
Abstract
Metastasis is a crucial hallmark of cancer progression, which involves numerous factors including the degradation of the extracellular matrix (ECM), the epithelial-to-mesenchymal transition (EMT), tumor angiogenesis, the development of an inflammatory tumor microenvironment, and defects in programmed cell death. Programmed cell death, such as apoptosis, autophagy, and necroptosis, plays crucial roles in metastatic processes. Malignant tumor cells must overcome these various forms of cell death to metastasize. This review summarizes the recent advances in the understanding of the mechanisms by which key regulators of apoptosis, autophagy, and necroptosis participate in cancer metastasis and discusses the crosstalk between apoptosis, autophagy, and necroptosis involved in the regulation of cancer metastasis.
Similar content being viewed by others
Introduction
Metastasis is a key step of cancer progression that indicates a more advanced stage and a poorer prognosis. Multiple cellular processes, including the degradation of the extracellular matrix (ECM), the epithelial-to-mesenchymal transition (EMT), tumor angiogenesis, the development of an inflammatory tumor microenvironment, and the dysfunction of programmed cell death machinery, have been demonstrated to be essential for cancer metastasis [1]. Any mistakes made by a metastatic cell during these cellular events may lead to cell death. Therefore, the regulation of cell death is critical for cancer cells to survive during metastasis.
Programmed cell death is defined as regulated cell death mediated by an intracellular program. Apoptosis was originally thought to be the only form of programmed cell death. However, in the last decade, programmed cell death has expanded to include autophagy and a form of necrosis termed necroptosis (programmed necrosis). Programmed cell death, especially apoptosis and necroptosis, are natural barriers that restrict malignant cells from surviving and disseminating. However, cancer cells evolve various strategies to evade programmed cell death by generating genetic mutations or epigenetic modifications in the key modulators of programmed cell death pathways.
In this review, we summarize the interplay (or the link) of the different form of program cell death with cancer metastasis, and we anticipate future challenges and unsolved questions related to these topics.
Review
An introduction to cancer metastasis
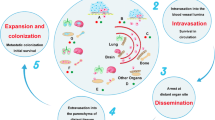
Cancer metastasis is a complex process that can be divided into five major steps: the first step, invasion, is characterized by increased cell motility caused by alterations in cell-cell and cell-ECM interactions [2]. The second step is intravasation, in which tumor cells escape from the primary site and migrate into circulation systems. The third step, dissemination, is the process in which malignant cells travel through the circulation systems to reach a capillary bed, where the cancer cells adhere to the vessel walls or are detained at these sites because of size constraints. The fourth step is extravasation, in which cancer cells permeate the vessels to enter their destination organs. Colonization is the final step, in which metastatic cells proliferate and form micrometastases or macrometastases [2]. Alternatively, metastasis can be considered as a two-phase process according to a new perspective [3]: the first phase involves the physical translocation of a cancer cell to a distant organ, whereas the second phase encompasses the process of the development of the cancer cells into a metastatic lesion at the distant site. Typically, the initial steps of metastasis (invasion, intravasation, dissemination, and extravasation) proceed at a very high efficiency, but the final step, colonization, is less efficient. It has been estimated that only ~0.01% of circulating tumor cells ultimately produce macrometastases [4]. This inefficiency may be closely related to the activation of cell death machinery by various stresses before or after the cells reach a new environment. Such stresses include the loss of cell-cell contacts, the recognition and destruction of the cancer cells by the immune system, and the lack of necessary growth factors, all of which may trigger programmed cell death, including apoptosis, autophagy and necroptosis [4].
Apoptosis and cancer metastasis
Apoptosis is a type of programmed cell death that is characterized by cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation [5,6]. There are two basic apoptotic signaling pathways: the extrinsic and the intrinsic pathways [7]. The intrinsic apoptotic pathway is activated by various intracellular stimuli, including DNA damage, growth factor deprivation, and oxidative stress. It relies on the formation of a complex termed the apoptosome, composed of procaspase-9, apoptotic protease-activating factor (Apaf-1), and cytochrome c. A series of Bcl-2 family members, such as Bax, Bak, Bcl-2, and Bcl-xL, control the release of cytochrome c by regulating mitochondrial membrane permeabilization. The extrinsic pathway of apoptosis is initiated by the binding of death ligands [e.g., Fas ligand (FasL), TNF-related apoptosis inducing ligand (TRAIL), and TNF-α] to death receptors of the TNF receptor superfamily. This interaction is followed by the assembly of the death-inducing signaling complex (DISC), which consists of the Fas-associated death domain (FADD) protein and procaspase-8/10. DISC then either activates downstream effector caspases (caspase-3, 6 and 7) to directly induce cell death or cleaves the Bcl-2 family member Bid into tBid to activate the mitochondria-mediated intrinsic apoptotic pathway [7]. Numerous factors, such as p53, cellular inhibitor of apoptosis proteins (cIAPs), and NF-κB, have been reported to be involved in the regulation of apoptotic pathways [2,8]. Many small molecules targeting apoptotic pathways have been developed for cancer therapy. For example, ABT-737, ABT-263, and GX15-070 have been reported to act on Bcl-2 family members; GDC-0152, birinapant, AT-406, and HGS-1029 have been designed to antagonize cIAPs, among the most promising targets for anti-cancer agent development; and Nutlins, MI-219 and MI-77301 have been shown to antagonize murine double-minute 2 (MDM2), a critical negative regulator of p53 that promotes p53 ubiquitination and degradation [9,10].
Apoptosis may block metastatic dissemination by killing misplaced cells. Thus, apoptosis serves as an important process for inhibiting metastasis. The success of the metastatic process relies on the ability of malignant cells to escape apoptosis. Apoptotic resistance is indispensable for all steps of metastatic progression, but the most critical step may be the resistance to cell death induced by the loss of cell-cell and cell-ECM contacts [8]. The detachment of cells from the ECM induces a type of apoptosis termed anoikis. Numerous reports have demonstrated that anoikis resistance is frequently observed in metastatic cells [11-14]. For instance, TrkB, a neurotrophic tyrosine kinase receptor, was found to act as a specific suppressor of caspase-associated anoikis in non-malignant epithelial cells. TrkB activates the phosphatidylinositol-3-OH kinase (PI3K)/protein kinase B (PKB) pathway to promote the formation of large cellular aggregates that survive and proliferate in suspension, and these cellular aggregates develop into rapidly growing tumors that infiltrate lymphatics and blood vessels to colonize a distant organ in mice [13]. In addition, signal transducer and activator of transcription 3 (STAT3) was found to play a role in conferring anoikis resistance to pancreatic cancer cells and in promoting metastasis. Enhanced STAT3 expression and phosphorylation at Tyr 705 have been associated with the anoikis resistance and the metastatic capacity of pancreatic cells [14].
In addition, metastatic cells must develop a mechanism to evade cell death resulting from recognition and destruction by cytotoxic lymphocytes such as natural killer (NK) cells. Furthermore, tumor cells must survive in the environment of reactive oxygen species (ROS) produced by endothelial cells when crossing the vessel or tissue barrier during the extravasation step [15,16]. Finally, malignant cells must tolerate hypoxic conditions and proliferate in an environment lacking the necessary cytokines for growth to achieve successful colonization at their destination sites [8,17].
Even after the successful formation of micrometastases, macroscopic tumors may not develop because of dormancy [18]. The nature of dormancy remains to be elucidated. One hypothesis states that the rate of proliferation is balanced by the rate of apoptosis such that no net tumor growth occurs [19], whereas another hypothesis states that dormant cells neither proliferate nor undergo apoptosis [20]. However, genetic variation and/or environmental stimulation may drive cells out of dormancy and into an anti-apoptotic, highly proliferative state [21].
In the Table 1, we summarize the roles of the known major apoptotic factors that participate in cancer metastasis.
Among these modulators of apoptosis in Table 1, the c-Jun N-terminal kinases (JNK), TGF-β, and matrix metalloproteinase (MMP) pathways play dual roles in apoptosis and metastasis. JNKs, a subgroup of the MAP kinase superfamily, are indispensable for both cell proliferation and apoptosis. Whether the activation of JNKs leads to cell proliferation or apoptosis is dependent on the cell type, the nature of the death stimulus, the duration of its activation and the activities of other signaling pathways [66]. In the absence of NF-κB activation, enhanced JNK activation contributes to TNF-α induced apoptosis. JNK is also a critical mediator of UV radiation-induced apoptosis. JNK promotes apoptosis via different mechanisms. Activated JNK translocates to the nucleus and transactivates c-Jun and other transcription factors (e.g., p53), which further transactivate various pro-apoptotic genes, such as Fas-L, Bak, and p53-upregulated modulator of apoptosis (PUMA) [67]. In addition, JNKs contribute to apoptosis by modulating the activities of mitochondrial pro- and antiapoptotic proteins via distinct phosphorylation events. However, JNK inhibits apoptosis in IL-3-dependent hematopoietic cells via the phosphorylation and antagonism of the proapoptotic Bcl-2 family protein BAD [66].
It was reported that JNK signaling prevented the progression of invasive adenocarcinoma in PTEN−/− prostate cancer. Mice exhibiting JNK deficiency in the prostate epithelium (JNK and PTEN double-deficient mice) develop androgen-independent metastatic prostate cancer more rapidly than control (PTEN-deficient) mice. In addition, JNK-deficient progenitor cells exhibited increased proliferation and tumorigenic capacity compared with progenitor cells from control prostate tumors [45].
A group reported that Notch and myocyte enhancer factor 2 (Mef2) cooperated to promote proliferation and metastasis via JNK signal activation and the consequent induction of the invasion marker MMP1 in a Drosophila model [46]. Another study showed that receptor for advanced glycation end products (RAGE) splice variant 1 inhibited tumor formation, cell invasion, and angiogenesis induced by RAGE ligand signaling, which was closely related to the strong suppression of JNK by this splice variant protein [47].
The transforming growth factor-β (TGF-β) family proteins bind to cell surface type I and type II serine/threonine kinase receptors (TGFβRI and TGFβRII, respectively) and SMAD mediators to regulate many biological processes. Numerous studies have reported roles of the TGF-β pathway in apoptosis. Many pro-apoptotic genes are under the control of the SMAD transcription-regulating complexes. For example, TGF-β-inducible early response gene (TIEG1) [56], death-associated protein kinase (DAPK) [57], and SH2 domain-containing inositol-5-phosphatase (SHIP) [58] have been shown to be essential for the suppression of cell proliferation and the induction of apoptosis in many cell types. Another mechanism of TGF-β-related apoptosis is the induction of the mislocalization of a mitochondrial septin family member ARTS. Upon translocation, ARTS binds to and inactivates XIAP, a key inhibitor of apoptosis, which leads to the activation of caspase-3 and apoptosis [59]. In addition, TGF-β signals induce the death associated protein (Daxx)-JNK pathway to induce apoptosis under certain circumstances [60].
The roles of the TGF-β signaling pathway in cancer development are uncertain [61]. Prior to tumor initiation and during the early stage of progression, TGF-β acts as a tumor suppressor via cell cycle arrest and the induction of apoptosis; however, at advanced stages, TGF-β often acts as a tumor promoter via the acceleration of the EMT, invasion, and angiogenesis, the maintenance of tumor stem cells, and the alteration of the tumor microenvironment [61]. Interestingly, TGF-β is expressed at high levels during the late stages of tumor progression in many human cancers, but paradoxically, the TGF-β pathway is frequently mutationally inactivated in cancer cells. Further studies revealed that elevated TGF-β expression played an important role in promoting stromal cells to secret cytokines that favor cancer cell metastasis [68].
Matrix metalloproteinases (MMPs) constitute one of the most prominent families of proteinases associated with tumor metastasis. MMPs interfere with the induction of apoptosis in malignant cells via the cleavage of ligands or receptors in apoptotic pathways [62]. For instance, MMP-7 was reported to cleave membrane-bound FasL on doxorubicin-treated cancer cells, thereby attenuating apoptosis and increasing the resistance of these cells to chemotherapy [63,64]. MMP-13 was shown to be involved in the shedding of nerve/glial antigen 2 (NG2), a novel anoikis receptor, thereby contributing to the attenuation of anoikis [65].
Autophagy and cancer metastasis
Autophagy is an evolutionarily conserved catabolic process in which intracellular membrane structures package protein complexes and organelles to degrade and renew these cytoplasmic components. It is thus critical for cell growth regulation and internal homeostasis [69]. Autophagy is physiologically a cellular strategy and mechanism for survival under stress conditions. When over-activated under certain circumstances, excess autophagy results in cell death. To date, three types of autophagy have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy. We only discuss macroautophagy in this review; therefore, henceforth, "autophagy" specifically refers to macroautophagy. Autophagy is a multi-step process that includes nucleation, elongation, and autophagosome and autolysosome formation and that is executed by a series of highly conserved genes termed autophagy-related genes (ATGs) [70]. Autophagy is often triggered by nutrient deprivation, ROS, hypoxia, drug stimuli, and endoplasmic reticulum (ER) stress via complex signal transduction pathways. Alterations in the autophagy machinery may lead to diverse pathological conditions, such as neurodegeneration, ageing, and cancer [71]. Mammalian target of rapamycin complex 1 (mTORC1), class I PI3K, AKT, class III PI3K, Beclin-1 and p53 are critical components of the autophagic pathway that have become major targets of autophagy-related drug design. Numerous small molecules have been found to target these components and to play a role in tumor treatment. For example, rapamycin and its derivatives (i.e., rottlerin, PP242 and AZD8055) target the PI3K/AKT/mTOR signaling pathway to induce autophagy; spautin-1 and tamoxifen regulate Beclin-1 activity to inhibit and promote autophagy, respectively; and oridonin and metformin trigger p53-mediated autophagy and cell death [72].
The role of autophagy in cancer metastasis is complex, as reports have indicated both pro-metastatic and anti-metastatic roles of autophagy. Stage-specificity may affect the cellular response to autophagy during cancer metastasis [73]. During the early stage of cancer metastasis, autophagy may act as a suppressor of metastasis by restricting tumor necrosis and inflammatory cell infiltration and by alleviating oncogene-induced senescence. These processes may help to reduce the invasion and dissemination of cancer cells from the primary site. During the advanced stages of metastasis, autophagy tends to act as a promoter of metastasis by promoting ECM-detached metastatic cell survival and colonization in a distant site and by inducing metastatic cells that fail to establish contact with the ECM in the new environment to enter dormancy.
The anti-metastatic role of autophagy
Necrosis frequently occurs inside a tumor due to hypoxia and metabolic stress, which enables inflammatory cells, especially macrophages, to infiltrate tumor sites and which generates a favorable microenvironment for tumor metastasis [74,75]. Autophagy facilitates the survival of tumor cells under metabolic stress and hypoxic conditions, thereby effectively reducing tumor necrosis and subsequent immune cell infiltration and metastasis [76]. In addition, autophagy regulates the selective release of the immune modulator high-mobility group B1 (HMGB1) by the tumor cells that are destined to die [77,78]. Once released, HMGB1 activates dendritic cells by engaging Toll-like receptor 4, which triggers an intense antitumor immune response and restricts metastasis [79,80]. Prophylactic treatment with the TLR4 and TLR9 agonist complex triggered anti-metastatic immunity and impaired tumor metastasis by inducing the autophagy-associated death of melanoma cells via IFN-γ/STAT1 activation. The induction of autophagy via the injection of rapamycin with or without the TLR4/9 agonist complex into the tumor attenuated metastasis [81].
ATG5, a key regulator of autophagy, was found to be downregulated in primary melanomas compared to benign nevi, and this decrease in ATG5 expression is accompanied by a reduction in the expression of LC3 and in basal autophagy. It was shown that patients expressing low levels of ATG5 in their tumors exhibited decreased progression-free survival according to a follow-up of 158 primary melanoma patients. Mechanically, reducing ATG5 expression may promote cell proliferation by preventing oncogene-induced senescence and may contribute to the progression of early-stage cutaneous melanoma [82].
The PI3K/Akt/mTOR pathway is a critical signaling pathway that negatively regulates autophagy and that promotes cancer progression. Recent studies have suggested that PI3K/Akt/mTOR signaling is upregulated in 30-50% of prostate cancers, often due to the loss of PTEN. It has been reported that molecular changes in the PI3K/Akt/mTOR signaling pathway are implicated in the elevation of the tumor stage and grade and the risk of recurrence [83]. PTEN mutations and deletion within primary tumors have been associated with an increased risk of metastasis, and early targeting of PTEN may prevent metastasis [84]. Genistein, an Akt inhibitor, has been shown to play a role in decreasing the incidence of lung metastasis in an orthotopic prostate model using PC-3 cells [85]. Mechanically, increasing autophagic flux by inhibiting the PI3K/Akt/mTOR pathway may promote the apoptosis of cancer cells [86,87].
In addition, a unique death modality termed autophagic cell death plays a role in impeding cancer metastasis. Autophagic cell death refers to cell death caused by autophagy rather than cell death with autophagy. Thus, the ultimate cell death process of autophagic cell death is executed by over-activated autophagic flux rather than apoptosis or necroptosis. The genetic or drug-based inhibitors of autophagy, but not apoptosis or necroptosis inhibitors, rescue this type of cell death [88,89]. It is known that there is a complex relationship between apoptosis and autophagy. Typically, autophagy antagonizes apoptosis, and as a feedback response, apoptosis-related caspase activation reduces the autophagic process. However, autophagy can also trigger apoptosis under certain circumstances via the activation of caspase-8 and the depletion of endogenous apoptosis inhibitors [90-92]. Autophagic cell death does not include autophagy-induced apoptosis or necroptosis. Several studies have suggested a potential relationship between autophagic cell death and cancer metastasis. For example, one study showed that blocking the CXCR4/mTOR signalling pathway induced autophagic cell death and the anti-metastatic properties of peritoneally disseminated gastric cancer cells [93].
The pro-metastatic role of autophagy
Acquiring the ability to survive and proliferate in the absence of the ECM while disseminating through the circulation systems and colonizing a distant site is necessary for cancer cell metastasis [94,95]. Otherwise, cancer cells die of anoikis (a specific type of apoptosis induced by the loss of ECM attachment). The constitutive activation of pro-survival signals such as PI3K, Ras–ERK, NF-κB, and Rho GTPase often occurs in cancer cells, thereby antagonizing anoikis. This antagonism can be achieved via the autocrine secretion of growth factors or the overexpression of receptor tyrosine kinases [13,96]. Accumulating evidence suggests that autophagy also provides a mechanism for matrix-detached pre-metastatic tumor cells to avoid anoikis [97,98]. In a hepatocellular carcinoma (HCC) lung metastasis model, the inhibition of autophagy (via the lentivirus-mediated silencing of BECN1 and ATG5) markedly decreased the pulmonary metastasis of HCC cells. Further investigation indicated that the inhibition of autophagy did not affect cell invasiveness, migration or the EMT but attenuated the anoikis resistance and lung colonization of HCC cells [99]. Another study showed that autophagy was induced by either matrix detachment or β1 integrin inhibition [100]. Autophagy in ECM-disrupted cells may compensate for the loss of extrinsic signals that promote and maintain nutrient and energy metabolism. Under these conditions, autophagy might delay the onset of apoptosis, providing cells with additional time to re-attach to an appropriate ECM. In a rapidly growing tumor with high energy and biosynthesis requirements, detachment-induced autophagy undoubtedly increases the survival of the cells deprived of ECM contact [96,97].
Aside from ECM disruption, increased metabolic and oxidative stresses in cancer cells and adverse environmental stresses are major barriers to the metastasis of cancer cells. Autophagy-defective KRAS-driven lung cancer cells exhibited impaired mitochondrial energy homoeostasis, oxidative stress and a constitutively active DNA damage response that were further mediated by p53 and that triggered apoptosis in malignant cells, suggesting that autophagy may play an important role in the maintenance of mitochondrial function and in the clearance of unfavorable factors the induce cell death, thus promoting tumor progression [101,102]. Autophagy may also promote the survival of HCC under hypoxic conditions via the activation of mitochondrial β-oxidation and intracellular ATP production [103].
Disseminated tumor cells that are unable to form firm ECM contacts in a foreign microenvironment may transform to enter dormancy [104], which may allow the tumor cells to survive for years or decades at distant sites without developing into secondary tumors while retaining the ability to metastasize under the appropriate conditions. Lu et al. reported that the tumor suppressor aplasia Ras homolog member I (ARHI) induced autophagy and enhanced the survival of dormant tumor cells in vivo, demonstrating an association between autophagy and the regulation of cancer cell dormancy for the first time [105]. Therefore, it is possible that the partially disseminated tumor cells that cannot successfully establish an interaction with the ECM may initiate autophagy, which drives the tumor cells into dormancy and promotes their survival.
Cancer stem cells (CSCs) are characteristically resistant to conventional anticancer therapy, which may contribute to treatment failure and tumor relapse. CSCs exhibit the potential to regenerate for an indefinite period, which may promote tumor metastasis [106]. Recently, autophagy has been shown to be a critical factor for CSC survival and drug resistance [107,108]. Malignant breast tissue contains a rare population of multi-potent cells exhibiting the capacity to self-renew; these cells are defined as CSCs. These mammary CSCs can propagate in culture as floating spherical colonies termed ‘mammospheres’. The key autophagy protein Beclin 1 was more strongly expressed in mammospheres established from human breast cancer samples or cell lines than in the parental adherent cells, resulting in a higher level of autophagy in mammospheres. This prosurvival autophagic flux was important for CSC maintenance and tumor progression [108]. In addition, one group reported that HIF-1a and autophagy played a role in modulating the conversion of non-stem pancreatic cancer cells to stem cells. This result suggested a role of HIF-1a and autophagy in sustaining the dynamic equilibrium between CSCs and non-CSCs [109]. The relationships between autophagy and cancer metastasis are depicted in Figure 1.

Contradictory effects of autophagy on cancer metastasis. The text in the wathet boxes summarizes the possible pro-metastatic mechanisms of autophagy, and the text in the yellow boxes depicts the potential anti-metastatic mechanisms of autophagy. HMGB1, high-mobility group B1; TLR4, Toll-like receptor 4; CSCs, cancer stem cells.
Necroptosis and metastasis
Necrosis was originally considered to be an accidental and unregulated cell death. Accumulating evidence has shown that necrosis can be induced and proceed in a regular manner like apoptosis, although in a caspase-independent fashion. Regulated necrosis is termed “programmed necrosis” or “necroptosis” to distinguish it from necrosis caused by physical trauma [110]. Necroptosis can be induced by the activation of the TNF receptor superfamily [111], T cell receptors [112], interferon receptors [113], Toll-like receptors (TLRs) [114], cellular metabolic and genotoxic stresses, or various anti-cancer agents. It can be pharmacologically inhibited by chemical compounds such as necrostatin-1 (Nec-1) [115]. The formation of the “necrosome” by receptor-interacting protein kinase 1 (RIP1) and RIP3 is one of the most critical characteristics of necroptosis. It is a multi-step process that contains three key checkpoints. For example, in TNF-α mediated necroptosis [110], at the first checkpoint, the E3 ligases cellular inhibitor of apoptosis 1 (cIAP1) and cIAP2 induce RIP1 ubiquitination [116], which blocks necroptosis via NF-κB-dependent or -independent mechanisms. The removal of ubiquitin chains from RIP1 by the deubiquitinase cylindromatosis (CYLD) is critical for the packaging of Complex IIa (including caspase-8, FADD, and RIP1) and Complex IIb [(including caspase-8, FADD, RIP1, RIP3, and mixed lineage kinase domain-like (MLKL)] [117]. At the second checkpoint, activated caspase-8 cleaves and abolishes the activities of RIP1, RIP3, and CYLD [118-120]. Cleaved RIP1 and RIP3 lose their capabilities of trans-phosphorylation and downstream substrate phosphorylation. At the third checkpoint, when the disruption of RIP1 and RIP3 is prevented by caspase-8 inhibitors (i.e., zVAD) or by the genetic inhibition of caspase-8 or FADD, the trans-phosphorylation of RIP1 and RIP3 promotes their aggregation into the filamentous-like necrosome [110]. MLKL is further phosphorylated by RIP3 and is recruited to the necrosome by its interaction with RIP3 [121]. MLKL forms a homotrimer via its amino-terminal coiled-coil domain and translocates to the plasma membrane during TNF-induced necroptosis, which leads to necrotic plasma membrane permeabilization [122]. In addition to MLKL, phosphoglycerate mutase 5 (PGAM5) is a downstream substrate of RIP3 [123].
Several molecules and pathways contribute to the execution of TNF receptor-mediated necroptosis. Some of these effectors are also involved in other receptor-mediated necroptosis pathways [124]. During the progression of necroptosis, ROS are generated [125,126], resulting in lipid peroxidation and increased mitochondrial membrane permeability. The cytosolic ATP levels are sharply reduced due to the attenuation of ATP transport from the mitochondria to the cytosol and ATP consumption by over-active poly(ADP-ribose) polymerase 1 (PARP1). Apoptosis-inducing factor (AIF) is released from mitochondria due to the increase in the mitochondrial membrane permeability and enters the nucleus to cleave DNA. Lysosomal membrane permeabilization (LMP) also occurs during necroptosis, resulting in the leakage of cytotoxic hydrolases into the cytosol [124]. In addition, dynamin-related protein l (Drp1), which acts downstream of PGAM5, is thought to regulate mitochondrial fission to execute necroptosis [123].
Necroptosis plays an indispensable role during normal development. Moreover, it has been implicated in the pathogenesis of a variety of human diseases, including cancer [127]. The necroptosis machinery is often impaired during tumorigenesis and tumor progression. For example, chronic lymphocytic leukemia (CLL) cells failed to undergo necroptosis upon stimulation using TNFα combined with the pan-caspase inhibitor zVAD. Two key components of necroptotic machinery, RIP3 and CYLD, were markedly downregulated in CLL [128]. In non-Hodgkin lymphoma, single nucleotide polymorphisms (SNPs) in the RIP3 gene were detected in 458 patients and correlated with increased risk of non-Hodgkin lymphoma, which indicates that genetic variations in the RIP3 gene may contribute to the onset of this disease [129]. There is a growing list of compounds and anticancer agents that have been shown to induce necroptosis in cancer cells. Shikonin was the first reported small molecule that induced necroptosis [130]. Numerous studies have shown that shikonin and its analogs not only are highly tumoricidal but also exhibit the ability to bypass drug resistance machineries [130-133]. In addition, compounds such as 5-benzylglycinyl-amiloride, obatoclax, and D-galactose employ necroptosis to kill malignant cells [134-136]. Notably, some traditionally pro-apoptotic anti-cancer agents have recently been demonstrated to share the ability to induce the necroptosis of tumor cells under certain circumstances. For example, interferon-β-armed oncolytic adenovirus (ZD55-IFN-β) induced both apoptosis and necroptosis in cancer cells. However, Nec-1 treatment converted ZD55-IFN-β-induced necroptosis to apoptosis [137]. In addition, although TRAIL is a well-known apoptosis inducer, one study showed that acidic extracellular pH converted TRAIL-induced apoptosis to necroptosis in human HT29 colon and HepG2 liver cancer cells via a process that involved RIPK1/RIPK3-dependent PARP-1 activation [138].
To date, few studies have associated necroptosis with metastasis. Fu et al. reported that shikonin greatly reduced the lung metastasis of osteosarcoma by inducing RIP1- and RIP3-dependent necroptosis [132]. The induction of a high level of ROS via necroptosis may represent one factor that restricts cancer cell metastasis [139]. As we have described above, metastatic cells in the circulation or at new sites must survive in an environment without interacting with the ECM; under these conditions, tumor cells face major difficulties in maintaining nutrient and energy equilibration and in antagonizing metabolic stresses, especially ROS. Disseminated tumor cells have evolved different strategies to restore their ATP levels and to restrict cellular ROS production, including the over-activation of pro-survival signals (such as PI3K, Ras-ERK, and NF-κB), the enhancement of antioxidant activity, the altered activation of metabolic pathways (preferentially the glycolysis and pentose phosphate pathways), and the initiation of autophagy. However, necroptosis represents another mechanism to eliminate metastatic cancer cells by triggering ROS bursts. RIP3 has been observed to be critical for regulating ROS production during necroptosis [126], and this finding is in agreement with another study showing that RIP3 activates several metabolic enzymes [including glycogen phosphorylase (PYGL), glutamate-ammonia ligase (GLUL), and glutamate dehydrogenase 1 (GLUD1)] to regulate TNF-induced ROS production. Silencing each of these enzymes results in decreased levels of ROS accumulation and cell death [125]. Therefore, it appears to be reasonable that necroptosis is an important mechanism that restricts tumor metastasis. In this case, tumor cells must overcome both anoikis and necroptosis to successfully metastasize.
Interaction between apoptosis, autophagy, necroptosis, and cancer metastasis
Programmed cell death in vivo involves the complex interaction between apoptosis, autophagy, and necroptosis [140]. In some cases, a specific stimulus triggers only one type of programmed cell death, but in other situations, the same stimulus may initiate multiple cell death processes. Different types of mechanisms may co-exist and interact with each other within a cell, but ultimately, one mechanism dominates the others. The decision taken by a cell to undergo apoptosis, autophagy, or necroptosis is regulated by various factors, including the energy/ATP levels, the extent of damage or stress, and the presence of inhibitors of specific pathways (e.g., caspase inhibitors). ATP depletion activates autophagy. However, if autophagy fails to maintain the energy levels, necroptosis occurs [141]. Slight/moderate damage and low levels of death signaling typically induce apoptosis, whereas severe damage and high levels of the death signaling often result in necroptosis [142]. Although apoptosis is often the first mode of cell death and although necroptosis is triggered only as a backup mechanism to ensure that cell death occurs, emerging evidence has shown that the necroptotic pathway may predominate under certain pathological conditions [142]. The complex relationships between different types of cell death and cancer metastasis are depicted in Figure 2.

Interaction between different types of programmed cell death and cancer metastasis. Disseminating metastatic cells must face many unfavorable conditions, including detachment from the ECM, attack by immune cells, hypoxia or a growth factor-lacking environment, that cause increased cellular ROS production and DNA damage and insufficient energy status. Low levels of death signals stimulate apoptosis, whereas high levels of death signals often result in necroptosis. Due to the activity of the apoptosis (anoikis) and necroptosis machineries, most metastatic cells from the primary tumor cannot successfully macrometastasize. Compared with apoptosis and necroptosis, autophagy appears to be fairly capricious, as on one hand, autophagy greatly improves the fitness of metastatic cells under stressful conditions to counteract apoptosis and necroptosis, but on the other hand, autophagy reduces metastasis by restricting tumor necrosis and by precluding inflammatory immune cell infiltration. Additionally, excess autophagy induces the death of metastasizing cells.
In the course of cancer metastasis, malignant cells must overcome a series of unfavorable conditions, including detachment from the ECM, attack by immune cells, hypoxia and a growth factor-lacking environment, which cause increased cellular ROS production and DNA damage and an insufficient energy status. Therefore, most metastatic cells from the primary tumor are unable to successfully macrometastasize and are killed via apoptosis or necroptosis. On one hand, autophagy greatly improves the fitness of cancer cells under stressful conditions and, thus, attenuates apoptosis and necroptosis, but on the other hand, autophagy antagonizes metastasis by restricting tumor necrosis and subsequent immune cell infiltration. Additionally, excess autophagy induces the death of metastasizing cells. Therefore, the interaction between different types of cell death and cancer metastasis is highly complex. In addition, cancer cells have evolved sophisticated mechanisms to antagonize apoptosis and necroptosis. However, defects in the machinery of one type of cell death may not affect that of another. Thus, triggering a single type of programmed cell death may not be sufficient for the treatment of cancer metastasis. The selection of different cell death inducers or the combined use of different cell death pathway inducers will help to overcome drug resistance to kill metastatic cells [140,143,144].
Conclusions and perspectives
Programmed cell death, are natural barriers that restrict malignant cells from surviving and disseminating. However, cancer cells evolve various strategies to evade programmed cell death by generating genetic mutations or epigenetic modifications in the key modulators of programmed cell death pathways. The role of autophagy in cancer metastasis is complex, as reports have indicated both pro-metastatic and anti-metastatic roles of autophagy. Stage-specificity may affect the cellular response to autophagy during cancer metastasis. Programmed cell death in vivo involves the complex interaction between apoptosis, autophagy, and necroptosis. Different types of mechanisms may co-exist and interact with each other within a cell. The decision taken by a cell to undergo apoptosis, autophagy, or necroptosis is regulated by various factors, including the energy/ATP levels, the extent of damage or stress, and the presence of inhibitors of specific pathways.
In this review, we summarized how apoptosis, autophagy, and necroptosis affect cancer metastasis based on the current literature. However, still many issues remain to be clarified.
(i) As we have discussed in this review, autophagy plays a dual role in tumor metastasis. Generally speaking, autophagy may exert an inhibitory effect during the early step of cancer metastasis by, for example, restricting necrosis and inflammation and by preventing oncogene-induced senescence, thereby limiting the invasion and dissemination of cancer cells from the primary site. Alternatively, autophagy tends to promote metastasis during advanced cancer stages by supporting ECM-detached metastatic cell survival and colonization at a distant site and by inducing metastatic cells to enter dormancy if they fail to establish a contact with the ECM in the new environment. As a delicate process, autophagy may play either pro- or anti-metastatic roles depending on the context. At present, the complex role of autophagy in metastasis remains unclear. It is necessary to determine how the dual role of autophagy in metastasis is regulated; i.e., what are the signals, molecules, and mechanisms that enable autophagy to play a dominant anti-metastatic role in one situation an opposite role in another situation.
(ii) At present, we know very little about the roles of necroptosis in cancer progression. As we have mentioned above, the necroptosis machinery may be impaired during tumorigenesis and tumor progression. For example, CLL leukemia cells failed to undergo necroptosis due to the downregulation of RIP3 and CYLD [128]. In non-Hodgkin lymphoma, genetic variations in the RIP3 gene were detected in 458 patients and correlated with increased risk of non-Hodgkin lymphoma [129]. However, according to our study (unpublished) using a series of cancer cell lines, only a small proportion (approximately 10%) of cancer cells undergo necroptosis in response to stimuli. It is necessary to determine why so many cancer cells lose their necroptotic machinery and the importance of this machinery in regulating tumorigenesis and cancer metastasis.
(iii) It is necessary to investigate the relationship between programmed cell death and cancer metastasis in the tumor microenvironment, including the tumor-infiltrating immune cells. For example, preclinical evidence has shown that chemotherapy-induced autophagy in cancer cells may contribute to the recruitment of myeloid cells to the tumors and the subsequent T lymphocyte-mediated suppression of tumor growth [145]. In addition, programmed cell death plays critical roles in the maintenance of proper innate and adaptive immune function [146]. Therefore, dysfunction of the programmed cell death machinery in the immune system may change its effect on cancer growth and metastasis. This evidence suggests that programmed cell death should be investigated simultaneously in both cancer cells and immune cells to understand the interaction between these cell types.
(iv) The regulation of programmed cell death and metastasis by non-coding RNAs (ncRNAs) is a novel direction of this research field. ncRNAs include multiple classes of RNA transcripts that are not translated into proteins but can regulate the transcription, stability or translation of protein-coding genes in the mammalian genome [147]. The most studied ncRNAs are microRNAs (typically consisting of 19–24 nucleotides), which are highly conserved small ncRNA molecules that function to regulate a wide variety of cellular processes by interfering with protein expression or mRNA degradation. Numerous miRNAs have been reported to be involved in the regulation of programmed cell death or cancer progression [148,149]. More recently, long none-coding RNAs (lncRNAs) have been found to play critical roles in transcriptional and translational regulation [150] and have been implicated in a wide range of human diseases, including cancer [151]. Some lncRNAs have been observed to target the apoptotic and autophagy pathways and to play a role in cancer development. For example, MEG3 inhibited the proliferation and induced the apoptosis of NSCLC cells by affecting p53 expression [152], and GAS5 regulated the apoptosis of NSCLC cells [153]. In addition, HULC was overexpressed in human gastric cancer (GC) cell lines and tissues compared with normal controls, and this overexpression correlated with lymph node metastasis, distant metastasis and advanced tumor node metastasis stage. Further investigation showed that HULC-induced autophagy was a major reason for GC cell survival and metastasis [154]. At present, the regulation of programmed cell death and cancer metastasis by ncRNAs, especially lncRNAs, remains largely unknown, and further investigation is required to clarify their mechanisms.
The more we understand the specific roles, mechanisms, and regulators of apoptosis, autophagy, and necroptosis and their interaction with cancer metastasis, the better therapeutic strategies can be developed for cancer treatment.
Abbreviations
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-to-mesenchymal transition
- Apaf-1:
-
Apoptotic protease-activating factor
- FasL:
-
Fas ligand
- TRAIL:
-
TNF-related apoptosis inducing ligand
- DISC:
-
Death-inducing signaling complex
- FADD:
-
Fas-associated death domain
- cIAPs:
-
Cellular inhibitor of apoptosis proteins
- MDM2:
-
Murine double-minute 2
- PI3K:
-
Phosphatidylinositol-3-OH kinase
- PKB:
-
Protein kinase B
- STAT3:
-
Signal transducer and activator of transcription 3
- NK:
-
Natural killer
- ROS:
-
Reactive oxygen species
- NSCLC:
-
Non-small-cell lung cancer
- XIAP:
-
X-linked inhibitor of apoptosis
- DAPK:
-
Death-associated protein kinase
- FADD:
-
Fas-associated death domain-containing protein
- sFas:
-
soluble Fas
- DcR3:
-
Decoy receptor 3
- DcR:
-
Decoy receptor
- OPG:
-
Osteoprotegerin
- DR4:
-
Death receptor 4
- TβR I/II:
-
TGF-β receptor I/II
- MMPs:
-
Matrix metalloproteinases
- JNK:
-
c-Jun N-terminal kinases
- PUMA:
-
p53-upregulated modulator of apoptosis
- Mef2:
-
Myocyte enhancer factor 2
- RAGE:
-
Receptor for advanced glycation end products
- TGF-β:
-
Transforming growth factor-β
- TIEG1:
-
TGF-β-inducible early response gene
- SHIP:
-
SH2 domain-containing inositol-5-phosphatase
- Daxx:
-
Death associated protein
- NG2:
-
Nerve/glial antigen 2
- ATGs:
-
Autophagy-related genes
- ER:
-
Endoplasmic reticulum
- mTORC1:
-
Mammalian target of rapamycin complex 1
- HMGB1:
-
High-mobility group B1
- HCC:
-
Hepatocellular carcinoma
- ARHI:
-
Aplasia Ras homolog member I
- CSCs:
-
Cancer stem cells
- TLRs:
-
Toll-like receptors
- Nec-1:
-
Necrostatin-1
- RIP1:
-
Receptor-interacting protein kinase 1
- CYLD:
-
Deubiquitinase cylindromatosis
- MLKL:
-
Mixed lineage kinase domain-like
- PGAM5:
-
Phosphoglycerate mutase 5
- PARP1:
-
Poly(ADP-ribose) polymerase 1
- LMP:
-
Lysosomal membrane permeabilization
- Drp1:
-
Dynamin-related protein l
- AIF:
-
Apoptosis-inducing factor
- CLL:
-
Chronic lymphocytic leukemia
- SNPs:
-
Single nucleotide polymorphisms
- PYGL:
-
Glycogen phosphorylase
- GLUL:
-
Glutamate-ammonia ligase
- GLUD1:
-
Glutamate dehydrogenase 1
- GC:
-
Gastric cancer
- ncRNAs:
-
Non-coding RNAs
- lncRNAs:
-
Long none-coding RNAs
References
Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–69.
Horak CE, Bronder JL, Bouadis A, Steeg PS. Metastasis-The Evasion of Apoptosis. Apoptosis, Cell Signaling, and Human Diseases: Molecular Mechanisms. 2007;1:63–96.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64.
Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–73.
Nagasaka A, Kawane K, Yoshida H, Nagata S. Apaf-1-independent programmed cell death in mouse development. Cell Death Differ. 2010;17:931–41.
Burgess DJ. Apoptosis: Refined and lethal. Nat Rev Canc. 2013;13:79–9.
Verbrugge I, Johnstone RW, Smyth MJ. SnapShot: extrinsic apoptosis pathways. Cell. 2010;143:1192-2. e1192.
Zörnig M, Hueber A-O, Baum W, Evan G. Apoptosis regulators and their role in tumorigenesis. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2001;1551:F1–37.
Wong R. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87.
Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med. 2014;65:139–55.
Kim Y, Koo K, Sung J, Yun U, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. International Journal of Cell Biology.2012; 2012. doi:10.1155/2012/306879
Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett. 2008;272:177–85.
Douma S, van Laar T, Zevenhoven J, Meuwissen R, van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–9.
Fofaria NM, Srivastava SK. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis. 2015;36:142–50.
Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–57.
Townson JL, Naumov GN, Chambers AF. The role of apoptosis in tumor progression and metastasis. Curr Mol Med. 2003;3:631–42.
Azab F, De La Puente P, Vij R, Azab AK. Tumor Hypoxia Promotes Dissemination and Tumor Colonization In Waldenström Macroglobulinemia. Blood. 2013;122:3011–1.
Hedley BD, Chambers AF. Tumor dormancy and metastasis. Adv Canc Res. 2009;102:67–101.
Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–53.
Naumov GN, MacDonald IC, Chambers AF, Groom AC. Solitary cancer cells as a possible source of tumour dormancy? Seminars in Cancer Biology. 2001. p. 271–6.
Folkman J. Dormancy. Encyclopedia of Cancer. 2009. p. 901–4.
Teitz T, Inoue M, Valentine MB, Zhu K, Rehg JE, Zhao W, et al. Th-MYCN mice with caspase-8 deficiency develop advanced neuroblastoma with bone marrow metastasis. Cancer Res. 2013;73:4086–97.
Shin MS, Kim HS, Lee SH, Lee JW, Song YH, Kim YS, et al. Alterations of Fas-pathway genes associated with nodal metastasis in non-small cell lung cancer. Oncogene. 2002;21:4129–36.
Koomägi R, Volm M. Relationship between the expression of caspase-3 and the clinical outcome of patients with non-small cell lung cancer. Anticancer Res. 1999;20:493–6.
Isobe N, Onodera H, Mori A, Shimada Y, Yang W, Yasuda S, et al. Caspase-3 expression in human gastric carcinoma and its clinical significance. Oncology. 2004;66:201–9.
Berezovskaya O, Schimmer AD, Glinskii AB, Pinilla C, Hoffman RM, Reed JC, et al. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005;65:2378–86.
Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64.
Chen H-Y, Lee Y-R, Chen R-H. The functions and regulations of DAPK in cancer metastasis. Apoptosis. 2014;19:364–70.
Umetani N, Fujimoto A, Takeuchi H, Shinozaki M, Bilchik AJ, Hoon DS. Allelic imbalance of APAF-1 locus at 12q23 is related to progression of colorectal carcinoma. Oncogene. 2004;23:8292–300.
Pinkas J, Martin SS, Leder P. Bcl-2-mediated cell survival promotes metastasis of EpH4 βMEKDD mammary epithelial cells. Mol Canc Res. 2004;2:551–6.
Sun T, Sun B, Zhao X, Zhao N, Dong X, Che N, et al. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl‐2 and Twist1: A study of hepatocellular carcinoma. Hepatology. 2011;54:1690–706.
Martin SS, Ridgeway AG, Pinkas J, Lu Y, Reginato MJ, Koh EY, et al. A cytoskeleton-based functional genetic screen identifies Bcl-xL as an enhancer of metastasis, but not primary tumor growth. Oncogene. 2004;23:4641–5.
Jansson A, Sun X-F. Bax expression decreases significantly from primary tumor to metastasis in colorectal cancer. J Clin Oncol. 2002;20:811–6.
Lee G, Yan C, Shin S, Hong S, Ahn T, Moon A, et al. BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene. 2010;29:2130–41.
Stark AM, Schem C, Maass N, Hugo H, Jonat W, Mehdorn HM, et al. Expression of metastasis suppressor gene maspin is reduced in breast cancer brain metastases and correlates with the estrogen receptor status. Neurol Res. 2010;32:303–8.
Yuan K, Xie K, Fox J, Zeng H, Gao H, Huang C, et al. Decreased levels of miR-224 and the passenger strand of miR-221 increase MBD2, suppressing maspin and promoting colorectal tumor growth and metastasis in mice. Gastroenterology. 2013;145:853–64. e859.
Pattje W, Melchers L, Slagter‐Menkema L, Mastik M, Schrijvers M, Gibcus J, et al. FADD expression is associated with regional and distant metastasis in squamous cell carcinoma of the head and neck. Histopathology. 2013;63:263–70.
Owen-Schaub LB, van Golen KL, Hill LL, Price JE. Fas and Fas ligand interactions suppress melanoma lung metastasis. J Exp Med. 1998;188:1717–23.
Huang G, Nishimoto K, Zhou Z, Hughes D, Kleinerman ES. miR-20a encoded by the miR-17–92 cluster increases the metastatic potential of osteosarcoma cells by regulating Fas expression. Cancer Res. 2012;72:908–16.
Wu Y, Han B, Sheng H, Lin M, Moore PA, Zhang J, et al. Clinical significance of detecting elevated serum DcR3/TR6/M68 in malignant tumor patients. Int J Canc. 2003;105:724–32.
Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. 2001;7:94–100.
Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010;29:4752–65.
Grosse-Wilde A, Voloshanenko O, Bailey SL, Longton GM, Schaefer U, Csernok AI, et al. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J Clin Invest. 2008;118:100–10.
Powell E, Piwnica-Worms D, Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov. 2014;4:405–14.
Hübner A, Mulholland DJ, Standen CL, Karasarides M, Cavanagh-Kyros J, Barrett T, et al. JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proc Natl Acad Sci. 2012;109:12046–51.
Pallavi S, Ho DM, Hicks C, Miele L, Artavanis‐Tsakonas S. Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in Drosophila. EMBO J. 2012;31:2895–907.
Kalea AZ, See F, Harja E, Arriero M, Schmidt AM, Hudson BI. Alternatively spliced RAGEv1 inhibits tumorigenesis through suppression of JNK signaling. Cancer Res. 2010;70:5628–38.
Bharti AC, Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Biochem Pharmacol. 2002;64:883–8.
Liu X, Xiang L, Zhang Y, Becker KG, Bera TK, Pastan I. CAPC negatively regulates NF-κB activation and suppresses tumor growth and metastasis. Oncogene. 2012;31:1673–82.
Bollrath J, Greten FR. IKK/NF‐κB and STAT3 pathways: central signalling hubs in inflammation‐mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–9.
Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Canc. 2002;2:594–604.
Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell. 2013;23:93–106.
Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011;18:1487–99.
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell. 2009;137:87–98.
Heldin C-H, Landström M, Moustakas A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial–mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–76.
Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, et al. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99:2365.
Jang C-W, Chen C-H, Chen C-C, Chen J-y, Su Y-H, Chen R-H. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol. 2001;4:51–8.
Valderrama-Carvajal H, Cocolakis E, Lacerte A, Lee E-H, Krystal G, Ali S, et al. Activin/TGF-β induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat Cell Biol. 2002;4:963–9.
Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004;23:1627–35.
Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. TGF-β-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol. 2001;3:708–14.
Drabsch Y, ten Dijke P. TGF-β signalling and its role in cancer progression and metastasis. Canc Metastasis Rev. 2012;31:553–68.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67.
Mitsiades N, W-h Y, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Canc Res. 2001;61:577–81.
Liu H, Zhang T, Li X, Huang J, Wu B, Huang X, et al. Predictive value of MMP‐7 expression for response to chemotherapy and survival in patients with non‐small cell lung cancer. Cancer Science. 2008;99:2185–92.
Joo NE, Miao D, Bermúdez M, Stallcup WB, Kapila YL. Shedding of NG2 by MMP-13 Attenuates Anoikis. DNA and Cell Biology. 2014;33:854–62.
Jing L, Anning L. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15:36–42.
Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–51.
Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Céspedes MV, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–84.
Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–20.
Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32.
Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62.
Tong XP, Chen Y, Zhang SY, Xie T, Tian M, Guo MR, et al. Key autophagic targets and relevant small‐molecule compounds in cancer therapy. Cell Prolif. 2015;48:7–16.
Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol. 2010;22:241–5.
DeNardo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Canc Metastasis Rev. 2008;27:11–8.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64.
Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009;16:175–83.
Thorburn J, Frankel AE, Thorburn A. Regulation of HMGB1 release by autophagy. Autophagy. 2009;5:247–9.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci. 2010;107:11942–7.
Yan J, Wang ZY, Yang HZ, Liu HZ, Mi S, Lv XX, et al. Timing is critical for an effective anti-metastatic immunotherapy: the decisive role of IFNγ/STAT1-mediated activation of autophagy. PloS One. 2011;6:e24705.
Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon H-U. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med. 2013;5:202ra123–3.
Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 2009;9:237.
Schmitz M, Grignard G, Margue C, Dippel W, Capesius C, Mossong J, et al. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int J Cancer. 2007;120:1284–92.
Lakshman M, Xu L, Ananthanarayanan V, Cooper J, Takimoto CH, Helenowski I, et al. Dietary genistein inhibits metastasis of human prostate cancer in mice. Canc Res. 2008;68:2024–32.
Singh BN, Kumar D, Shankar S, Srivastava RK. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem Pharmacol. 2012;84:1154–63.
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7:176.
Shen H-M, Codogno P. Autophagic cell death. Autophagy. 2011;7:457–65.
Clarke P, Puyal J. Autophagic cell death exists. Autophagy. 2012;8:867–9.
Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjørkøy G, Johansen T, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–31.
Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–68.
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94.
Hashimoto I, Koizumi K, Tatematsu M, Minami T, Cho S, Takeno N, et al. Blocking on the CXCR4/mTOR signalling pathway induces the anti-metastatic properties and autophagic cell death in peritoneal disseminated gastric cancer cells. Eur J Canc. 2008;44:1022–9.
Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Canc. 2009;9:274–84.
Vanharanta S, Massagué J. Origins of Metastatic Traits. Cancer Cell. 2013;24:410–21.
Guadamillas MC, Cerezo A, del Pozo MA. Overcoming anoikis–pathways to anchorage-independent growth in cancer. J Cell Sci. 2011;124:3189–97.
Lock R, Debnath J. Extracellular matrix regulation of autophagy. Curr Opin Cell Biol. 2008;20:583–8.
Debnath J. Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy. 2008;4:351–3.
Peng Y-F, Shi Y-H, Ding Z-B, Ke A-W, Gu C-Y, Hui B, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9:2056–68.
Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806.
Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2013;5:3056–6.
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Gene Dev. 2013;27:1447–61.
Toshima T, Shirabe K, Matsumoto Y, Yoshiya S, Ikegami T, Yoshizumi T, et al. Autophagy enhances hepatocellular carcinoma progression by activation of mitochondrial β-oxidation. J Gastroenterol. 2013;49:1–10.
Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611–22.
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118:3917.
Ojha R, Bhattacharyya S, Singh SK. Autophagy in Cancer Stem Cells: A Potential Link Between Chemoresistance, Recurrence, and Metastasis. BioResearch Open Access. 2015;4:97–108.
Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor–induced cell death in Philadelphia chromosome–positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–23.
Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2012;32:2261–72.
Zhu H, Wang D, Liu Y, Su Z, Zhang L, Chen F, et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013;13:119.
Chan FK-M. Programmed Necrosis/Necroptosis: An Inflammatory Form of Cell Death, Cell Death: Mechanism and Disease. 2013. p. 211–28.
Lu J, Chen H, Walsh C. Necroptotic signaling in adaptive and innate immunity. Semin Cell Dev Biol. 2014;35:33–9.
Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;208:633–41.
Dempsey LA. Interferon-induced necroptosis. Nat Immunol. 2013;14:892–2.
He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc Natl Acad Sci. 2011;108:20054–9.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael‐Tomasevic A, Phu L, et al. c‐IAP1 and UbcH5 promote K11‐linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010;29:4198–209.
Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–36.
O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–42.
Lin Y, Devin A, Rodriguez Y, Liu Z-g. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Gene Dev. 1999;13:2514–26.
Feng S, Yang Y, Mei Y, Ma L, Zhu D-e, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cellular Signalling. 2007;19:2056–67.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Cai Z, Jitkaew S, Zhao J, Chiang H-C, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65.
Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–43.
Vandenabeele P, Galluzzi L, Berghe TV, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14.
Zhang D-W, Shao J, Lin J, Zhang N, Lu B-J, Lin S-C, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6.
Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
Fulda S. The mechanism of necroptosis in normal and cancer cells. Canc Biol Ther. 2013;14:999–1004.
Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, et al. Dysregulation of TNFα-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2011;26:1293–300.
Cerhan JR, Ansell SM, Fredericksen ZS, Kay NE, Liebow M, Call TG, et al. Genetic variation in 1253 immune and inflammation genes and risk of non-Hodgkin lymphoma. Blood. 2007;110:4455–63.
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Canc Therapeut. 2007;6:1641–9.
Kawano Y, Fujiwara S, Kikukawa Y, Okuno Y, Mitsuya H, Hata H. A Small Molecule, Shikonin, Dually Functions As a Proteasome Inhibitor and a Necroptosis Inducer In Multiple Myeloma Cells. Blood. 2013;122:3172–2.
Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13:580.
Xuan Y, Hu X. Naturally-occurring shikonin analogues–a class of necroptotic inducers that circumvent cancer drug resistance. Cancer Lett. 2009;274:233–42.
Pasupuleti N, Leon L, Carraway KL, Gorin F. 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase-independent necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp Ther. 2013;344:600–15.
Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–73.
Li N, He Y, Wang L, Mo C, Zhang J, Zhang W, et al. D‐galactose induces necroptotic cell death in neuroblastoma cell lines. J Cell Biochem. 2011;112:3834–44.
Huang H, Xiao T, He L, Ji H, Liu X-Y. Interferon-β-armed oncolytic adenovirus induces both apoptosis and necroptosis in cancer cells. Acta Biochim Biophys Sin. 2012;44:737–45.
Jouan-Lanhouet S, Arshad M, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19:2003–14.
Buchheit C, Rayavarapu R, Schafer Z. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev Biol. 2012;23:402–11.
Long J, Ryan K. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene. 2012;31:5045–60.
Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Canc Res. 2007;13:7271–9.
Carrasco-García E, Martínez-Lacaci I, Ferragut JA, Mayor-López L, Rocamora-Reverte L, Menéndez-Gutiérrez MP, et al. Chapter 4. Cell death and cancer, novel therapeutic strategies. In: Apoptosis and Medicine, InTech. 2012.
Hu X, Han W, Li L. Targeting the weak point of cancer by induction of necroptosis. Autophagy. 2007;3:490.
Hu X, Xuan Y. Bypassing cancer drug resistance by activating multiple death pathways–a proposal from the study of circumventing cancer drug resistance by induction of necroptosis. Cancer Lett. 2008;259:127–37.
Weiner LM, Lotze MT. Tumor-cell death, autophagy, and immunity. N Engl J Med. 2012;366:1156–8.
Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev. 2012;249:205–17.
Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet. 2009;5:e1000459.
Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH. MicroRNA regulation of core apoptosis pathways in cancer. Eur J Canc. 2011;47:163–74.
Liu B, Wen X, Cheng Y. Survival or death: disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013;4:e892.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14.
Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen X, et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41:D983–6.
K-h L, Li W, Liu X-h, Sun M, Zhang M-l, Wu W-q, et al. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13:461.
Shi X, Sun M, Liu H, Yao Y, Kong R, Chen F, et al. A critical role for the long non‐coding RNA GAS5 in proliferation and apoptosis in non‐small‐cell lung cancer. Mol Carcinog. 2013; doi:10.1002/mc.22120
Zhao Y, Guo Q, Chen J, Hu J, Wang S, Sun Y. Role of long non-coding RNA HULC in cell proliferation, apoptosis and tumor metastasis of gastric cancer: a clinical and in vitro investigation. Oncol Rep. 2014;31:358–64.
Acknowledgements
This research was supported in part by grants from the National Natural Science Foundation of China (No. 81460440, No. 31300666, No. 81260322, and No. 81372322), the first Class General Financial Grant from the China Postdoctoral Science Foundation (No. 2013M542478), the Natural Science Foundation of Yunnan Province (No. 2012FB163), the Joint Special Funds for the Department of Science and Technology of Yunnan Province-Kunming Medical University (No. 2014FB059), and the Specialty Fund of High-level Talents Medical Personnel Training of Yunnan Province (No. D-201242).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZYS drafted the manuscript; ZZY conceived the structure of this manuscript and wrote a part of it; YQX completed the diagrams; YBC revised the manuscript; QY improved the structure and languages of the manuscript. All authors read and approved the final manuscript.
Authors’ information
Zhenyi Su, Zuozhang Yang, Yongqing Xu, Yongbin Chen and Qiang Yu: co-first authors.
Zhenyi Su, Zuozhang Yang, Yongqing Xu, Yongbin Chen and Qiang Yu contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Su, Z., Yang, Z., Xu, Y. et al. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer 14, 48 (2015). https://doi.org/10.1186/s12943-015-0321-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-015-0321-5