
Abstract
Background
Infections caused by linezolid-resistant enterococci (LRE) are clinically difficult to treat and threaten patient health. However, there is a lack of studies on long time-span LRE strains in China. For this reason, our study comprehensively revealed the resistance mechanisms of LRE strains collected in a Chinese tertiary care hospital from 2011 to 2022.
Methods
Enterococcal strains were screened and verified after retrospective analysis of microbial data. Subsequently, 65 LRE strains (61 Enterococcus faecalis and 4 Enterococcus faecium, MIC ≥ 8 µg/ml), 1 linezolid-intermediate Enterococcus faecium (MIC = 4 µg/ml) and 1 linezolid-susceptible Enterococcus faecium (MIC = 1.5 µg/ml) were submitted for whole-genome sequencing (WGS) analysis and bioinformatics analysis.
Results
The optrA gene was found to be the most common linezolid resistance mechanism in our study. We identified the wild-type OptrA and various OptrA variants in 98.5% of LRE strains (61 Enterococcus faecalis and 3 Enterococcus faecium). We also found one linezolid-resistant Enterococcus faecium strain carried both optrA and cfr(D) gene, while one linezolid-resistant Enterococcus faecium only harbored the poxtA gene. Most optrA genes (55/64) were located on plasmids, with impB-fexA-optrA, impB-fexA-optrA-erm(A), fexA-optrA-erm(A), and fexA-optrA segments. A minority of optrA genes (9/64) were found on chromosomes with the Tn6674-like platform. Besides, other possible linezolid resistance-associated mechanisms (mutations in the rplC and rplD genes) were also found in 26 enterococcal strains.
Conclusions
Our study suggested that multiple mechanisms of linezolid resistance exist among clinical LRE strains in China.
Similar content being viewed by others


Introduction
Enterococcus species (enterococci) are Gram-positive bacteria widely spread in the environment and hospital. They are regarded as opportunistic pathogens, which can colonize the gut of humans or animals as well as can lead to healthcare-associated infections. Enterococci may be responsible for various infections, including bacteremia, urinary tract infection, endocarditis, surgical site infections, and root canal failure [1]. Concerningly, enterococci show intrinsic resistance to commonly used antibiotics, including cephalosporins, aminoglycosides, clindamycin, and trimethoprim-sulfamethoxazole [2]. Additionally, the emergence and rapid expansion of vancomycin-resistant enterococci (VRE) strains has narrowed the therapeutic options [3]. Even worse, the property of enterococci to acquire resistance genes through plasmids or other genetic elements makes infections difficult to control [4].
Linezolid is the first oxazolidinone antibiotic approved by the U.S. Food and Drug Administration (FDA) for clinical use. Linezolid has demonstrated clinical benefits in treating severe Gram-positive bacterial infections caused by VRE, multidrug-resistant Streptococcus pneumoniae, and the challenging methicillin-resistant Staphylococcus aureus (MRSA) [5]. Unfortunately, in the last few years, reports of linezolid-resistant enterococci (LRE) have begun to appear and increase worldwide [6,7,8,9,10]. From 2000 to 2016, the bloodstream infections caused by linezolid-resistant Enterococcus faecium (E. faecium) and Enterococcus faecalis (E. faecalis) increased globally from 0.8% and 0.3% to 3% and 2%, respectively [11]. Although the detection rate of LRE is not high at present, the threat posed by its spread may make enterococcal infections uncontrollable.
Several specific mechanisms have been reported to be associated with LRE. Since linezolid inhibits polypeptide synthesis and elongation by binding to 23S rRNA, mutations in 23S rRNA can reduce the susceptibility to linezolid in enterococci [12]. Among these, G2505A or G2576U substitutions in domain V on 23S rRNA were more common [13]. Alternatively, mutations in the ribosomal proteins L3 (rplC) and L4 (rplD) could also confer resistance to linezolid [14]. The cfr gene was initially identified on the multi-resistance plasmid from a bovine strain of Staphylococcus sciuri, which encodes the 23S rRNA methyltransferase that confers multi-resistance against phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A (PhLOPSA phenotype) [15]. Furthermore, cfr(B) and cfr(D), as variants of cfr, have also been observed as mobile genetic elements in enterococci [16, 17]. In addition, it has been reported that the optrA gene encodes the ATP-binding cassette (ABC) protein that may confer transferable resistance to oxazolidinones and phenicols through a ribosomal protection mechanism [18]. The optrA gene was first identified in China in E. faecalis and E. faecium strains of human and animal origin but was subsequently detected in enterococci from more than 20 countries [10, 19, 20]. Another novel phenicol-oxazolidinone-tetracycline resistance gene, poxtA, was characterized in the chromosome of a MRSA of clinical origin [21]. The poxtA gene has been reported to be detected in enterococci isolated from humans, animals, and environmental sources [18]. In terms of the genomic context, the conserved structure constituted by the flanking IS1216 of the poxtA gene is associated with its mobility [22].
There are relatively few reports on LRE in China. These studies have collectively pointed out that the co-existence of multiple resistance mechanisms in LRE, which may be a worrisome issue [23,24,25,26]. Moreover, the clonal relatedness and genetic context of clinical LRE strains in China have not been fully elucidated. Existing studies mostly used polymerase chain reaction (PCR) or Sanger sequencing to detect the mechanism of linezolid resistance in enterococci [27, 28]. However, these studies may need more precise identification for the resistance mechanism. On this basis, this study retrospectively analyzed the prevalence of LRE isolated from our healthcare institution from 2011 to 2022 and used whole-genome sequencing (WGS) to explore these isolated strains’ clonal correlations and resistance mechanisms.
Materials and methods
Bacterial strains and antimicrobial susceptibility testing (AST)
The overall design of this study is displayed in Figure S1. A total of 5779 enterococci strains were isolated at Peking Union Medical College Hospital, a tertiary care hospital in Beijing, from 2011 to 2022. These strains were continuously isolated from non-repeat clinical patients and were all pathogenic or conditioned pathogens. The species of the strains were identified using the matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS; bioMérieux, Lyons, France). In clinical practice at our institution, the Kirby-Bauer disk diffusion method or broth microdilution method was routinely used to determine the susceptibility of enterococci to linezolid. The linezolid susceptibility testing results of all enterococci were retrospectively analyzed by the Laboratory Information System (LIS) and 65 LRE strains were screened. Furthermore, all screened LRE strains were revalidated using the E-test method. Therefore, all minimum inhibitory concentration (MIC) values of LRE strains against linezolid in this study were derived from the E-test method. Antimicrobial susceptibility profiles of 65 LRE strains were also subsequently determined using the broth microdilution method. The MIC values of 65 LRE strains against various antibiotics, including PEN (Penicillin), AMP (Ampicillin), TGC (Tigecycline), ERY (Erythromycin), TEC (Teicoplanin), VAN (Vancomycin), LEV (Levofloxacin), FOS (Fosfomycin), NIT (Nitrofurantoin), TCY (Tetracycline), DAP (Daptomycin), CHL (Chloramphenicol), RIF (Rifampicin) and MI (Minocycline), were determined and interpreted according to the Clinical and Laboratory Standards Institute (CLSI) and European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines. E. faecalis ATCC 29212 was used as the quality control strain. Patient characteristics and disease information corresponding to the strains were obtained from the hospital information system (HIS). This study was conducted in accordance with the guidelines of the Helsinki Declaration and was approved by Ethics Committee of Peking Union Medical College Hospital (Approval No. I-23PJ1724).
Detection of linezolid resistance-associated genes using PCR
Two non-LRE E. faecium strains (L1 and L49) were submitted to detect whether linezolid resistance genes [optrA, cfr, cfr(B), cfr(D) and poxtA] were present by using PCR. The sequences of the primers were derived from a previous literature [9]. Since the Tm values of all primers are close to 59 °C, we chose the same annealing condition for the different PCR reactions targeting various resistance genes. The detailed amplification conditions were: pre-denaturation (94 °C for 30s), 30 amplification cycles (94 °C for 30s, 56 °C for 30s, 72 °C for 30s), extension (72 °C for 5 min).
DNA extraction, library construction, and whole-genome sequencing
65 LRE strains and 2 non-LRE strains (L1 and L49) were submitted for whole genome sequencing (WGS) analysis. Genomic DNA was extracted from pure cultures using a commercial kit (QIAGEN, United States) and quantified by Qubit 4.0 (USA Invitrogen ABI). Whole-genome shotgun DNA was used for library preparation using either PCR-based protocol (MGIEasy FS DNA Library Prep Set, containing PCR-amplification steps after second bead purification). One portion of the genomic DNA was fragmented to about 5–10 kbp. Sequencing libraries were constructed using the MGI Easy Universal DNA Library Prep Set. All libraries were then sequenced on the MGISEQ-2000 platform with the PE150 model.
Data preprocessing, genome assembly, gene prediction, and type determination
Low quality, PCR duplication and adapter sequence were removed from raw data by Trim Galore (version 0.6.7, https://github.com/FelixKrueger/TrimGalore). Genome assembly and gene prediction of each strain were performed by SPAdes [29] (version 3.15.4) and Prokka [30] (version 1.14.6), respectively. Taxonomy classification of each strain was performed by Kraken [31] (version 2.1.2) and Bracken [32] (version 2.6.1). The completeness and contamination of final assemblies were evaluated using CheckM [33] (version 1.2.2). Genome statistics were evaluated by QUAST [34] (version 5.2.0). Multi-locus sequence typing (MLST) of each strain was determined by using mlst (version 2.23.0, https://github.com/tseemann/mlst) based on the PubMLST website (https://pubmlst.org/).
Other bioinformatics analyses
The whole-genome phylogenetic tree of strains was built using PhyloPhlan [35] (version 3.0.67) and RaxML [36] (version 8.2.12). PhyloPhlan used the parameters “--diversity low --fast -d phylophlan” and RaxML used the parameters “-f a -x 12345 -p 12345 -# 1000 -m PROTGAMMAAUTO”. LRE-Finder [13] (version 1.0.0) was applied to detect linezolid resistance genes [optrA, cfr, cfr(B), and poxtA] and common mutations in the V domain of the 23S rRNA (G2576U or G2505A) in enterococci. The Resistance Gene Identifier (version 4.0.3) was used to predict other linezolid resistance genes [cfr(D)] based on the reference data from the Comprehensive Antibiotic Resistance Database (CARD) [37] (version 3.2.6). All identified linezolid resistance genes were verified through the blastp (DIAMOND [38], version 2.0.15.153) alignment against the non-redundant protein sequences database (NR) (screening parameters: identity ≥ 99%, e-value < 1e-10). The location of linezolid resistance genes was predicted by Plasmer [39] (version 0.1). The predicted plasmid sequences (greater than 10 kp in length) containing linezolid resistance genes were annotated through the online BLAST software (https://blast.ncbi.nlm.nih.gov/Blast.cgi) with default parameters. The multiple sequence alignments among optrA genes were performed by MAFFT [40] (version 7.520) with parameters: ‘‘--localpair --maxiterate 1000’’. The phylogenetic tree of OptrA proteins was constructed by RaxML [36] (version 8.2.12) with default parameters and chose the wild-type OptrA sequence (NG_048023.1) as the outgroup. Phylogenetic trees were visualized using ggtree [41] (version 3.2.1). The protein variants of optrA genes were identified by alignments with NG_048023.1 as a reference. The PROVEAN tool [42] (http://provean.jcvi.org/) was used to predict amino acid mutation effects. To identify the genetic variation in the rplC (ribosomal protein L3) and rplD (ribosomal protein L4) genes, the multiple sequence alignments of those genes were also performed by MAFFT [40] (version 7.520) with CP003583.1 and CP008816.1 as references. In gene structure analysis, IslandViewer 4 [43] (http://www.pathogenomics.sfu.ca/islandviewer/) was applied in the prediction of genomic islands. The ISfinder [44] platform (http://www-is.biotoul.fr) was used for annotating insertion sequences (IS). The comparison of gene clusters was visualized by Easyfig [45] (version 2.2.5).
Results
Prevalence, clinical characteristics, antimicrobial susceptibility, and genomic data statistics of LRE
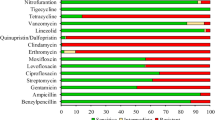
From 2011 to 2022, a total of 5779 enterococci isolates were isolated. Of these, E. faecalis (3514/5779, 60.8%) and E. faecium (2114/5779, 36.6%) accounted for the majority, while Enterococcus avium (69/5779, 1.2%) and other enterococci (82/5779, 1.4%) represented a relatively small proportion. Regarding linezolid resistance, a total of 61 E. faecalis and 4 E. faecium strains were recognized as LRE (MIC ≥ 8 µg/ml). The overall LRE prevalence rate is 1.1% (65/5779). Specifically, the prevalence of LRE in E. faecalis and E. faecium was 1.7% (61/3514) and 0.2% (4/2114), respectively. The MIC range of these LRE strains against linezolid was 8–48 µg/ml. Most of them were isolated in 2020 and 2021. The clinical characteristics of the patients who isolated these strains were presented in Table S1. Most of these patients were female and ranged from newborn to 89 years old. Patients were usually admitted to Surgery (SUR), Pediatrics (PED), or General Medicine (MED) departments. There was a wide range of sample types, including drainage fluid, wound secretions, swabs, and peripheral blood. In terms of patients’ conditions, it is of note that patients with malignant tumors, infections, and preterm deliveries carried LRE in this study. The antimicrobial susceptibility results of 65 LRE strains are displayed in Table 1. All LRE strains were susceptible to TGC, FOS, and NIT, whereas all were resistant to TCY. Additionally, most of the strains were also susceptible to TEC, VAN, PEN, and AMP. For CHL, ERY, and LEV, most strains exhibit resistance. Moreover, 58.5%, 52.3% and 46.2% of LRE strains exhibited intermediate to MI, DAP, and RIF, respectively.
Whole-genome sequencing was performed on 65 LRE and 2 non-LRE strains. Non-LRE strains contained E. faecium strain L1 (intermediate, MIC = 4 µg/ml) and E. faecium strain L49 (susceptible, MIC = 1.5 µg/ml). The genomes of 67 enterococcal strains had an average genome size of 2.96 Mbp (range from 2.69 to 3.28 Mb), an average scaffold number of 96 (range from 31 to 495), and an average N50 of 0.46 Mb (range from 35.7 kb − 1.47 Mb). Detailed quality assessment results can be found in Table S2. Based on the PubMLST website (https://pubmlst.org/), 67 strains were classified into 15 sequence types (STs). ST16 was most prevalent among 61 E. faecalis strains (52.5%). ST547, ST555, and ST1693 were identified among E. faecium strains. Besides, STs of 4 E. faecalis and 3 E. faecium strains were not identified. The detailed distribution of sequence types is shown in Figure S2.
Phylogenetic tree of enterococci
We constructed a phylogenetic tree based on whole genomes to determine the evolutionary relationships among 65 LRE and 2 non-LRE strains (Fig. 1). Overall, strains with the same ST type tended to cluster in the same clade, such as the ST16, ST476, ST480, and ST585 of E. faecalis. In addition, we noted that the evolutionary relationships of strains did not correlate significantly with the time and department of isolation. For example, E. faecalis strain s4 (isolated from Surgery in 2012) and E. faecalis strain L6 (isolated from Dermatology in 2019) were in the same clade, and E. faecalis strain s1 (isolated from Obstetrics and Gynecology in 2012) was closely evolutionarily related to E. faecalis strain L48 (isolated from Surgery in 2021).

Phylogenetic tree of 67 enterococcal strains. Different circles represent different types of annotations. From the outermost to the innermost layer, different colors represent the species information, sequence type, department, and isolation time of the strain. Nodes with the same color indicate that the corresponding strain was isolated from the same individual at different times. Pentagrams and triangles represent different non-LRE strains, the rest of strains are all linezolid-resistant. Bootstrap values of 1000 repetitions of sampling are labelled on the evolutionary tree nodes
Identifying the mechanism of linezolid resistance
Linezolid resistance-associated mutations in the 23S rRNA, rplC, and rplD genes, as well as the carriage of linezolid resistance genes [optrA, cfr, cfr(B), cfr(D), and poxtA], have been reported to be the main mechanisms of linezolid resistance in enterococci [16, 46]. We comprehensively identified these mechanisms in 65 LRE and 2 non-LRE strains based on the next-generation sequencing (NGS) technique. The results revealed that carriage of the optrA gene was the most common linezolid resistance mechanism (Table S3). Collectively, the optrA genes were detected in 100% of linezolid-resistant E. faecalis strains (n = 61, MIC: 8–48 µg/ml) and 75% of linezolid-resistant E. faecium strains (n = 3, MIC: 16–24 µg/ml). Notably, no other resistance genes associated with linezolid resistance were detected in E. faecalis strains. Among four linezolid-resistant E. faecium strains, two only carried the optrA gene (MIC = 16 µg/ml and MIC = 24 µg/ml, respectively), one had both the optrA gene and the cfr(D) gene (MIC = 16 µg/ml), and one only carried the poxtA gene (MIC = 32 µg/ml). No resistance genes associated with linezolid resistance were detected in one linezolid-intermediate E. faecium strain (L1, MIC = 4 µg/ml) and one linezolid-susceptible E. faecium (L49, MIC = 1.5 µg/ml) strain. To further investigate whether there were linezolid resistance-associated genes in these two non-LRE strains, we validated the WGS results using the PCR assay. The PCR results showed that the optrA, cfr, cfr(B), cfr(D) and poxtA genes were not detected in non-LRE strains. Based on the pre-developed machine learning model [39], we also predicted the plasmids in the assembled genomes of enterococcal strains to infer the location of the resistance genes optrA, cfr(D), and poxtA. The results showed that cfr(D), poxtA, and 85.9% (55/64) of the optrA genes were located on plasmids while only 14.1% (9/64) of the optrA genes were located on the chromosomes. Detailed location information can be found in Table S3.
In addition to the resistance genes, we detected several mutations in rplC and rplD genes, but none of these mutations resulted in alterations in the corresponding amino acid sequences. Overall, 7.5% (5/67) and 35.8% (24/67) of enterococcal strains contained mutations in rplC and rplD genes, respectively. The mutations on the rplC gene were mainly C369T (1 strain), T600C (3 strains), and C606T (1 strain). Moreover, A75T, T93G, T495C, C537A, T585G and T600C mutations were identified in the linezolid-intermediate E. faecium strain L1 (MIC = 4 µg/ml). The mutations on the rplD gene were predominantly the C348T type (20 strains), which has been reported previously [24]. Moreover, the common mutations (G2576U, G2505A) on the 23S rRNA associated with linezolid resistance were not detected in any of the 67 enterococcal strains. The identification results of linezolid resistance mechanisms in enterococci are detailed in Table S3.
Bioinformatic analysis of optrA genes
Given the optrA gene was found to be the primary resistance mechanism to linezolid of enterococci in this study, we conducted further studies on these optrA genes with different sources. By comparing the OptrA proteins from 64 enterococcal strains with reference sequence in the database (NCBI Reference Sequence: NG_048023.1), we constructed a phylogenetic tree of OptrA proteins (Fig. 2). The phylogenetic tree demonstrated the topology between the wild-type OptrA and different variants. Overall, we identified the wild-type OptrA (16 strains) and ten OptrA variants (48 strains). Of all OptrA variants, the D variant was closest to the wild-type OptrA in terms of evolutionary relationship. In contrast, the RDK variant was furthest away from the wild-type OptrA in the phylogenetic tree. Through alignments, we found a total of four unreported optrA variants, namely the D and KDS variants in E. faecalis (both MIC = 8 µg/ml) and the KLDD and EDS variants in E. faecalis (MIC = 16 and MIC = 24 µg/ml, respectively). More information can be found in Table S3. To further explore the potential effects of amino acid mutations, we performed the mutation effects prediction analysis (Table S4). Of all amino acid substitutions on OptrA proteins, Y176D and G393D were predicted to be deleterious to protein function, whereas other substitutions were neutral. Except for the D variant (G394D), the other nine OptrA variants had the Y176D amino acid mutation, covering 44 E. faecalis and 3 E. faecium strains. The G393D mutation was only present in one E. faecalis strain (EDD variant) and one E. faecalis strain (KLDD variant). Additionally, we noted that the type of OptrA variants did not correlate with their location on the genome. Specifically, nine optrA genes on the chromosome were wild-type, while the other genes of the wild-type OptrA and OptrA variants were on the plasmid. After annotating the plasmids, we found that different genes of OptrA variants were present on different plasmids (Table S3). The newly discovered D, KDS, and KLDD variants were on plasmids pAR_0780, pEFs17-1, and pDY28-optrA, respectively, while 5 DP variants were located on the plasmid p661-b. Furthermore, the isolation time of strains with the same OptrA variant type varied. RDK variant, the most common OptrA variant in this study (25 strains), originated from strains isolated in 2012 and 2014 and those isolated in 2020 and 2021.

Phylogenetic tree of OptrA proteins. The first appeared OptrA protein (NG_048023.1) represents an outgroup of the evolutionary tree. Different circles represent different types of annotations. From the outermost to the innermost layer: the species information of the corresponding strain, the location of the optrA gene, the isolation time of the corresponding strain, and the type of the OptrA variant. Branches with the same color indicate that the corresponding strain was isolated from the same individual at different times. Bootstrap values for 1000 repetitions of sampling are labelled on the evolutionary tree nodes
Since the optrA gene was the most common linezolid resistance gene in this study, we analyzed the genetic context of all chromosomal optrA (n = 9) and some optrA plasmids (greater than 10 kp in length) (n = 15) (Fig. 3). We found that the upstream and downstream structures of chromosomal optrA genes were significantly different from those present on plasmids. On chromosomal sequences from 9 strains, clusters of genes carrying optrA (from folC to rnjA) were predicted to be genomic islands, indicating that this region might be associated with horizontal gene transfer. In this optrA-carrying region, we identified the Tn6674-like platform, consisting of genes tnpA, tnpB, and tnpC (encoding proteins involved in the transposition of transposon Tn554), spc (resistance to spectinomycin), erm(A) (resistance to macrolides, lincosamides, and streptogramin B antibiotics), met (encoding methyltransferase), fexA (resistance to phenicols), and optrA (resistance to oxazolidinones and phenicols). The structures of chromosomal optrA genes were basically the same, whereas the genetic context of optrA plasmids was diverse. The impB-fexA-optrA plasmid segment was identified in 2 strains of wild-type OptrA (E. faecalis s3 and s14), insertion sequences ISVlu1 and IS1297 may affect the transfer and expression of this fragment. The impB-fexA-optrA-erm(A) arrangement was identified in 5 strains of DP variant (E. faecalis L33, L40, L58, L65, and L74). We observed the fexA-optrA-erm(A) arrangement in 3 strains, involving the D variant (E. faecalis s5), KLDD variant (E. faecium L28), and wild-type OptrA (E. faecalis L48). In 4 strains of wild-type OptrA (E. faecalis L59, s6, s7, and s12) and 1 strain of KDS variant (E. faecalis L53), we also identified the fexA-optrA plasmid segment.

Genomic upstream and downstream structures of the optrA gene. Red indicates resistance genes, blue indicates mobile genetic elements, dark grey indicates genes encoding hypothetical proteins, and brown indicates other genes
Discussion
Linezolid has been an essential antibiotic for the treatment of Gram-positive bacterial infections in clinical settings. However, infections caused by LRE have become global public health challenges. A meta-analysis estimated the global prevalence of linezolid-resistant E. faecalis and linezolid-resistant E. faecium to be 2.2% and 1.1%, respectively [47]. A 6-year surveillance from a teaching hospital in China showed that 3.93% (31/789) and 0.24% (2/834) of E. faecalis and E. faecium, respectively, exhibited resistance to linezolid [23]. In this study, we retrospectively analyzed microbial data from 2011 to 2022 and revealed that 1.7% (61/3514) and 0.2% (4/2114) of E. faecalis and E. faecium were resistant to linezolid, respectively. Although there may be variations in the prevalence of LRE, both our study and above studies implied that E. faecalis could be more likely to be resistant to linezolid than E. faecium. In terms of antimicrobial susceptibility testing results, most of the LRE strains isolated at our institution were susceptible to tigecycline, fosfomycin, nitrofurantoin, teicoplanin, vancomycin, penicillin, and ampicillin while resistant to tetracycline, chloramphenicol, erythromycin, and levofloxacin. Analogous to our findings, Li et al. observed that all LRE strains isolated at their institution were resistant to tetracycline, kanamycin, erythromycin, and ciprofloxacin but all were susceptible to vancomycin and teicoplanin [9]. In another report, all 22 LRE strains were confirmed to be susceptible to vancomycin, ampicillin, teicoplanin, and penicillin [23]. Taken together, vancomycin appears to be an effective option for the treatment of LRE, but intensive surveillance is necessary to prevent the emergence and rapid expansion of enterococci which resistant to both vancomycin and linezolid.
In this study, carriage of the optrA gene was the primary resistance mechanism of LRE strains (98.5%, 64/65) at our institution. However, this phenomenon was different from other researches. Zhang et al. analyzed 33 LRE strains collected from a teaching hospital in Wenzhou, China, and found that 54.6% carried both cfr and optrA genes [23]. Egan et al. studied 154 strains of LRE strains collected from 14 hospitals in Ireland and found that most of the strains had the G2576U 23S rRNA mutation or carried the poxtA gene, and only 9.7% of the strains carried the optrA gene [48]. Moure et al. studied different hospitals from Spain on 97 LRE strains and found that most strains carried the optrA gene or G2576U mutation [49]. Sassi et al. analyzed nine LRE strains collected from French hospitals between 2006 and 2016 and found that eight carried the optrA gene [8]. Overall, there may be variations in the primary resistance mechanism of LRE across different regions, which could be associated with discrepancies in healthcare conditions as well as antibiotic application strategies in various regions. Two other resistance genes [cfr(D) and poxtA] associated with linezolid resistance were identified in two E. faecium strains respectively in this study. Interestingly, one E. faecium strain harboring the cfr(D) gene also carried the optrA gene. This co-occurrence pattern was also reported in Streptococcus parasuis and Vagococcus lutrae [50, 51]. Remarkably, the E. faecium strain carrying the poxtA gene was highly resistant to linezolid (MIC = 32 µg/ml). However, no other resistance genes or 23S rRNA mutations associated with linezolid were detected in this strain. Additionally, this study also identified several rplC and rplD mutations in enterococci that may be associated with linezolid resistance. Among these, the C348T mutation in rplD was most frequently detected in linezolid-resistant E. faecalis strains (32.8%, 20/61). The C348T mutation in the rplD gene has been reported and may be associated with low-level linezolid resistance in enterococci [24]. In the linezolid-intermediate E. faecium strain L1 (MIC = 4 µg/ml), we only identified the rplC (A75T, T93G, T495C, C537A, T585G, T600C) and rplD (C174T, C180T) mutations. These mutations may be responsible for the elevated level of resistance to linezolid in E. faecium strain L1, but further experimental validation is needed.
As optrA gene was the most common resistance mechanism identified in the LRE strains in this study, further bioinformatics analyses could be of interest. We found that RDK variant dominated in all variants, but its evolutionary relationship was most distant from the wild-type in the phylogenetic tree. The MIC values of these strains carrying the RKD variant for linezolid were in the range of 8–32 µg/ml, whereas the strains carrying the wild-type optrA gene showed the MIC range for linezolid being 8–48 µg/ml. A previous study demonstrated that enterococci strains (isolated from asymptomatic healthy humans) carrying the wild-type optrA gene or the RDK variant exhibited relatively high levels of resistance to linezolid compared to other variants [52]. Another study demonstrated by transformation experiments that recipient carrying the RDK variant (MIC = 4 µg/ml) increased the MIC of linezolid relative to the original recipient strain (MIC = 2 µg/ml), but failed to reach the MIC of wild-type optrA gene recipient strain (MIC = 8 µg/ml) [9]. Thus, distinct variants of the optrA gene may confer differential resistance to linezolid in enterococci. On the other hand, it also highlighted the critical role of optrA gene in the resistance mechanism of linezolid. Importantly, we have identified four novel variants (D, KDS, KLDD, and EDS, MIC: 8–24 µg/ml) which have not been reported in the previous study. To fully understand the functions of these new variants, plasmids carrying the above variants can be constructed and transfected into the recipient organisms for further studies in the future.
In previous reports, the optrA gene was detected in E. faecalis at a higher rate than in E. faecium [52]. It has been found that the optrA gene can integrate into the genome (plasmids or chromosomes) of enterococci [53, 54], Vagococcus lutrae [51], Clostridium perfringens [55], Streptococcus suis [56], and other species. In this study, we identified that the chromosomal optrA was present in the Tn6674-like platform, which had been reported in the linezolid-resistant E. faecalis isolated from food-producing animals [57] and surface water [58]. Other optrA-carrying plasmid arrangements [impB-fexA-optrA, impB-fexA-optrA-erm(A), fexA-optrA-erm(A), and fexA-optrA] identified in this study was also appeared in the linezolid-resistant E. faecalis in previous studies [52, 57, 59]. Furthermore, we observed the Tn6674-like platform and insertion sequences ISVlu1 and IS1297 around the optrA gene. However, in a large-scale study of optrA-positive enterococci in Hangzhou, China, Cai et al. indicated that Tn554, Tn558 transposon, and IS1216E may be associated with the transmission of the optrA gene in enterococci [52]. These results suggest that the transmission mechanism of the optrA gene is more complex and not fixed. Through analyzing the upstream and downstream genes, we found that the optrA gene was adjacent to multiple resistance genes such as fexA, aadE, spc, and erm(A) (Fig. 3). It implies that LRE carrying the optrA gene may also have potential phenicol (fexA-mediated), streptomycin (aadE-mediated), spectinomycin (spc-mediated), streptogramin, lincosamide, and macrolide [erm(A)-mediated] resistant property, which warrants vigilance.
However, this study still has some shortcomings. Due to read length limitations, whole-genome sequencing of bacteria based on the NGS technique may not generate fined genome maps (chromosome or complete genome), thus preventing in-depth and comprehensive analyses of the location of linezolid resistance genes and transmission mechanism. Furthermore, we identified new OptrA variants and mutation sites in rplC and rplD genes. But the association between these findings and the level of linezolid resistance needs to be verified by further experiments. In summary, our study suggested that multiple mechanisms of linezolid resistance exist among clinical LRE strains in China. This study also addressed knowledge gaps and provided data support for monitoring linezolid resistance in enterococci.
Data availability
Genomes of 67 enterococcal strains analyzed in this study has been deposited in the NCBI database under accession number PRJNA1008900.
References
Fiore E, Van Tyne D, Gilmore MS. Pathogenicity of Enterococci. Microbiol Spectr 2019, 7(4).
Garcia-Solache M, Rice LB. The Enterococcus: a model of adaptability to its environment. Clin Microbiol Rev 2019, 32(2).
Tacconelli E, Cataldo MA. Vancomycin-resistant enterococci (VRE): transmission and control. Int J Antimicrob Agents. 2008;31(2):99–106.
Torres C, Alonso CA, Ruiz-Ripa L, Leon-Sampedro R, Del Campo R, Coque TM. Antimicrobial Resistance in Enterococcus spp. of animal origin. Microbiol Spectr 2018, 6(4).
Leach KL, Brickner SJ, Noe MC, Miller PF. Linezolid, the first oxazolidinone antibacterial agent. Ann N Y Acad Sci. 2011;1222:49–54.
Gargis AS, Spicer LM, Kent AG, Zhu W, Campbell D, McAllister G, Ewing TO, Albrecht V, Stevens VA, Sheth M, et al. Sentinel Surveillance reveals emerging daptomycin-resistant ST736 Enterococcus faecium and multiple mechanisms of Linezolid Resistance in Enterococci in the United States. Front Microbiol. 2021;12:807398.
Ruiz-Ripa L, Feßler AT, Hanke D, Eichhorn I, Azcona-Gutiérrez JM, Pérez-Moreno MO, Seral C, Aspiroz C, Alonso CA, Torres L et al. Mechanisms of Linezolid Resistance among Enterococci of Clinical Origin in Spain-Detection of Optra- and cfr(D)-Carrying E. faecalis. Microorganisms 2020, 8(8).
Sassi M, Guerin F, Zouari A, Beyrouthy R, Auzou M, Fines-Guyon M, Potrel S, Dejoies L, Collet A, Boukthir S, et al. Emergence of optra-mediated linezolid resistance in enterococci from France, 2006-16. J Antimicrob Chemother. 2019;74(6):1469–72.
Li P, Yang Y, Ding L, Xu X, Lin D. Molecular investigations of Linezolid Resistance in Enterococci OptrA variants from a hospital in Shanghai. Infect Drug Resist. 2020;13:2711–6.
Hu Y, Won D, Nguyen LP, Osei KM, Seo Y, Kim J, Lee Y, Lee H, Yong D, Choi JR et al. Prevalence and genetic analysis of Resistance mechanisms of Linezolid-Nonsusceptible Enterococci in a Tertiary Care Hospital examined via whole-genome sequencing. Antibiot (Basel) 2022, 11(11).
Jabbari Shiadeh SM, Pormohammad A, Hashemi A, Lak P. Global prevalence of antibiotic resistance in blood-isolated Enterococcus faecalis and Enterococcus faecium: a systematic review and meta-analysis. Infect Drug Resist. 2019;12:2713–25.
Leach KL, Swaney SM, Colca JR, McDonald WG, Blinn JR, Thomasco LM, Gadwood RC, Shinabarger D, Xiong L, Mankin AS. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol Cell. 2007;26(3):393–402.
Hasman H, Clausen P, Kaya H, Hansen F, Knudsen JD, Wang M, Holzknecht BJ, Samulioniene J, Roder BL, Frimodt-Moller N, et al. LRE-Finder, a web tool for detection of the 23S rRNA mutations and the optrA, cfr, cfr(B) and poxtA genes encoding linezolid resistance in enterococci from whole-genome sequences. J Antimicrob Chemother. 2019;74(6):1473–6.
Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther. 2014;12(10):1221–36.
Long KS, Poehlsgaard J, Kehrenberg C, Schwarz S, Vester B. The cfr rRNA methyltransferase confers resistance to Phenicols, Lincosamides, Oxazolidinones, pleuromutilins, and Streptogramin a antibiotics. Antimicrob Agents Chemother. 2006;50(7):2500–5.
Guerin F, Sassi M, Dejoies L, Zouari A, Schutz S, Potrel S, Auzou M, Collet A, Lecointe D, Auger G, et al. Molecular and functional analysis of the novel cfr(D) linezolid resistance gene identified in Enterococcus faecium. J Antimicrob Chemother. 2020;75(7):1699–703.
Deshpande LM, Ashcraft DS, Kahn HP, Pankey G, Jones RN, Farrell DJ, Mendes RE. Detection of a New Cfr-Like Gene, cfr(B), in Enterococcus faecium isolates recovered from human specimens in the United States as Part of the SENTRY Antimicrobial Surveillance Program. Antimicrob Agents Chemother. 2015;59(10):6256–61.
Brenciani A, Morroni G, Schwarz S, Giovanetti E. Oxazolidinones: mechanisms of resistance and mobile genetic elements involved. J Antimicrob Chemother. 2022;77(10):2596–621.
Wang Y, Lv Y, Cai J, Schwarz S, Cui L, Hu Z, Zhang R, Li J, Zhao Q, He T, et al. A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J Antimicrob Chemother. 2015;70(8):2182–90.
Schwarz S, Zhang W, Du XD, Krüger H, Feßler AT, Ma S, Zhu Y, Wu C, Shen J, Wang Y. Mobile Oxazolidinone Resistance genes in Gram-positive and Gram-negative Bacteria. Clin Microbiol Rev. 2021;34(3):e0018820.
Antonelli A, D’Andrea MM, Brenciani A, Galeotti CL, Morroni G, Pollini S, Varaldo PE, Rossolini GM. Characterization of poxtA, a novel phenicol-oxazolidinone-tetracycline resistance gene from an MRSA of clinical origin. J Antimicrob Chemother. 2018;73(7):1763–9.
Dejoies L, Sassi M, Schutz S, Moreaux J, Zouari A, Potrel S, Collet A, Lecourt M, Auger G, Cattoir V. Genetic features of the poxtA linezolid resistance gene in human enterococci from France. J Antimicrob Chemother. 2021;76(8):1978–85.
Zhang Y, Dong G, Li J, Chen L, Liu H, Bi W, Lu H, Zhou T. A high incidence and coexistence of multiresistance genes cfr and optrA among linezolid-resistant enterococci isolated from a teaching hospital in Wenzhou, China. Eur J Clin Microbiol Infect Dis. 2018;37(8):1441–8.
Chen H, Wu W, Ni M, Liu Y, Zhang J, Xia F, He W, Wang Q, Wang Z, Cao B, et al. Linezolid-resistant clinical isolates of enterococci and Staphylococcus cohnii from a multicentre study in China: molecular epidemiology and resistance mechanisms. Int J Antimicrob Agents. 2013;42(4):317–21.
Jia X, Ma W, Xu X, Yang S, Zhang L. Retrospective analysis of hospital-acquired linezolid-nonsusceptible enterococci infection in Chongqing, China, 2011–2014. Am J Infect Control. 2015;43(12):e101–106.
Tian Y, Li T, Zhu Y, Wang B, Zou X, Li M. Mechanisms of linezolid resistance in staphylococci and enterococci isolated from two teaching hospitals in Shanghai, China. BMC Microbiol. 2014;14:292.
Ngbede EO, Sy I, Akwuobu CA, Nanven MA, Adikwu AA, Abba PO, Adah MI, Becker SL. Carriage of linezolid-resistant enterococci (LRE) among humans and animals in Nigeria: coexistence of the cfr, optrA, and poxtA genes in Enterococcus faecium of animal origin. J Glob Antimicrob Resist. 2023;34:234–9.
Zou J, Xia Y. Molecular characteristics and risk factors associated with linezolid-resistant Enterococcus faecalis infection in Southwest China. J Glob Antimicrob Resist. 2020;22:504–10.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15(3):R46.
Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci 2017, 3.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–55.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5.
Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, Zhu Q, Bolzan M, Cumbo F, May U, et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun. 2020;11(1):2500.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Alcock BP, Huynh W, Chalil R, Smith KW, Raphenya AR, Wlodarski MA, Edalatmand A, Petkau A, Syed SA, Tsang KK, et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023;51(D1):D690–9.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60.
Zhu Q, Gao S, Xiao B, He Z, Hu S. Plasmer: an accurate and sensitive bacterial plasmid prediction Tool Based on Machine Learning of Shared k-mers and genomic features. Microbiol Spectr. 2023;11(3):e0464522.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Yu G. Using ggtree to visualize data on Tree-Like structures. Curr Protoc Bioinf. 2020;69(1):e96.
de Brevern AG, Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7(10).
Bertelli C, Laird MR, Williams KP, Lau BY, Hoad G, Winsor GL, Brinkman FSL. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017;45(W1):W30–5.
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34(Database issue):D32–36.
Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–10.
Bender JK, Cattoir V, Hegstad K, Sadowy E, Coque TM, Westh H, Hammerum AM, Schaffer K, Burns K, Murchan S, et al. Update on prevalence and mechanisms of resistance to linezolid, tigecycline and daptomycin in enterococci in Europe: towards a common nomenclature. Drug Resist Updat. 2018;40:25–39.
Dadashi M, Sharifian P, Bostanshirin N, Hajikhani B, Bostanghadiri N, Khosravi-Dehaghi N, van Belkum A, Darban-Sarokhalil D. The global prevalence of Daptomycin, Tigecycline, and linezolid-resistant Enterococcus faecalis and Enterococcus faecium strains from human clinical samples: a systematic review and Meta-analysis. Front Med (Lausanne). 2021;8:720647.
Egan SA, Shore AC, O’Connell B, Brennan GI, Coleman DC. Linezolid resistance in Enterococcus faecium and Enterococcus faecalis from hospitalized patients in Ireland: high prevalence of the MDR genes optrA and poxtA in isolates with diverse genetic backgrounds. J Antimicrob Chemother. 2020;75(7):1704–11.
Moure Z, Lara N, Marin M, Sola-Campoy PJ, Bautista V, Gomez-Bertomeu F, Gomez-Dominguez C, Perez-Vazquez M, Aracil B, Campos J, et al. Interregional spread in Spain of linezolid-resistant Enterococcus spp. isolates carrying the optrA and poxtA genes. Int J Antimicrob Agents. 2020;55(6):105977.
Zhu Y, Yang Q, Schwarz S, Yang W, Xu Q, Wang L, Liu S, Zhang W. Identification of a Streptococcus parasuis isolate co-harbouring the oxazolidinone resistance genes cfr(D) and optrA. J Antimicrob Chemother. 2021;76(11):3059–61.
Zhu Y, Yang W, Schwarz S, Xu Q, Yang Q, Wang L, Liu S, Zhang W. Characterization of the novel optra-carrying pseudo-compound transposon Tn7363 and an Inc18 plasmid carrying cfr(D) in Vagococcus lutrae. J Antimicrob Chemother. 2022;77(4):921–5.
Cai J, Schwarz S, Chi D, Wang Z, Zhang R, Wang Y. Faecal carriage of optra-positive enterococci in asymptomatic healthy humans in Hangzhou, China. Clin Microbiol Infect. 2019;25(5):e630631–6.
Tian T, Yang X, Liu S, Han Z, Qiao W, Li J, Yang M, Zhang Y. Hyper-thermophilic anaerobic pretreatment enhances the removal of transferable oxazolidinone and phenicol cross-resistance gene optrA in enterococci. Waste Manag. 2023;167:92–102.
Shen W, Zhang R, Cai J. Co-occurrence of multiple plasmid-borne linezolid resistance genes-optrA, cfr, poxtA2 and cfr(D) in an Enterococcus faecalis isolate from retail meat. J Antimicrob Chemother. 2023;78(7):1637–43.
Wu K, Li Z, Fang M, Yuan Y, Fox EM, Liu Y, Li R, Bai L, Zhang W, Zhang WM, et al. Genome characteristics of the optra-positive Clostridium perfringens strain QHY-2 carrying a novel plasmid type. mSystems. 2023;8(4):e0053523.
Yang Q, Zhu Y, Schwarz S, Zhang W, Wang X. Characterization of an optra-harbouring unconventional circularizable structure located on a novel ICESa2603 family-like integrative and conjugative element ICESsuHN38 in Streptococcus suis. J Antimicrob Chemother. 2023;78(8):2066–9.
Elghaieb H, Tedim AP, Abbassi MS, Novais C, Duarte B, Hassen A, Peixe L, Freitas AR. From farm to fork: identical clones and Tn6674-like elements in linezolid-resistant Enterococcus faecalis from food-producing animals and retail meat. J Antimicrob Chemother. 2020;75(1):30–5.
Nüesch-Inderbinen M, Raschle S, Stevens MJA, Schmitt K, Stephan R. Linezolid-resistant Enterococcus faecalis ST16 harbouring optrA on a Tn6674-like element isolated from surface water. J Global Antimicrob Resist. 2021;25:89–92.
Freitas AR, Tedim AP, Novais C, Lanza VF, Peixe L. Comparative genomics of global optra-carrying Enterococcus faecalis uncovers a common chromosomal hotspot for optrA acquisition within a diversity of core and accessory genomes. Microb Genomics 2020, 6(6).
Funding
This work was supported by the National Key Research Program of China [Grant No. 2021YFF0703805].
Author information
Authors and Affiliations
Contributions
ZRW, JJZ, LLL, CL, and HLS provided samples and performed the experiments. DPL and ZLH analyzed the data. ZRW and DPL wrote the manuscript. ZLH and HLS conceptualized and designed the study. DPL, ZMZ, SNH, and LHW prepared the data, figures, and tables. All authors reviewed, edited, and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the guidelines of the Helsinki Declaration and was approved by Ethics Committee of Peking Union Medical College Hospital (Approval No. I-23PJ1724). This study did not require written informed consent for participation. All privacy data involving patients in this study are strictly confidential in accordance with the national legislation and the institutional requirements.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.


12941_2024_689_MOESM6_ESM.png
Figure S2: The sequence typing of 67 enterococcal strains. (A) ST statistics of E. faecalis. (B) ST statistics of E. faecium. The colors in the bar graph represent the departments of strains
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Wang, Z., Liu, D., Zhang, J. et al. Genomic epidemiology reveals multiple mechanisms of linezolid resistance in clinical enterococci in China. Ann Clin Microbiol Antimicrob 23, 41 (2024). https://doi.org/10.1186/s12941-024-00689-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-024-00689-0