Trolox and NAC can rescue ATP deficiency independent of glucose uptake, further demonstrating the role of AMPK in anoikis resistance [22]. In addition, AKT is activated upon ECM detachment by TKR activation, and it inhibits cell death mainly by promoting glucose uptake and upregulating anti-apoptotic pathways such as BCL2 signaling. AMPK and AKT are both activated in the resistance of anoikis, but AMPK activation seems to be dominant. It is reported that mTOR, the downstream of both AMPK and AKT, was inhibited in aniokis resistant cells [99, 101, 102].
Drug resistance
Many chemotherapy agents and radiotherapy kill cancer cells through ROS-mediated cell death, and ROS resistance is one way for cancer cells to develop drug resistance. Tumor stem cells, which are considered the seed of relapse, have superior resistance to anti-tumor agent [103]. Due to enhanced antioxidant systems, these cells tend to have a lower ROS load than to their non-tumorigenic progeny [18, 104, 105]. Increased autophagy is important for chemotherapy resistance. AMPK, as the inhibitor of mTOR, has been reported to induce autophagy-mediated drug resistance [106–108]. Additionally, replenishing NADPH through AMPK-mediated glucose uptake and the PPP also contributes to drug resistance.
Targeting ROS as anti-cancer therapy
Even though cancer cells exhibit an enhanced antioxidant system, they still maintain higher ROS levels than normal cells [20, 109]. ROS load increases with the tumor progression [23]. Compared to normal cells, cancer cells are closer to the threshold of ROS toxicity. Interrupting redox homeostasis may be a potential target to inhibit tumor metastasis and mitigate the drug resistance. This could be achieved by inhibition of ROS detoxification or stimulation of ROS production [18] (Fig. 5c-f).
Metabolic inhibition is one way to cause ROS accumulation. 2-deoxyglucose (2-DG), the glucose analogue, competes with the glucose transporter and inhibits HK2 activity and classically used to mimic metabolic stress. 2-DG not only activates AMPK, but also induces AKT phosphorylation [110, 111]. It is designed to have anti-tumor activity both in vivo and in vitro, but the clinical trials fail to reach better patient outcomes after a single use of 2-DG [112]. However, 2-DG combined with chemotherapy or radiotherapy has had better results, as it renders cancer cells more sensitive to apoptosis by further elevation of ROS [113, 114]. The inability of AKT to inhibit ROS-mediated cell apoptosis also provides a strategy to treat cancer. In tumor with hyperactivated AKT, further ROS production sensitizes cells to ROS induced apoptosis. This has been achieved by combined use of ROS inducer phenylethyl isothiocyanate (PEITC) and mTOR inhibitor rapamycin, which completely eradicate tumor growth in cells with hyperactivated AKT both in vitro and in vivo [14]. Since mTOR elicits a negative feedback loop to suppress AKT, rapamycin could lead to further AKT activation through mTOR1 inhibition [115]. In addition, RV Pusapati et al. found that 2-DG-induced glycolytic inhibition could be enhanced by active mTOR signaling via increased glutamine uptake and pentose phosphate flux [111], which suggested metabolic inhibition alone was not sufficient for cancer treatment. Promisingly, the combination of glycolytic inhibition and ROS inducer may exert a more synergistic effect for cancer therapy.
Depletion of antioxidant is another way to induce ROS toxicity. GSH is the most abundant antioxidant in cells and its recyclability is dependent on NADPH production. Targeting GSH dramatically breaks down the redox balance, as demonstrated by the dramatic inhibition of the thioredoxin (TXN) pathway by the combination use of buthioine sulfoximine (BSO) and auranofin (AUR) [28]. PEITC, an inhibitor of glutathione peroxidase (GPX), also depletes GSH and shows anti-tumor effects in multiple cancers. Further, sulphasalazine (SSA) decreases GSH levels by inhibiting the cysteine transport, reducing the growth and viability in cancer cells [116, 117]. A newly synthetized enzyme cysteinase could effectively lead to ROS elevation and cell death by depleting extracellular L-cysteine, which is essential for cellular GSH synthesis [118]. However, there are controversies about antioxidant depletion to treat cancer. Previous studies stress that antioxidant can decrease carcinogenesis and tumor development [119], but recent studies show that antioxidant can relieve the anoikis-mediated oxidative stress and promote cell survival and metastasis [120–122]. Some antidiabetic drugs like saxagliptin, sitagliptin and antineuropathic α-lipoic acid (ALA) also have antioxidant properties, and are reported to promote metastasis by increasing antioxidant capacity [123].
Conclusions
During cancer progression, cancer cells are frequently confronted with metabolic stress, which is accompanied with redox disequilibrium. The rewiring of glucose metabolism and antioxidant maintenance intersect at the response to the stress. In this process, AMPK and AKT play significant roles. AMPK mediates metabolic change to resist the ROS accumulation, while AKT renders cells more susceptible to ROS mediated cell death. This review summarizes the cross-talk between these two kinases, mainly through antagonistic regulation on their downstream effectors, mTOR and FOXO. We suggest that under metabolic stress, the role of AMPK and AKT might be varied and context-specific to influence the cell fate.
The tight relationship between the AMPK and AKT acting on ROS homeostasis is closely related to the tumor progression and treatment. The resilience of ROS toxicity is one of the main mechanisms of tumor metastasis and drug resistance. Studying the dynamic change of ROS in tumor progression deepens the understanding of aniokis and metastatic colonization. Further, modulation of oxidative stress by targeting ROS detoxification or stimulation provides new strategies to treat cancers. Compared to traditional chemotherapy or radiotherapy, combined utilization of ROS inducer (or antioxidant inhibitors) with traditional therapies may exert synergistic effect through enhanced ROS-mediated cell death. Meanwhile, this strategy may cause less harm to normal cells, which have more potential capacities to resist ROS than cancer cells. Therein, targeting ROS may be a promising way for anticancer therapy, and their regulatory mechanisms need more detailed research.
Abbreviations
- 2-DG:
-
2-deoxyglucose
- 4E-BP:
-
Eukaryotic translation initiation factor 4E-binding protein
- 6PG:
-
6-phosphogluconate
- ACC:
-
Acetyl-CoA carboxylase
- AICAR:
-
5-aminoimidazole-4-carboxamide1-β-D-ribofuranoside
- AKT:
-
Protein kinase B
- ALA:
-
α-lipoic acid
- AMPK:
-
AMP-activated protein kinase
- ATG:
-
Autophagy-related gene
- AUR:
-
Auranofin
- BAD:
-
Bcl2-associated agonist of cell death
- BSO:
-
Buthioine sulfoximine
- CARM1:
-
Coactivator-associated arginine methyltransferase 1
- CAT:
-
Catalase
- CBP:
-
CREB-binding protein
- CTC:
-
Circulation tumor cell
- Deptor:
-
DEP domain-containing mTOR-interacting protein
- ECM:
-
Extracellular matrix
- eIF4B/E:
-
Eukaryotic translation initiation factor 4B/E
- F6P:
-
Fructose-6-phosphate
- FOXO:
-
Forkhead box protein O
- G3P:
-
Glycerol-3-phosphate
- G6P:
-
Glucose-6-phosphate
- G6Pase:
-
Glucose-6-phosphatase
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GLUT:
-
Glucose transporter
- GPAT:
-
Glycerol phosphate acyltransferase
- GPX:
-
Glutathione peroxidase
- GR:
-
Glutathione reductase
- GSH:
-
Glutathione
- GSK:
-
Glycogen synthase kinase
- GSSG:
-
Oxidized glutathione
- GYS:
-
Glycogen synthase
- HDAC:
-
Histone deacetylase
- HK:
-
Hexokinase
- HMGR:
-
3-hydroxy-3-methylglutaryl-CoA reductase
- IKK:
-
NF-kappa-B essential modulator
- IRS1:
-
Insulin receptor substrate 1
- LKB1:
-
Liver kinase B1
- MDM2:
-
E3 ubiquitin-protein ligase Mdm2
- MLST8:
-
Target of rapamycin complex subunit LST8
- mTOR:
-
Mammalian target of rapamycin
- NAC:
-
N-acetylcysteine
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- OXPHOS:
-
Oxidative phosphorylation
- PDK1:
-
3-phosphoinositide-dependent protein kinase 1
- PEITC:
-
Phenylethyl isothiocyanate
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- PFK:
-
ATP-dependent 6-phosphofructokinase
- PFKFB:
-
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase
- PGC1α:
-
Peroxisome proliferator-activated receptor γ coactivator 1α
- PI3K:
-
Phosphoinositide 3 kinase
- PK:
-
Pyruvate kinase
- PML:
-
Promyelocytic leukemia
- PPP:
-
Pentose phosphate pathway
- PRAS40:
-
Proline-rich AKT1 substrate of 40 kDa
- PTEN:
-
Phosphatase and tensin homolog
- PTP:
-
Protein tyrosine phosphatases
- Raptor:
-
Regulatory associated protein of mTOR
- ROS:
-
Reactive oxygen species
- RRPA:
-
Reverse phase protein array
- S6K:
-
Ribosomal protein S6 kinase
- SKP2:
-
S-phase kinase-associated protein 2
- SOD:
-
Superoxide dismutase
- SREBP1C:
-
Sterol regulatory element-binding protein 1C
- SSA:
-
Sulfasalazine
- ST loop:
-
Serine/threonine-rich loop
- TCA:
-
Tricarboxylic acid
- TIFIA:
-
Transcription initiation factor IA
- TKR:
-
Tyrosine kinase receptor.
- TSC2:
-
Tuberous sclerosis 2
- TXN:
-
Thioredoxin
- ULK:
-
UNC-51-like kinase
- α-KG:
-
α-ketoglutarate
References
Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28–31.
Gimbrone MA, Leapman SB, Cotran RS, Folkman J. Tumor dormancy in vivo by prevention of neovascularization. J Exp Med. 1972;136:261–76.
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–121.
Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, Jain RK. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res. 2013;73:3833–41.
Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16(10):635–49.
Cairns RA, Mak TW. The current state of cancer metabolism. Nat Rev Cancer. 2016;16:613–4.
Vincent EE, Sergushichev A, Griss T, Gingras M-C, Samborska B, Ntimbane T, Coelho PP, Blagih J, Raissi TC, Choinière L. Mitochondrial phosphoenolpyruvate carboxykinase regulates metabolic adaptation and enables glucose-independent tumor growth. Mol Cell. 2015;60:195–207.
Panieri E, Santoro M. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7:e2253.
Marengo B, Nitti M, Furfaro AL, Colla R, De Ciucis C, Marinari UM, Pronzato MA, Traverso N, Domenicotti C. Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxid Med Cell Longev. 2016;2016:6235641.
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–56.
Los M, Maddika S, Erb B, Schulze‐Osthoff K. Switching Akt: from survival signaling to deadly response. Bioessays. 2009;31:492–5.
Coloff JL, Mason EF, Altman BJ, Gerriets VA, Liu T, Nichols AN, Zhao Y, Wofford JA, Jacobs SR, Ilkayeva O. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J Biol Chem. 2011;286:5921–33.
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9.
Nogueira V, Park Y, Chen C-C, Xu P-Z, Chen M-L, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–70.
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–50.
Ray PD, Huang B-W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–90.
Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605.
Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–47.
Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14:443–51.
Kaynar H, Meral M, Turhan H, Keles M, Celik G, Akcay F. Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu–Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer Lett. 2005;227:133–9.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Kenneth HY, Yeo CJ, Calhoun ES. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9.
Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–13.
Miar A, Hevia D, Muñoz-Cimadevilla H, Astudillo A, Velasco J, Sainz RM, Mayo JC. Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radic Biol Med. 2015;85:45–55.
Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P. The Warburg effect: insights from the past decade. Pharmacol Ther. 2013;137:318–30.
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308.
Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–54.
Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–22.
Desideri E, Filomeni G, Ciriolo MR. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy. 2012;8:1769–81.
Chen Z, Odstrcil EA, Tu BP, McKnight SL. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science. 2007;316:1916–9.
Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377–88.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47.
Zhang S, Yang C, Yang Z, Zhang D, Ma X, Mills G, Liu Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am J Cancer Res. 2015;5:928.
Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, Buettner GR, Venkataraman S, Mackey MA, Flanagan SW, Oberley LW. Mitochondrial and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem. 2005;280:4254–63.
Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–12.
Moley K, Mueckler M. Glucose transport and apoptosis. Apoptosis. 2000;5:99–105.
Graham NA, Tahmasian M, Kohli B, Komisopoulou E, Zhu M, Vivanco I, Teitell MA, Wu H, Ribas A, Lo RS. Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Mol Syst Biol. 2012;8:589.
Lee YJ, Galoforo SS, Berns CM, Chen JC, Davis BH, Sim JE, Corry PM, Spitz DR. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J Biol Chem. 1998;273:5294–9.
Blackburn R, Spitz D, Liu X, Galoforo S, Sim J, Ridnour L, Chen J, Davis B, Corry P, Lee Y. Metabolic oxidative stress activates signal transduction and gene expression during glucose deprivation in human tumor cells. Free Radic Biol Med. 1999;26:419–30.
Gao M, Liang J, Lu Y, Guo H, German P, Bai S, Jonasch E, Yang X, Mills GB, Ding Z. Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene. 2014;33:745–55.
Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313:459–65.
Martinelle K, Doverskog M, Jacobsson U, Chapman BE, Kuchel PW, Häggström L. Elevated glutamate dehydrogenase flux in glucose‐deprived hybridoma and myeloma cells: Evidence from 1H/15N NMR. Biotechnol Bioeng. 1998;60:508–17.
Yang C, Sudderth J, Dang T, Bachoo RG, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69:7986–93.
Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–42.
Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008;118:3835–7.
Jeon S-M, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–5.
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–9.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci. 2007;104:19345–50.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62.
Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2015;33:1–7.
Lin L, Huang H, Liao W, Ma H, Liu J, Wang L, Huang N, Liao Y. MACC1 supports human gastric cancer growth under metabolic stress by enhancing the Warburg effect. Oncogene. 2015;34:2700–10.
Yang T, He W, Cui F, Xia J, Zhou R, Wu Z, Zhao Y, Shi M. MACC1 mediates acetylcholine-induced invasion and migration by human gastric cancer cells. Oncotarget. 2016;7(14):18085–94.
Chaube B, Malvi P, Singh SV, Mohammad N, Viollet B, Bhat MK. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015;1:15063.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74.
Robey RB, Hay N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19(1):25–31.
Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47.
Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–18.
Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol. 2015;33:55–66.
Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–62.
Schieke SM, Phillips D, McCoy JP, Aponte AM, Shen R-F, Balaban RS, Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–52.
Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon S-O, Cantley LC, Blenis J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell. 2010;38:487–99.
Kelly CJ, Hussien K, Fokas E, Kannan P, Shipley RJ, Ashton TM, Stratford M, Pearson N, Muschel RJ. Regulation of O 2 consumption by the PI3K and mTOR pathways contributes to tumor hypoxia. Radiother Oncol. 2014;111:72–80.
Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97.
Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle. 2010;9:1091–6.
Méndez-Lucas A, Hyroššová P, Novellasdemunt L, Viñals F, Perales JC. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. J Biol Chem. 2014;289:22090–102.
Montal ED, Dewi R, Bhalla K, Ou L, Hwang BJ, Ropell AE, Gordon C, Liu W-J, DeBerardinis RJ, Sudderth J. PEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol Cell. 2015;60:571–83.
Wallace M, Metallo CM. PGC1 [alpha] drives a metabolic block on prostate cancer progression. Nat Cell Biol. 2016;18:589–90.
Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287–301.
Chiacchiera F, Simone C. Inhibition of p38α unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy. 2009;5:1030–3.
Wang F, Marshall CB, Yamamoto K, Li G-Y, Gasmi-Seabrook GM, Okada H, Mak TW, Ikura M. Structures of KIX domain of CBP in complex with two FOXO3a transactivation domains reveal promiscuity and plasticity in coactivator recruitment. Proc Natl Acad Sci. 2012;109:6078–83.
Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A. 2005;102:11278–83.
Shin H-JR, Kim H, Oh S, Lee J-G, Kee M, Ko H-J, Kweon M-N, Won K-J, Baek SH. AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534(7608):553–7.
Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–19.
Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L, Moschetta A, Simone C. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ. 2009;16:1203–14.
Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of PML and the nuclear body. Nat Cell Biol. 2000;2:E85–90.
Shackelford DB, Shaw RJ. The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75.
Klotz L-O, Sánchez-Ramos C, Prieto-Arroyo I, Urbánek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72.
Burgering BM, Medema RH. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukoc Biol. 2003;73:689–701.
Huang H, Tindall DJ. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim Biophys Acta (BBA)-Mol Cell Res. 2011;1813:1961–4.
Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase α-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281:5335–40.
Soltys C-LM, Kovacic S, Dyck JR. Activation of cardiac AMP-activated protein kinase by LKB1 expression or chemical hypoxia is blunted by increased Akt activity. Am J Phys Heart Circ Phys. 2006;290:H2472–9.
Valentine RJ, Coughlan KA, Ruderman NB, Saha AK. Insulin inhibits AMPK activity and phosphorylates AMPK Ser 485/491 through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch Biochem Biophys. 2014;562:62–9.
Hawley SA, Ross FA, Gowans GJ, Tibarewal P, Leslie NR, Hardie DG. Phosphorylation by Akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem J. 2014;459:275–87.
Ning J, Xi G, Clemmons DR. Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology. 2011;152:3143–54.
Berggreen C, Gormand A, Omar B, Degerman E, Göransson O. Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am J Physiol Endocrinol Metab. 2009;296:E635–46.
Kuznetsov JN, Leclerc GJ, Leclerc GM, Barredo JC. AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Mol Cancer Ther. 2011;10:437–47.
King TD, Song L, Jope RS. AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem Pharmacol. 2006;71:1637–47.
Tzatsos A, Tsichlis PN. Energy depletion inhibits phosphatidylinositol 3-kinase/Akt signaling and induces apoptosis via AMP-activated protein kinase-dependent phosphorylation of IRS-1 at Ser-794. J Biol Chem. 2007;282:18069–82.
Ning J, Clemmons DR. AMP-activated protein kinase inhibits IGF-I signaling and protein synthesis in vascular smooth muscle cells via stimulation of insulin receptor substrate 1 S794 and tuberous sclerosis 2 S1345 phosphorylation. Mol Endocrinol. 2010;24:1218–29.
Zakikhani M, Blouin M-J, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;123:271–9.
Bertrand L, Ginion A, Beauloye C, Hebert AD, Guigas B, Hue L, Vanoverschelde J-L. AMPK activation restores the stimulation of glucose uptake in an in vitro model of insulin-resistant cardiomyocytes via the activation of protein kinase B. Am J Phys Heart Circ Phys. 2006;291:H239–50.
Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE. 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem. 2001;276:46912–6.
Tao R, Gong J, Luo X, Zang M, Guo W, Wen R, Luo Z. AMPK exerts dual regulatory effects on the PI3K pathway. J Mol Signal. 2010;5:1.
Gual P, Le Marchand-Brustel Y, Tanti J-F. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109.
Leclerc GM, Leclerc GJ, Fu G, Barredo JC. AMPK-induced activation of Akt by AICAR is mediated by IGF-1R dependent and independent mechanisms in acute lymphoblastic leukemia. J Mol Signal. 2010;5:15.
Priebe A, Tan L, Wahl H, Kueck A, He G, Kwok R, Opipari A, Liu JR. Glucose deprivation activates AMPK and induces cell death through modulation of Akt in ovarian cancer cells. Gynecol Oncol. 2011;122:389–95.
He N, Kim N, Jeong E, Lu Y, Mills GB, Yoon S. Glucose starvation induces mutation and lineage-dependent adaptive responses in a large collection of cancer cell lines. Int J Oncol. 2016;48:67–72.
Kim Y-N, Koo KH, Sung JY, Yun U-J, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol. 2012;2012:306879.
Ng T, Leprivier G, Robertson M, Chow C, Martin M, Laderoute K, Davicioni E, Triche T, Sorensen P. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012;19:501–10.
Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806.
Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, Brugge JS. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–39.
Avivar-Valderas A, Bobrovnikova-Marjon E, Diehl JA, Bardeesy N, Debnath J, Aguirre-Ghiso JA. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene. 2013;32:4932–40.
Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:1.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3.
Di Francesco AM, Toesca A, Cenciarelli C, Giordano A, Gasbarrini A, Puglisi MA. Metabolic modification in gastrointestinal cancer stem cells: characteristics and therapeutic approaches. J Cell Physiol. 2016;231(10):2081–7.
Sanduja S, Feng Y, Mathis R, Sokol E, Reinhardt F, Halaban R, Gupta P. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene. 2016;35(40):5295–303.
Zhao C, Zhang Q, Yu T, Sun S, Wang W, Liu G. Hypoxia promotes drug resistance in osteosarcoma cells via activating AMP-activated protein kinase (AMPK) signaling. J Bone Oncol. 2016;5:22–9.
Harhaji‐Trajkovic L, Vilimanovich U, Kravic‐Stevovic T, Bumbasirevic V, Trajkovic V. AMPK‐mediated autophagy inhibits apoptosis in cisplatin‐treated tumour cells. J Cell Mol Med. 2009;13:3644–54.
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91.
Zhong D, Liu X, Schafer-Hales K, Marcus AI, Khuri FR, Sun S-Y, Zhou W. 2-Deoxyglucose induces Akt phosphorylation via a mechanism independent of LKB1/AMP-activated protein kinase signaling activation or glycolysis inhibition. Mol Cancer Ther. 2008;7:809–17.
Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, Baudy AR, Hatzivassiliou G, Evangelista M, Settleman J. mTORC1-dependent metabolic reprogramming underlies escape from glycolysis addiction in cancer cells. Cancer Cell. 2016;29:548–62.
Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30:671–8.
Bhardwaj R, Sharma PK, Jadon S, Varshney R. A combination of 2-deoxy-D-glucose and 6-aminonicotinamide induces cell cycle arrest and apoptosis selectively in irradiated human malignant cells. Tumor Biol. 2012;33:1021–30.
Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67:3364–70.
Bhaskar PT, Hay N. The two TORCs and AKT. Dev Cell. 2007;12:487–502.
Lo M, Ling V, Low C, Wang Y, Gout P. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol. 2010;17:9–16.
Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–8.
Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S. Systemic depletion of L-cyst (e) ine with cyst (e) inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23(1):120–127.
Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371:177–8.
Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyürek LM, Lindahl P, Nilsson J. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re308.
Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527(7577):186–91.
Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra215.
Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, Zheng Y, Liao X, Wang Y, Liao Q. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med. 2016;8:334ra351.
Acknowledgments
Not applicable.
Funding
This work is supported by the National Natural Science Foundation of China (81472317 to Min Shi), CSCO Merck Serono Oncology Research Fund (Y-MX2014-048 to Min Shi), and the Special Foundation for National Clinical Specialties of China (to The Department of Oncology, Nanfang Hosptial).
Availability of data and materials
Not applicable
Authors’ contributions
MS provided direction and guidance throughout the preparation of this manuscript. YZ and XH conducted the literature review and drafted the manuscript. WH and SD drafted and revised the figures in the article. YL, ZW, SZ, QH reviewed and made significant revisions on the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhao, Y., Hu, X., Liu, Y. et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer 16, 79 (2017). https://doi.org/10.1186/s12943-017-0648-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-017-0648-1