
Abstract
Background
Previous studies identified microRNAs (miRNAs) and messenger RNAs with significantly different expression between normal pancreas and pancreatic cancer (PDAC) tissues. Due to technological limitations of microarrays and real-time PCR systems these studies focused on a fixed set of targets. Expression of other RNA classes such as long intergenic non-coding RNAs or sno-derived RNAs has rarely been examined in pancreatic cancer. Here, we analysed the coding and non-coding transcriptome of six PDAC and five control tissues using next-generation sequencing.
Results
Besides the confirmation of several deregulated mRNAs and miRNAs, miRNAs without previous implication in PDAC were detected: miR-802, miR-2114 or miR-561. SnoRNA-derived RNAs (e.g. sno-HBII-296B) and piR-017061, a piwi-interacting RNA, were found to be differentially expressed between PDAC and control tissues. In silico target analysis of miR-802 revealed potential binding sites in the 3′ UTR of TCF4, encoding a transcription factor that controls Wnt signalling genes. Overexpression of miR-802 in MiaPaCa pancreatic cancer cells reduced TCF4 protein levels. Using Massive Analysis of cDNA Ends (MACE) we identified differential expression of 43 lincRNAs, long intergenic non-coding RNAs, e.g. LINC00261 and LINC00152 as well as several natural antisense transcripts like HNF1A-AS1 and AFAP1-AS1. Differential expression was confirmed by qPCR on the mRNA/miRNA/lincRNA level and by immunohistochemistry on the protein level.
Conclusions
Here, we report a novel lncRNA, sncRNA and mRNA signature of PDAC. In silico prediction of ncRNA targets allowed for assigning potential functions to differentially regulated RNAs.
Similar content being viewed by others

Background
A five year survival of around 4% makes pancreatic ductal adenocarcinoma (PDAC) the fourth leading cause of cancer-related deaths worldwide [1]. To better understand the aggressive growth and the poor response of PDAC to chemotherapeutic agents, studies are required that focus on molecular mechanisms underlying pancreatic cancer development and progression [2].
Somatic mutations, alterations in coding- and non-coding RNA expression as well as in the methylome of pancreatic cancer cells have been intensively studied [3]. The application of NGS technologies such as RNA-seq, whole exome sequencing or bisulfite sequencing to PDAC samples provided an unbiased view on genetic and epigenetic alterations [4-6]. However, studies investigating the small ncRNAome of PDAC tissues and healthy pancreas utilizing NGS methods are rare.
Studies utilizing microarrays revealed that deregulated microRNAs (miRNAs) have an impact on coding-gene expression in PDAC [7-12]. MiRNAs are a class of small non-coding RNAs (sncRNAs) that can repress gene expression [13]. Due to the known technical limitations of microarrays or real-time PCR most studies were focused on a fixed set of miRNA targets and many other sncRNA types have not been implicated in PDAC, but their deregulation and contribution to cancer progression has been described for other cancer types [14,15]. These sncRNAs include small nucleolar-derived RNAs (sno-derived RNAs, sdRNAs), functioning like miRNAs, or regulating splicing and translation [16], as well as piwi-interacting RNAs (piRNAs), that are associated with chromatin organization, messenger RNA stability and genome structure [17].
Similarly, only few studies have reported altered expression of long non-coding RNAs (lncRNAs) in PDAC [18]. LncRNAs represent a diverse class of modestly conserved, polyadenylated, non-protein-coding RNAs with essential roles in tumorigenesis [19]. LncRNAs comprise, among others, long intergenic non-coding RNAs (lincRNAs) and natural antisense transcripts (NATs). NATs, which are transcribed from the opposite (“anti-sense”) strand of a protein-coding gene can either stabilize or destabilize the expression of their sense partner [20], lincRNAs act as competitive endogenous RNAs (ceRNAs) and sequester miRNAs (“miRNA sponge”) [21], target chromatin modification complexes or RNA-binding proteins to alter gene expressing programs [22].
Here, we used high-throughput NGS-based technologies, namely Massive Analysis of cDNA Ends (MACE) and small RNA-sequencing (sRNA-seq), to characterize the complete coding- and non-coding transcriptomes of tissues from six PDAC patients and five normal controls. We merged the expression pattern obtained by the two techniques to gain insights into the altered miRNA regulation of coding gene expression in PDAC as compared to normal pancreatic tissues. Additionally, we provide evidence for differential expression of a piRNA and several sdRNAs, lincRNAs and NATs in PDAC.
Results
Differences in the coding transcriptome, lncRNAs and sncRNAs between pancreatic cancer tissue from six patients and normal pancreatic tissue from five controls were assessed by MACE and sRNA-seq. Sequencing results were experimentally validated by quantitative real time PCR (qPCR) for seven genes (CD1A, CTHRC1, FOXL1, GPR87, KLK7, TCF4, ZEB1), four miRNAs (miR-103a-3p, 135b-5p, 145-5p, 802) and two lincRNAs (LINC00152, LINC00261).
Overall, 396,542,460 sequences were generated. The amount of high-quality reads used for the analysis ranged from 4,150,706 reads in the MACE library for cancer patient P3 to 11,007,400 in the sRNA-seq normal pancreas library N2 (see Table 1). Robust expression was detected for 13,614 coding genes and 432 lncRNAs (Additional file 1: Table S1) whereas 1,961 mRNAs and 43 lncRNAs of these were significantly differentially expressed between cancer and control tissues (Additional file 2: Table S2). Unsupervised hierarchical clustering, principal component analysis (PCA) and Pearson’s moment product correlation coefficients (PCCs) revealed a clear separation between the groups based on the sequencing results (Figure 1A-C). Notably, 70% of all reads across control libraries mapped to 25 genes (PNLIPRP1, CELA3B, CPA2, CTRL, GP2, CPB1, CTRC, RBPJL, KLK1, PLA2G1B, CELA2A, CEL, GNMT, CELA3A, CPA1, PRSS3P2, PRSS3, CLPS, PNLIP, SLC39A5, SPINK1, CTRB1, TMED11P, PRSS1, GATM), encoding pancreatic acinar cell secretory- and related proteins (Table 2). In cancer libraries, only 3% of reads were annotated to these genes. Their homogenous, robust expression across all controls and strong downregulation in all PDACs underlines the existence of normal pancreas function in control libraries, which is lost in PDAC tissues. The significantly up-/downregulated lncRNAs, as determined by MACE, are listed in Table 3.

NGS MACE profiles discriminate PDAC from healthy control tissues. A) Unsupervised hierarchical cluster analysis of differentially expressed genes with euclidean distance measure clearly separates healthy controls (N) and diseased patients (P). (B) Principle component analysis (PCA) of all genes from the eleven samples under scrutiny. The first (x-axis) and second principal component (y-axis) account for 32% and 18%, respectively, of the total variation in the data. (C) Pearson product–moment correlation coefficient (PCC) for all samples compared within the control and patient group as well as between both groups.
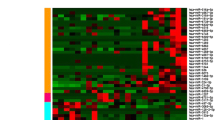
From 921 sncRNAs with robust expression (Additional file 3: Table S3), 123 were significantly differentially expressed between the two conditions (Additional file 4: Table S4). Similar to MACE, sRNA-seq allowed a clear separation of cancer and control tissues by PCA, PCCs, and unsupervised clustering (Additional file 5: Figure S1). The twenty most significantly up-/downregulated sncRNAs are listed in Table 4. Among all differentially regulated sncRNAs, we found 104 mature miRNAs, 18 sdRNAs and one piRNA. An overview of differential RNA expression across the genome is given in Figure 2.

Circos plot incorporating differential gene and miRNA expression. Chromosome numbers and bands are identified in the outer-most ring. Other tracks from outer to inner represent: Chromosomal positions of genes implicated in PDAC tumorigenesis; differential mRNA expression (FDR < 0.05) between controls and PDAC (Up in PDAC: red, down in PDAC: green); differential miRNA expression (FDR < 0.05) between control and PDAC tissues.
Enrichment analysis of protein-coding genes for identification of biological and functional differences
Gene ontology (GO)-enrichment analysis of downregulated genes in PDAC revealed a total of 39 significantly enriched GO terms (Additional file 6: Table S5) across all three GO categories. Most of these terms were linked to normal pancreas function, as e.g. “Digestion” (FDR = 1E-5), “Steroid metabolic process” (0.002) or “Triacylglycerol lipase activity” (0.014). In addition, other GO terms corresponding to translation of mRNAs into proteins, as e.g. “Translational elongation” (5.6E-15), “Cytosolic ribosome” (3.2E-8) or “Ribosomal subunit” (2E-5) were enriched, indicating a loss of normal pancreas function.
Upregulated genes in PDAC were enriched in 208 GO terms (Additional file 7: Table S6), including “Immune response” (1.2E-17), “Cell proliferation” (4.45E-5) and “Cell migration” (9E-5). The second set represents an enhanced proliferative potential, the third a high metastatic potential of the cancer cells. The most significantly enriched GO term was “Extracellular matrix” (2.2E-30), corresponding to genes involved in fibrogenesis, such as collagens and fibronectin, as well as TGFB and genes related to this pathway. Several categories were related to a sustained angiogenesis, like “Vasculature development” (2E-8) or “Blood vessel development” (8E-8). Taken together, GO-analysis confirms a loss of normal pancreatic function in the tumor tissues, in combination with increased proliferative potential, extracellular matrix (ECM) activation, blood vessel formation, metastatic spread and the potential to escape the immune system.
LncRNA expression in PDAC
Of 432 lncRNAs with robust expression, eleven were significantly up- and 32 downregulated in PDAC samples (Table 3). Other studies have already examined three lincRNAs (SNHG8, PVT1, H19), one NAT (HOTAIRM1) and two lncRNAs (MIAT, GAS5) which we identified as differentially expressed in PDAC [23]. Furthermore, the differential expression of two lincRNAs (LINC00261, LINC00152) [24] and two NATs (HNF1A-AS1, AFAP1-AS1) [25,26] were implicated in other cancer types.
SncRNA expression analysis
Of 921 measured sncRNAs, 45 (30 miRNAs, fourteen snoRNAs, one piRNA) were significantly downregulated in PDAC tissues. Previous microarray or qPCR studies reported downregulation for 25 of these (Table 4). The most significantly deregulated sncRNA without previous implication in pancreatic cancer was miR-802, which was highly expressed in normal pancreas but not in PDAC tissues (log2fc = 11, FDR = 9E-29). Beside sdRNAs (e.g. a 34 bp fragment from sno-HBII-296B) and miRNAs, piR-017061, a piRNA located within the HBII-296A snoRNA, was significantly downregulated in PDAC compared to normal pancreas tissues (log2fc = 2.3, FDR = 0.0001).
A total of 78 sncRNAs (74 miRNAs, 4 sdRNAs) showed significant upregulation in PDAC. Several of these were previously implicated in pancreatic cancer development (e.g. miR-21, 143, 222, 155, 10a, 874) others have not been shown to be upregulated in PDAC before (e.g. miR-31, 511, 2355). The expression of all differential miRNAs is visualized in Additional file 8: Figure S2.
In silico target analysis of miR-802
We used omiRas [27] to decipher potential interactions between miR-802 and genes significantly upregulated in PDAC, as detected by MACE. In total, 16 genes (AMPD3, CDH11, IGFBP5, ITPR3, HOXA5, MMD, PGM2L1, SLC4A7, ST8SIA4, TCF4, TMEM92, TRIB2, TSHZ3, RAI14, ZFHX4, ZNF521) were predicted to be upregulated due to loss of post-transcriptional silencing of miR-802 (for details see Additional file 9: Table S7). Enrichment analysis of miR-802 targets with starbase [28] revealed a significant enrichment of targets in Wnt signalling (p = 0.006), suggesting that loss of miR-802 might lead to increased Wnt activity in PDAC.
The TCF4 transcript (3.3-fold upregulated in PDAC, FDR = 0.001), encoding a transcription factor in the Wnt-signalling pathway, has the highest number of three mir-802 binding sites in its 3′ UTR (positions 3813, 4110, 5046, Figure 3A), and the interaction is predicted by all six interaction databases used for analysis. Co-expression analysis of all upregulated transcription factors (Additional file 10: Figure S3) revealed that the expression of TCF4 is significantly correlated with ZEB1 expression (PCC = 0.92, p = 5.2E-05). In addition, their expression is highly correlated with the expression of miR-21 (PCC = 0.88, p = 0.0003974) and inversely correlated with miR-802 expression (PCC = −0.83, p = 0.0015) (Figure 3B, C).

Co-expression of miR-802, ZEB1, TCF4 and miR-21. A) Alignment of miR-802 to the predicted binding sites in the 3′ UTR of TCF4. B) Co-expression analysis of significantly upregulated transcription factors that harbour predicted miRNA binding sites in their 3′ UTRs for one of the ten most significantly upregulated miRNAs (miRNAs that have no seed sequence for a TF UTR not shown). Significant (p < 0.01) correlations are indicated by a dot, positive correlations are marked in blue, negative correlations in red. The more significant the correlation, the larger the dot size. Sequence complementarity between an UTR and a miRNA is indicated by an “S”. C) Expression of TCF4, ZEB1 and miR-21 across all control (C) and PDAC (P) samples (significant positive correlation) as well as the expression of miR-802 (significantly inversely correlated). The normalized expression for each gene/miRNA is given in log2-scale for each sample.
Overexpression of miR-802 decreased TCF4 protein expression
Considering the absent expression of miR-802 in PDAC tissues and the in silico predicted binding sites of TCF4, we re-expressed miR-802 in MiaPaCa PDAC cells and assessed TCF4 expression. First, we induced miR-802 re-expression in MiaPaCa cells transfected with PCMV-MIR-802 (Figure 4a). Highly elevated levels of miR-802 were observed 24 h after transfection. To assess the effect of miR-802 on TCF4 protein levels, we harvested transfected MiaPaCa cells and analysed the proteins by western blot analysis (Figure 4b). Here, TCF4 protein levels decreased to 67% as compared with samples transfected with the negative control (Figure 4c).

Re-expression of miRNA induces downregulation of TCF4. A) RT-qPCR of miRNA isolated from MiaPaCa cells 24 h after transfection with PCMV-MIR-802 or PCMV-MIR-Control. miRNA-802 expression: fold-change with standard deviation (n = 3). B) TCF4 expression was analyzed 24 h after expression of PCMV-MIR-802 and PCMV-MIR-Control in MiaPaCa cells. Β-Actin was used as a loading control and for normalization (n = 3). C) LI-COR quantification of TCF4 protein levels in miRNA-treated cells. TCF4 levels were normalized to β-Actin (n = 3).
Validation of ZEB1 expression at the protein level
Since we hypothesize that miR-802 regulates ZEB1 expression via TCF4, we analysed expression levels of ZEB1 by immunohistochemistry in samples of human PDAC (n = 10) and normal pancreatic tissues (n = 10). In normal pancreatic tissue, ZEB1 was sparsely seen in periacinar cells (e.g. stellate cells). As depicted in Figure 5, we detected ZEB1 in PDAC samples in stromal cells within desmoplastic areas, but epithelial tumor cells did not express ZEB1. In accordance with previous observations [29] we detected ZEB1 in all analyzed pancreatic cancers only in the stromal compartment but not in epithelial cells. This observation emphasizes ZEB1 as a mesenchymal differentiation marker. We speculate that the newly identified miRNA802 might be involved in the regulation of ZEB1 and consequently might promote the mesenchymal character of pancreatic cancer.

Immunohistochemistry of ZEB1 protein expression. Immunohistochemical detection of ZEB1 in human pancreatic tissue samples. Representative images of ZEB1 expression: Upper panels, ZEB1 is expressed in periacinar cells in normal pancreatic tissue samples. Lower panels, detection of ZEB1 in stromal cells, but not in epithelial tumor cells in PDAC samples.
MiRNA-mRNA interaction network analysis of differentially expressed genes
To test whether miRNAs are involved in tumor-specific functional categories detected by GO-enrichment analysis, we exemplarily created a miRNA-mRNA interaction network for the term “Cell motion” (Figure 6). The network contains interactions between gene products of upregulated transcripts in PDAC, as well as downregulated miRNAs whose loss might cause the upregulation of their target genes, predicted by at least three independent miRNA-mRNA interaction tools.

Influence of miRNAs on metastatic potential. Interaction network between gene products of upregulated genes from GO category: “Cell motion” and downregulated miRNAs in PDAC. Receptors are indicated in green, transcription factors in orange, and other proteins in blue. Predicted miRNA binding of the mRNAs encoding the protein is encoded by a grey line, protein-protein interactions by bold blue lines.
Of 63 differentially regulated genes involved in cell motion, 43 form a highly connected protein-protein interaction network with 164 connections.
From this network, twelve genes (COL5A1, FN1, ITGA4, ITGB1, MET, NRP2, PDGFRB, ROBO1/SLIT, SEMA3A, SEMA3C, SPOCK1/SPARC, THBS1) are connected to the loss of seven miRNAs (miR-29c-3p, 30a, 130b-3p, 148-3p, 216, 217, 219-5p). Five genes (MET, NRP2, ROBO1/SLIT, SEMA3A, THBS1) are under potential post-transcriptional control of two or more of these miRNAs. This crosstalk predicts miR-130b to regulate the expression of five genes from the network (MET, SPOCK1/SPARC, THBS1, ITGA4, NRP2).
Confirmation of differentially expressed genes, lincRNAs and miRNAs by qPCR
To validate sequencing results obtained by sRNA-seq and MACE, the expression of seven candidate genes, upregulated in PDAC (TCF4, ZEB1, CD1A, FOXL1, GPR87, KLK7, CTHRC1), one down- and three upregulated miRNAs (miR-802, 135b-5p, 145-5p, 103a-3p) as well as two lincRNAs (LINC00152, LINC00261) was verified by qPCR. The expression of HPRT1 and miR-16 was used for normalization between both samples. The results of the qPCRs are given in Figure 7.

QPCR validation of sequencing results. The relative expression (Y-axis) for all candidate miRNAs/genes/lincRNAs (X-axis) is shown using boxplots for each condition. The bold black line represents the median expression across patients within a condition. *indictates a p-value < 0.05, **a p-value <0.01.
Except for miR-103a-3p, all tested RNAs were below a significance threshold of p < 0.05 (Wilcoxon’s rank sum test) when comparing expression of control and tumor samples. This is consistent with sequencing results, where similarly only miR-103a-3p did not reach the level of significance.
CD1A, FOXL1, GPR87 and KLK7, mRNA expression was undetectable by qPCR in normal pancreatic tissue. This is consistent with sequencing results, where no or at most two reads (normalized) were annotated to these transcripts. Similar to sequencing, all four genes were robustly expressed in PDAC tissues, with Ct values ranging from 24.8 to 31.1 and a median Ct of 27.3.
Consistent with MACE results, CTHRC1, TCF4 and ZEB1 were significantly higher expressed in PDAC compared to normal pancreas tissues, with median normalized Ct (ΔCt) values between −1.8 and 1.8 in cancer- and 3.1-5.1 in normal tissues. The expression differences (log2 ratio normal/tumor tissue) measured by qPCR (negative ΔΔCt) highly agree with the log2 fold-changes of the sequencing results (PCC = 0.86).
Similarly, downregulation of miR-802 was validated by qPCR, with a median negative normal/tumor ΔΔCt of 8.2 and p = 0 (Wilcoxon’s rank sum test), which is consistent with the log2fc and p-value estimated by sRNAseq (10.9, 0). Similar agreements between sequencing and qPCR results were obtained for the other three miRNAs.
Furthermore, the upregulation of LINC00152 (MACE log2fc: 2.3, qPCR: 1.5) and downregulation of LINC00261 (MACE log2fc: 5.3, qPCR: 4.4) in PDAC tissues was confirmed by qPCR.
External validation of MACE data by microarray
Badea and colleagues [30] investigated 36 whole tumor- and adjacent normal pancreatic tissue samples by coding-gene microarray analysis. We compared the 53 most significantly upregulated genes in tumors from their publication with the MACE sequencing results based on logarithmic fold-change (Additional file 11: Figure S4) and statistical significance (p-value). Only five of the genes (DCBLD1, PGM2L1, PDGFC, COX7A1, LY6E), were not significantly upregulated (p < 0.05) in the MACE data, whereas 48 genes showed a significant upregulation as detected by both methods. The PCC between log2fc of MACE and microarray data (0.61) indicates a strong correlation between the results and underlines the reliability of the approach.
Discussion
Our study investigated the coding- and non-coding transcriptomes of six PDAC patients and five healthy pancreatic control tissues. We detected 1,961 mRNAs, 43 lncRNAs and 123 sncRNAs as differentially expressed between the groups. Among these are several coding and non-coding RNAs without previous implication in pancreatic cancer development, most prominently miR-802 which is strongly downregulated in PDAC. Bioinformatic and functional analysis revealed post-transcriptional regulation of TCF4 protein levels by miR-802. Differential regulation of four miRNAS (miR-802, miR-135b-5p, miR-145-5p, 103a-3p), seven genes (CD1A, FOXL1, GPR87, KLK7, CTHRC1, TCF4, ZEB1) and two lincRNAs was confirmed by qPCR.
MiR-802 is the third most significantly repressed miRNA in PDAC, besides the tumor suppressor miRNAs miR-216 and miR-217 that - among others - target KRAS, PTEN, and SMAD7 [29]. MiR-802 is mainly expressed in pancreatic acinar cells [31], which may be the cells of origin for pancreatic preneoplastic lesions and pancreatic cancer [32]. A significant downregulation of miR-802 is observed in mice with ethanol-induced chronic pancreatitis, which predisposes to pancreatic cancer [33]. In contrast, sRNA-seq of pancreatic cyst fluids from low-grade benign and high-grade invasive lesions revealed thirteen enriched miRNAs, among these miR-216, miR-217, and miR-802, in the cyst fluids derived from invasive carcinomas [29]. The reason for the inverse correlation between the expression levels of these tumor suppressor miRNAs in body fluids and tumors currently remains unexplained.
Since no previous studies have reported downregulation of miR-802 in pancreatic cancer, validated targets are rare. Nevertheless, miR-802 targets were identified in two other cancer-types: osteosarcoma and lung cancer [34,35]. In contrast to PDAC, miR-802 is upregulated in both cancers. MiR-802 elevation promotes proliferation of lung carcinoma cell lines by targeting the tumor suppressor gene MEN1. Similarly, cell proliferation was promoted by miR-802 in osteosarcoma, where the gene encoding p27, a negative cell-cycle regulator, is a direct target. In hepatocellular carcinoma miR-802 is more than 100-fold downregulated, but no targets have yet been identified [36].
Bioinformatic in silico prediction points to Wnt signalling related transcription factor TCF4 mRNA (~12-fold upregulated in PDAC, FDR = 5E-7) as a direct target of miR-802. To validate the generated in silico predictions, we re-expressed miR-802 in the PDAC cell line MiaPaCa and analysed TCF4 protein expression. After re-expression of miR-802, we observed a 30% reduction of TCF4, indicating a direct impact of miR-802 on TCF4 regulation.
TCF4 activates miR-21 transcription by direct binding to its promoter in epithelial cancer [37]. Other oncomiRs with TCF4 binding sites proximal to their promoter include miR-10a, miR-424, miR-935 and miR615 [38].
Furthermore, the regulation of miR-181a/b expression has been associated with TCF4 expression in hepatocellular carcinoma [38]. Consistent with these observations, our study validates the upregulation (4.6-79-fold, FDR 0.002-4.9E-7) of these six miRNAs with important functions in PDAC development [39,40] that have TCF4 binding sites in their promoter.
Additionally, Sanchez-Tillo and colleagues showed that TCF4 induces ZEB1 (zinc finger E-box binding homeobox 1) transcription [41]. ZEB1 is an epithelial-to-mesenchymal transition (EMT)-activator that promotes PDAC tumorigenesis and metastasis [42]. ZEB1 is, concordant to TCF4, 3.5-fold upregulated in PDAC in this study (FDR = 0.03). Consistent with these data, we detected ZEB1 protein expression in the mesenchymal compartment of all analysed PDAC samples.
To test whether specific miRNAs contribute to the metastatic potential of pancreatic cancers, we used omiRas to predict interactions between downregulated miRNAs and upregulated genes from the GO term “Cell motion”. The miRNA with the highest number of targets was miR-130b. Previous studies confirmed downregulation of miR-130b in pancreatic cancer and functional tests revealed that overexpression of miR-130b remarkably inhibited the invasiveness of pancreatic cancer cells [43]. MiR-130b loss (8-fold downregulated in PDAC, FDR = 8.2E-7) might lead to the upregulation of metastasis associated key oncogenes MET, NRP2, ITGA4, THBS1 and SPOCK1 [44,45]. Recently published data describe the impact of miR-130b in metastasis formation [46] and therefore validates the approach of in silico prediction of miRNAs.
In contrast to miRNAs, lncRNAs have just recently moved into the focus of cancer research [47,48]. Nevertheless, studies examining the role of lncRNAs in specific oncogenic processes are limited to date.
Liu and colleagues reported an increased expression of the lncRNA MALAT1 in PDAC compared to adjacent normal pancreatic tissues [49]. Furthermore, they described a significant correlation between MALAT1 expression levels and tumor size, tumor stage, invasion, and disease-free survival [49]. In our analysis, however, MALAT1 upregulation in PDAC was not found. This is further supported by the pancreatic expression database (PED), where MALAT1 was also not reported as differentially expressed [50].
Similar to MALAT1, an increased expression of HOX antisense intergenic RNA HOTAIR has been found in pancreatic tumors [51]. According to the PED, no significant upregulation of HOTAIR is found in PDAC tissues, which is consistent with our results. Overexpression of PVT1 in the pancreatic cancer cell line ASPC-1 resulted in decreased gemcitabine sensitivity [52]. In this regard, we found an approximately 6-fold upregulation of lncRNA PVT1 in pancreatic cancer.
Besides PVT1 we provide evidence for a deregulation of other 42 lncRNAs in PDAC. Among these LINC00152 is, like in PDAC, overexpressed in gastric cancer (GCC), and its high expression correlates with increased invasion [53]. Xia and colleagues speculate about LINC00152 functioning as a competing endogenous RNA (ceRNA) that sequesters miR-18a-5p, 195-5p, 139-5p and miR-31-5p in GCC and thereby influences i.a. THBS1 expression [21]. We report LINC00152 overexpression in pancreatic cancer (5-fold, FDR = 7.1E-7), whereas all miRNAs with binding sites in the transcript are not significantly deregulated compared to control tissues. This indicates that increased LINC00152 expression might decrease the availability of particular miRNAs by competing for their binding and consequently lead to an upregulation of the miRNA target genes, such as THBS1 (5-fold upregulated in PDAC, FDR = 7.7E-6).
Overexpression of snoRNAs is a common feature in breast and prostate cancer [15], and contributes to tumorigenicity in vitro and in vivo. SnoRNA U50 is downregulated in prostate cancer and potentially functions as a tumor suppressor in other cancer types [54]. Several reports describe that snoRNAs are further processed to generate smaller fragments (sdRNAs) with miRNA-like functionality [55]. Currently, there is no evidence for snoRNAs/sdRNAs involved in pancreatic cancer development. To our best knowledge, this is the first report of differential regulation of snoRNAs/sdRNAs in PDAC. The most significantly regulated sdRNA (34 bps long) is from sno-HBII-296B (SNORD91B), which is approximately 5-fold downregulated in PDAC (FDR = 5.2E-5). However, its functional role and that of other differentially expressed snoRNAs/sdRNAs remains unclear; similarly, the functions of piRNAs has not been fully elucidated. PiRNAs are usually involved in germline development, silencing of transposons, and maintenance of DNA integrity [56]. Upregulated expression of piR-651 has been described in several cancer cell lines, where it promotes cell growth and might serve as a marker for cancer diagnosis [56]. Here we report the downregulation of piR-017061 in PDAC, a piRNA that is located within the sno-HBII-296A snoRNA.
Conclusion
This study underlines the role of miRNAs in PDAC and provides evidence for differentially regulated miRNAs that have not been previously implicated in PDAC. Additionally, we provide evidence that novel sncRNA classes, snoRNAs and piRNAs are differentially regulated between normal pancreas and PDAC tissues. Using a bioinformatics approach, we connect mRNA sequencing data with miRNA expression to assign potential functions to miR-802 and other miRNAs. Furthermore, we provide evidence for the differential expression of a variety of lncRNAs in pancreatic cancer.
Methods
Tissue samples were obtained from six patients with PDAC who underwent resection at the Department of Surgery, Technical University of Munich, Germany.
Normal pancreatic tissue samples from five patients without pancreatic ductal adenocarcinoma were used as controls.
Tissue collection was approved by the Ethics Committee of the Technical University of Munich and informed consent was obtained from all patients. Tissue were collected directly in the operating theatre and were immediately stored at −80°C until analysis.
Isolation of RNA
20 mg of frozen tissue were disrupted and homogenized (TissueLyser, Qiagen) and RNA was isolated (NucleoSpin miRNA Kit, Macherey-Nagel) in two fractions (small RNA < 200 nt and large RNA > 200 nt).
Preparation of small RNA libraries
For preparation of small RNA libraries, 5 μg RNA (small RNA fraction) was size-selected (<40 nt) by polyacrylamide gel electrophoresis (FlashPAGE, Life Technologies) and precipitated. About 30 ng small RNA (<40 nt) was successive ligated (T4 RNA Ligase 1 and T4 RNA Ligase 2, NEB) to modified 3′ and 5′ adapters (TrueQuant RNA adapters, GenXPro). Adapter-ligated RNA was reverse transcribed (SuperScript III, Life Technologies) and amplified by PCR (KAPA HiFi Hot-Start Polymerase, KAPA Biosystems). Amplified libraries were size-selected by polyacrylamide gel electrophoresis (PAGE) and sequenced (HiSeq2000, Illumina).
Preparation of massive analysis of cDNA ends (MACE) libraries
MACE libraries were prepared as described by Müller et al. [57]. Briefly, poly-adenylated RNA was extracted (Dynabeads mRNA Purification Kit, Life Technologies) from 5 μg RNA (large RNA fraction) and reverse transcribed (SuperScript Double-Stranded cDNA Synthesis Kit, Life Technologies) with biotinylated poly(dT) primers. cDNA was fragmented (Bioruptor, Diagenode) to an average size of 250 bp. Biotinylated ends were captured by streptavidin beads (Dynabeads M-270 Streptavidin Beads, Life Technologies) and ligated (T4 DNA Ligase 1, NEB) to modified adapters (TrueQuant DNA adapter, GenXPro). The libraries were amplified by PCR (KAPA HiFi Hot-Start Polymerase, KAPA Biosystems), purified by SPRI beads (Agencourt AMPure XP, Beckman Coulter) and sequenced (HiSeq2000, Illumina).
Cell culture and transfection
The pancreatic cancer cell line MiaPaCa was maintained at 37°C in a humified atmosphere of 5% CO2 and 95% air in Dulbecco’s modified Eagle Medium (Life Technologies, Inc, Darmstadt, Germany). The cells were transfected with Lipofectamine2000 (Life Technologies, Inc, Darmstadt, Germany) according to the manufacturer’s protocol with either PCMV-MIR-802 or PCMV-MIR-Control (OriGene, Rockville, USA).
RT-qPCR
MiRNA was extracted from MiaPaCa cells with the miRNeasy Mini Kit (Qiagen, Hilden, Germany). Reverse transcription was performed using the RevertAidH Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Braunschweig, Germany) using specific hsa-miR-802 and as control hsa-miR-16 primer. Amplification of cDNA was performed using the TaqMan Small RNA Assays Applied Biosystems (Life Technologies, Inc, Darmstadt, Germany). The primers for the cDNA synthesis and for the TaqMan analysis were included in the kit from TaqMan Small RNA Assays Applied Biosystems (Life Technologies, Inc, Darmstadt, Germany). For quantification of miRNA-802 expression the results were normalized against miR-16 expression.
Western blot
For quantification of protein levels after miRNA expression, transfected MiaPaCa cells were fractionated using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Braunschweig, Germany). 20 μg of protein from the nuclear fraction was loaded onto a 10% polyacrylamide gel and was then electrophoretically transferred to a nitrocellulose membrane. The membrane was blocked with Tween-20 (0.05%)-TBS (pH 7.4; 0.1 M Tris Base, 1.4 M NaCl) containing 5% milk, followed by incubation with respective primary antibody α-TCF4 (LS-Bio, Eching, Germany) at a concentration of 1:1000 or as a control α-β-Actin (Abcam, Cambridge, UK) with a concentration of 1:2000. Membranes were washed with Tween-20 (0.05%)-TBS and were incubated with a horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (1:5000). Signals were detected using the enhanced chemiluminescence system (ECL, Amersham Life Science Ltd., Bucks, UK). Films were scanned with a CanoScan 9900F scanner (Canon, Japan). Protein levels were quantified using the Odyssey software LI-COR and normalized to the β-Actin control.
Confirmation of MACE results by qRT-PCR of selected genes
LincRNA and mRNA expression analysis was carried out with the QuantiTect Multiplex PCR Kit (Qiagen) in combination with Superscript III reverse transcriptase (Life Technologies) and PrimeTime qPCR Assays (IDT). For miRNA detection, we used the miRCURY LNA Universal RT microRNA PCR system (Exiqon) according to the recommendations of the manufacturer. Reverse transcription and PCR-amplification for mRNA expression studies were performed with 50 ng of the large total RNA fraction. All quantitative real-time PCR reactions were carried out on the Lightcycler 480 II (Roche). For mRNAs/lincRNAs the expression of housekeeping gene HPRT1 was measured for data normalization, while miR-16 served as endogenous control for miRNA quantification. Differential expression between control and tumor tissues was assessed using the ∆∆Ct method, p-values were calculated with Wilcoxon’s rank sum test.
Immunohistochemistry analysis
Immunohistochemistry was performed using the Dako Envision System (Dako Cytomation GmbH, Hamburg, Germany). Consecutive paraffin-embedded tissue sections (3 μm thick) were deparaffinized and rehydrated using routine methods. Antigen retrieval was performed in citrate buffer (pH 6.0; 10 mM citric acid) in a microwave oven for 10 minutes. Endogenous peroxidase activity was quenched by incubation in TBS (pH 7.6; 0.2 M Tris Base; 1.4 M NaCl) containing 3% hydrogen peroxide at room temperature for 10 minutes. After permeabilization with 0.5% TritonX, nonspecific reactivity was blocked with TBS containing 5% BSA. Sections were incubated with the ZEB1 antibody (ZEB1: Atlas Antibodies #AMAb90510 (1:400)) at 4°C overnight followed by incubation with horseradish peroxidase-linked goat anti-mouse antibody, followed by a color-reaction with diaminebenzidine and counterstaining with Mayer’s hematoxylin.
Bioinformatics analysis of MACE data
To remove any PCR-bias, all duplicate reads detected by the TrueQuant technology were removed from the raw datasets. The remaining reads were additionally quality trimmed and the poly(A)-tail was clipped off. After raw data processing, reads were aligned to the human genome with novoalign (http://www.novocraft.com). Annotations for genomic mapping positions were derived by the RefSeq annotation track that includes coding genes as well as lncRNAs (http://genome.ucsc.edu/cgi-bin/hgTables) and only uniquely mapped reads were taken into account. Normalization and test for differential gene expression between normal and tumor tissue were calculated using the DESeq R/Bioconductor package [58]. To account for multiple testing, the false discovery rate (FDR) was estimated. Genes with FDR < 0.05 and |log2fc| > 1.6 were considered as differentially expressed.
Bioinformatics analysis of small RNA-seq data
The sRNA-seq data was quantified and tested for differential expression with omiRas [27]. Briefly, for each small RNA-seq library, data processing started with 3′ adapter clipping by a local alignment of the adapter sequence to each read. Subsequently, Illumina’s marked quality region was removed and the reads were summarized to UniTags. Singletons were removed from the data set and the remaining tags were mapped to the human genome (hg19) with bowtie. The mapped tags were annotated with the help of various models of coding and non-coding RNAs retrieved from the UCSC table browser. Tags mapping to exonic regions of coding genes were excluded from further analysis. Non-coding RNAs were quantified in each library. For tags mapping to multiple genomic loci the number of reads corresponding to the tag was divided by the number of mapping loci. Normalization and test for differential expression was performed in the same way as described for mRNAs.
Evaluation of normal pancreas function in control libraries
To underline the usability of apparently normal pancreas tissue samples to serve as healthy controls, all genes were sorted in ascending order according to their normalized mean expression in normal sequencing libraries. To determine genes encoding pancreas specific proteins, functional annotations of the fifty most highly expressed genes were extracted from genecards [59].
Enrichment analysis
Differentially expressed genes were uploaded to the Database for Annotation, Visualization and Integrated Discovery (DAVID) resource [60] using an enrichment cutoff of FDR < 0.05.
Co-expression analysis
A list of all human transcription factors (TFs) was received from AnimalTFDB [61]. Individual expression values of significantly upregulated (in PDAC) transcription factors were clustered using the k-means method with PCC as a distance measure and six initial clusters. The median expression of all transcription factors for each sample was taken as the representative expression for the cluster. To detect the influence of significant miRNA loss on transcription factor upregulation, PCCs were calculated for each TF/TF, miRNA/miRNA and miRNA/TF pair.
Network analysis
Interactions between miRNAs and transcripts with negatively correlated differential expression were detected with omiRas, using a minimum database overlap of three required interaction databases. Additionally, the STRING database v 9.0.5 [62] was used with standard parameters to detect interactions between gene products of differentially expressed genes.
Availability of supporting data
The data sets supporting the results of this article are available in the ArrayExpress expository, accession E-MTAB-3494.
The data sets supporting the results of this article are available in the GEO repository, [GEO: will be inserted after publication].
Abbreviations
- PDAC:
-
Pancreatic ductal adenocarcinoma
- MACE:
-
Massive Analyis of cDNA Ends
- sncRNA:
-
Small non-coding RNA
- miRNA:
-
microRNA
- sdRNA:
-
small nucleolar-derived RNA
- piRNA:
-
Piwi-interacting RNA
- lncRNA:
-
Long non-coding RNA
- lincRNA:
-
Long intergenic non-coding RNA
- NAT:
-
Natural antisense transcript
- ceRNA:
-
Competitive endogenous RNA
- sRNA-seq:
-
Small RNA-Sequencing
- qPCR:
-
Quantitative real time PCR
- PCA:
-
Principal component analysis
- PCC:
-
Pearson’s moment product correlation coefficient
- TCF4:
-
Transcription factor 4
- GO:
-
Gene ontology
- ZEB1:
-
Zinc finger E-box binding homeobox 1
- GEO:
-
Gene Expression Omnibus
- FDR:
-
False discovery rate
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- TF:
-
Transcription factor
References
Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–20. doi:10.1016/s0140-6736(10)62307-0.
Logsdon CD, Simeone DM, Binkley C, Arumugam T, Greenson JK, Giordano TJ, et al. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003;63(10):2649–57.
Costello E, Greenhalf W, Neoptolemos JP. New biomarkers and targets in pancreatic cancer and their application to treatment. Nat Rev Gastroenterol Hepatol. 2012;9(8):435–44. doi:10.1038/nrgastro.2012.119.
Rosty C, Ueki T, Argani P, Jansen M, Yeo CJ, Cameron JL. Overexpression of S100A4 in pancreatic ductal adenocarcinomas is associated with poor differentiation and DNA hypomethylation. Am J Pathol. 2002;160(1):45–50. doi:10.1016/s0002-9440(10)64347-7.
Wang L, Tsutsumi S, Kawaguchi T, Nagasaki K, Tatsuno K, Yamamoto S, et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012;22(2):208–19. doi:10.1101/gr.123109.111.
Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates Wnt signalling in metastasis. Nature. 2012;487(7408):510–3. doi:10.1038/nature11217.
Bauer AS, Keller A, Costello E, Greenhalf W, Bier M, Borries A, et al. Diagnosis of pancreatic ductal adenocarcinoma and chronic pancreatitis by measurement of microRNA abundance in blood and tissue. PLoS One. 2012;7(4):e34151. doi:10.1371/journal.pone.0034151.
Jamieson NB, Morran DC, Morton JP, Ali A, Dickson EJ, Carter CR, et al. MicroRNA molecular profiles associated with diagnosis, clinicopathologic criteria, and overall survival in patients with resectable pancreatic ductal adenocarcinoma. Clin Cancer Res. 2012;18(2):534–45. doi:10.1158/1078-0432.ccr-11-0679.
Munding JB, Adai AT, Maghnouj A, Urbanik A, Zollner H, Liffers ST, et al. Global microRNA expression profiling of microdissected tissues identifies miR-135b as a novel biomarker for pancreatic ductal adenocarcinoma. Int J Canc. 2012;131(2):E86–95. doi:10.1002/ijc.26466.
Piepoli A, Tavano F, Copetti M, Mazza T, Palumbo O, Panza A, et al. Mirna expression profiles identify drivers in colorectal and pancreatic cancers. PLoS One. 2012;7(3):e33663. doi:10.1371/journal.pone.0033663.
Szafranska AE, Davison TS, John J, Cannon T, Sipos B, Maghnouj A, et al. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26(30):4442–52. doi:10.1038/sj.onc.1210228.
Zhang Y, Li M, Wang H, Fisher WE, Lin PH, Yao Q, et al. Profiling of 95 microRNAs in pancreatic cancer cell lines and surgical specimens by real-time PCR analysis. World J Surg. 2009;33(4):698–709. doi:10.1007/s00268-008-9833-0.
Djuranovic S, Nahvi A, Green R. MiRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science (New York, NY). 2012;336(6078):237–40. doi:10.1126/science.1215691.
Mei Y, Clark D, Mao L. Novel dimensions of piRNAs in cancer. Cancer Lett. 2013;336(1):46–52. doi:10.1016/j.canlet.2013.04.008.
Su H, Xu T, Ganapathy S, Shadfan M, Long M, Huang TH, et al. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene. 2014;33(11):1348–58. doi:10.1038/onc.2013.89.
Martens-Uzunova ES, Olvedy M, Jenster G. Beyond microRNA–novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett. 2013;340(2):201–11. doi:10.1016/j.canlet.2012.11.058.
Cheng J, Deng H, Xiao B, Zhou H, Zhou F, Shen Z. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. 2012;315(1):12–7. doi:10.1016/j.canlet.2011.10.004.
Tahira AC, Kubrusly MS, Faria MF, Dazzani B, Fonseca RS, Maracaja-Coutinho V, et al. Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol Cancer. 2011;10:141. doi:10.1186/1476-4598-10-141.
Huarte M, Rinn JL. Large non-coding RNAs: missing links in cancer? Hum Mol Genet. 2010;19(R2):R152–61. doi:10.1093/hmg/ddq353.
Werner A. Biological functions of natural antisense transcripts. BMC Biol. 2013;11:31. doi:10.1186/1741-7007-11-31.
Xia T, Liao Q, Jiang X, Shao Y, Xiao B, Xi Y, et al. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci Rep. 2014;4:6088. doi:10.1038/srep06088.
Tsai MC, Spitale RC, Chang HY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res. 2011;71(1):3–7. doi:10.1158/0008-5472.can-10-2483.
Dayem Ullah AZ, Cutts RJ, Ghetia M, Gadaleta E, Hahn SA, Crnogorac-Jurcevic T, et al. The pancreatic expression database: recent extensions and updates. Nucleic Acids Res. 2014;42(Database issue):D944–9. doi:10.1093/nar/gkt959.
Cao WJ, Wu HL, He BS, Zhang YS, Zhang ZY. Analysis of long non-coding RNA expression profiles in gastric cancer. World J Gastroenterol. 2013;19(23):3658–64. doi:10.3748/wjg.v19.i23.3658.
Wu W, Bhagat TD, Yang X, Song JH, Cheng Y, Agarwal R, et al. Hypomethylation of noncoding DNA regions and overexpression of the long noncoding RNA, AFAP1-AS1, in Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterology. 2013;144(5):956–66. doi:10.1053/j.gastro.2013.01.019. 5.
Yang X, Song JH, Cheng Y, Wu W, Bhagat T, Yu Y, et al. Long non-coding RNA HNF1A-AS1 regulates proliferation and migration in oesophageal adenocarcinoma cells. Gut. 2014;63(6):881–90. doi:10.1136/gutjnl-2013-305266.
Muller S, Rycak L, Winter P, Kahl G, Koch I, Rotter B. OmiRas: a Web server for differential expression analysis of miRNAs derived from small RNA-Seq data. Bioinformatics (Oxford, England). 2013;29(20):2651–2. doi:10.1093/bioinformatics/btt457.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92–7. doi:10.1093/nar/gkt1248.
Wang J, Paris PL, Chen J, Ngo V, Yao H, Frazier ML. Next generation sequencing of pancreatic cyst fluid microRNAs from low grade-benign and high grade-invasive lesions. Cancer Lett. 2015;356(2 Pt B):404–9. doi:10.1016/j.canlet.2014.09.029.
Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato-Gastroenterology. 2008;55(88):2016–27.
Prevot PP, Augereau C, Simion A, Van den Steen G, Dauguet N, Lemaigre FP, et al. Let-7b and miR-495 stimulate differentiation and prevent metaplasia of pancreatic acinar cells by repressing HNF6. Gastroenterology. 2013;145(3):668–78. doi:10.1053/j.gastro.2013.05.016. 3.
Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(6):737–50. doi:10.1016/j.ccr.2012.10.025.
Charrier A, Chen R, Chen L, Kemper S, Hattori T, Takigawa M, et al. Connective tissue growth factor (CCN2) and microRNA-21 are components of a positive feedback loop in pancreatic stellate cells (PSC) during chronic pancreatitis and are exported in PSC-derived exosomes. J Cell Commun Signal. 2014;8(2):147–56. doi:10.1007/s12079-014-0220-3.
Cao ZQ, Shen Z, Huang WY. MicroRNA-802 promotes osteosarcoma cell proliferation by targeting p27. Asian Pac J Cancer Prev. 2013;14(12):7081–4.
Wang LQ, Chen G, Liu XY, Liu FY, Jiang SY, Wang Z. microRNA802 promotes lung carcinoma proliferation by targeting the tumor suppressor menin. Mol Med Rep. 2014;10(3):1537–42. doi:10.3892/mmr.2014.2361.
Wang C, Ren R, Hu H, Tan C, Han M, Wang X, et al. MiR-182 is up-regulated and targeting Cebpa in hepatocellular carcinoma. Chin J Cancer Res. 2014;26(1):17–29. doi:10.3978/j.issn.1000-9604.2014.01.01.
Lan F, Yue X, Han L, Shi Z, Yang Y, Pu P, et al. Genome-wide identification of TCF7L2/TCF4 target miRNAs reveals a role for miR-21 in Wnt-driven epithelial cancer. Int J Oncol. 2012;40(2):519–26. doi:10.3892/ijo.2011.1215.
Ji J, Yamashita T, Wang XW. Wnt/beta-catenin signaling activates microRNA-181 expression in hepatocellular carcinoma. Cell & Bioscience. 2011;1(1):4. doi:10.1186/2045-3701-1-4.
Liu J, Xu D, Wang Q, Zheng D, Jiang X, Xu L. LPS induced miR-181a promotes pancreatic cancer cell migration via targeting PTEN and MAP2K4. Dig Dis Sci. 2014;59(7):1452–60. doi:10.1007/s10620-014-3049-y.
Sicard F, Gayral M, Lulka H, Buscail L, Cordelier P. Targeting miR-21 for the therapy of pancreatic cancer. Mol Ther. 2013;21(5):986–94. doi:10.1038/mt.2013.35.
Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. Beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci U S A. 2011;108(48):19204–9. doi:10.1073/pnas.1108977108.
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–95. doi:10.1038/ncb1998.
Zhao G, Zhang JG, Shi Y, Qin Q, Liu Y, Wang B, et al. MiR-130b is a prognostic marker and inhibits cell proliferation and invasion in pancreatic cancer through targeting STAT3. PLoS One. 2013;8(9):e73803. 10.1371/journal.pone.0073803.
Cao Y, Hoeppner LH, Bach S, E G, Guo Y, Wang E. Neuropilin-2 promotes extravasation and metastasis by interacting with endothelial alpha5 integrin. Cancer Res. 2013;73(14):4579–90. doi:10.1158/0008-5472.can-13-0529.
Li C, Wu JJ, Hynes M, Dosch J, Sarkar B, Welling TH, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141(6):2218–27. doi:10.1053/j.gastro.2011.08.009. 6.
Chen Q, Zhao X, Zhang H, Yuan H, Zhu M, Sun Q, et al. MiR-130b suppresses prostate cancer metastasis through down-regulation of MMP2. Molecular carcinogenesis. 2014. doi:10.1002/mc.22204
Mitra SA, Mitra AP, Triche TJ. A central role for long non-coding RNA in cancer. Front Genet. 2012;3:17. doi:10.3389/fgene.2012.00017.
Serviss JT, Johnsson P, Grander D. An emerging role for long non-coding RNAs in cancer metastasis. Front Genet. 2014;5:234. doi:10.3389/fgene.2014.00234.
Liu JH, Chen G, Dang YW, Li CJ, Luo DZ. Expression and prognostic significance of lncRNA MALAT1 in pancreatic cancer tissues. Asian Pac J Cancer Prev. 2014;15(7):2971–7.
Chelala C, Hahn SA, Whiteman HJ, Barry S, Hariharan D, Radon TP, et al. Pancreatic Expression database: a generic model for the organization, integration and mining of complex cancer datasets. BMC Genomics. 2007;8:439. doi:10.1186/1471-2164-8-439.
Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R, et al. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene. 2013;32(13):1616–25. doi:10.1038/onc.2012.193.
You L, Chang D, Du HZ, Zhao YP. Genome-wide screen identifies PVT1 as a regulator of Gemcitabine sensitivity in human pancreatic cancer cells. Biochem Biophys Res Commun. 2011;407(1):1–6. doi:10.1016/j.bbrc.2011.02.027.
Pang Q, Ge J, Shao Y, Sun W, Song H, Xia T, et al. Increased expression of long intergenic non-coding RNA LINC00152 in gastric cancer and its clinical significance. Tumour Biol. 2014;35(6):5441–7. doi:10.1007/s13277-014-1709-3.
Dong XY, Rodriguez C, Guo P, Sun X, Talbot JT, Zhou W, et al. SnoRNA U50 is a candidate tumor-suppressor gene at 6q14.3 with a mutation associated with clinically significant prostate cancer. Hum Mol Genet. 2008;17(7):1031–42. doi:10.1093/hmg/ddm375.
Brameier M, Herwig A, Reinhardt R, Walter L, Gruber J. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res. 2011;39(2):675–86. doi:10.1093/nar/gkq776.
Cheng J, Guo JM, Xiao BX, Miao Y, Jiang Z, Zhou H. PiRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin Chim Acta. 2011;412(17–18):1621–5. doi:10.1016/j.cca.2011.05.015.
Müller S, Rycak L, Afonso-Grunz F, Winter P, Zawada AM, Damrath E, et al. APADB: a database for alternative polyadenylation and microRNA regulation events. Database. 2014; bau076.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi:10.1186/gb-2010-11-10-r106.
Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: a novel functional genomics compendium with automated data mining and query reformulation support. Bioinformatics (Oxford, England). 1998;14(8):656–64.
Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(Web Server issue):W169–75. doi:10.1093/nar/gkm415.
Zhang HM, Chen H, Liu W, Liu H, Gong J, Wang H, et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res. 2012;40(Database issue):D144–9. doi:10.1093/nar/gkr965.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–8. doi:10.1093/nar/gkq973.
Acknowledgements
We thank Nadja Maeritz and Nadine Wagner for excellent technical assistance. This work was supported by the Bundesministerium für Bildung und Forschung (BMBF; FKZ0316043).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SM wrote the manuscript, analysed the NGS expression data, created visualizations. SR carried out the immunoassay, did the overexpression experiments, coordinated the study and drafted the manuscript. PB analysed the data. CJ was responsible for administrative and material support. AP prepared the small RNA libraries. KH coordinated the study and supervised the MACE and sRNA-seq experiments. RT performed mRNA qPCR analysis. FAG performed miRNA/lincRNA qPCR analysis. AMS analysed and interpreted the data. BK analysed and interpreted the data. IR analysed and interpreted the data. WKR supervised the experiments. BR and PW supervised the experiments and revised the manuscript. GK and IK revised the manuscript. FT revised the manuscript. JK interpreted the data, revised the manuscript, and supervised the study. CWM designed the study, coordinated the study and drafted the manuscript. All authors read and approved the final manuscript.
Jörg Kleeff, Peter Winter and Christoph W Michalski contributed equally to this work.
An erratum to this article is available at http://dx.doi.org/10.1186/s12943-015-0401-6.
Additional files
Additional file 1: Table S1.
Expression of all mRNAs and lncRNAs detected with MACE.
Additional file 2: Table S2.
Differentially expressed mRNAs and lncRNAs between controls and PDAC tissues.
Additional file 3: Table S3.
Expression of all sncRNAs detected with sRNA-seq.
Additional file 4: Table S4.
Differentially expressed sncRNAs between control and PDAC tissues.
Additional file 5: Figure S1.
NGS sncRNA profiles discriminate PDAC from healthy control tissues. Unsupervised hierarchical cluster analysis with Euclidean distance measure of differentially expressed sncRNAs clearly separates healthy controls and diseased patients. (B) Principle component analysis (PCA) of all sncRNAs from the eleven examined samples. (C) Pearson product–moment correlation coefficient (PCC) for all samples compared within the control and patient group as well as between both groups.
Additional file 6: Table S5.
Enriched GO terms of significantly downregulated genes in PDAC.
Additional file 7: Table S6.
Enriched GO terms of significantly upregulated genes in PDAC.
Additional file 8: Figure S2.
Boxplot of differential miRNAs for control and PDAC libraries. For each differentially expressed miRNA (FDR < 0.05) the normalized expression in log2 scale for each group is visualized as a boxplot.
Additional file 9: Table S7
Predicted, significantly upregulated targets of downregulated miR-802.
Additional file 10: Figure S3
Transcription factor co-expression analysis. The normalized expression of differentially upregulated transcription factors is given for each library in each of the eight clusters determined by k-means clustering with PCC as a distance measure. The median expression in each cluster is indicated by a bold black line, the expression for the transcription factors in the cluster is indicated by different colours.
Additional file 11: Figure S4
Comparison of MACE and Microarray data. For 53 genes (colored dots) the log2 fold-change between control and PDAC tissues as determined by microarray (X-axis) and NGS-based MACE (Y-axis) is given. The blue line represents the linear regression line.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Müller, S., Raulefs, S., Bruns, P. et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol Cancer 14, 94 (2015). https://doi.org/10.1186/s12943-015-0358-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-015-0358-5