
Abstract
Background
Recently, extensively drug-resistant Pseudomonas aeruginosa (XDR-PA) isolates have been increasingly detected and posed great challenges to clinical anti-infection treatments. However, little is known about extensively resistant hypervirulent P. aeruginosa (XDR-hvPA). In this study, we investigate its epidemiological characteristics and provide important basis for preventing its dissemination.
Methods
Clinical XDR-PA isolates were collected from January 2018 to January 2023 and identified using matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry; antibiotic susceptibility testing was performed by broth microdilution method, and minimum inhibitory concentrations (MICs) were evaluated. Virulence was evaluated using the Galleria mellonella infection model; molecular characteristics, including resistance genes, virulence genes, and homology, were determined using whole-genome sequencing.
Results
A total of 77 XDR-PA strains were collected; 47/77 strains were XDR-hvPA. Patients aged > 60 years showed a significantly higher detection rate of XDR-hvPA than of XDR-non-hvPA. Among the 47 XDR-hvPA strains, 24 strains carried a carbapenemase gene, including blaGES−1 (10/47), blaVIM−2 (6/47), blaGES−14 (4/47), blaIMP−45 (2/47), blaKPC−2 (1/47), and blaNDM−14 (1/47). ExoU, exoT, exoY, and exoS, important virulence factors of PA, were found in 31/47, 47/47, 46/47, and 29/47 strains, respectively. Notably, two XDR-hvPA simultaneously co-carried exoU and exoS. Six serotypes (O1, O4–O7, and O11) were detected; O11 (19/47), O7 (13/47), and O4 (9/47) were the most prevalent. In 2018–2020, O4 and O7 were the most prevalent serotypes; 2021 onward, O11 (16/26) was the most prevalent serotype. Fourteen types of ST were detected, mainly ST235 (14/47), ST1158 (13/47), and ST1800 (7/47). Five global epidemic ST235 XDR-hvPA carried blaGES and showed the MIC value of ceftazidime/avibactam reached the susceptibility breakpoint (8/4 mg/L).
Conclusions
The clinical detection rate of XDR-hvPA is unexpectedly high, particularly in patients aged > 60 years, who are seemingly more susceptible to contracting this infection. Clonal transmission of XDR-hvPA carrying blaGES, which belongs to the global epidemic ST235, was noted. Therefore, the monitoring of XDR-hvPA should be strengthened, particularly for elderly hospitalized patients, to prevent its spread.
Similar content being viewed by others

Background
Pseudomonas aeruginosa (PA) is a leading cause of healthcare-associated infections, including pneumonia, intra-abdominal infection, urinary tract infection, surgical site infection, and bloodstream infections [1, 2]. The drug resistance rate of P. aeruginosa has recently increased because of the global spread of extensively drug-resistant (XDR) and multidrug-resistant (MDR) isolates, which are associated with treatment failure and increased mortality [3,4,5]. In China, the resistance rate of P. aeruginosa to carbapenems was ∼30%, and the related 30-day crude mortality was ∼40.0% [5, 6]. Therefore, in 2017, carbapenem-resistant P. aeruginosa was classified as a “priority one” pathogen for new antibiotics by the World Health Organization (WHO) [7]. Intrinsic resistance, chromosomal gene mutations, and transferable resistance determinants are responsible for this increasing threat, such as carbapenemases (GES, KPC, VIM, and IMP enzymes) and co-transferred aminoglycoside-modifying enzyme determinants [e.g., AAC (3′), AAC (6′), and ANT (2′)-I] [4].
Notably, recent clinical studies have reported the emergence of hypervirulent P. aeruginosa (hvPA). Zhang et al. reported the emergence and recurrence of KPC-producing hvPA ST697 and ST463 between 2010 and 2021 in China [8]. In vivo acquisition of blaKPC-2 in blaAFM-1-expressing hvPA ST463 was also reported in a patient with hematologic malignancy [9]. Early detection of a KPC-2-producing hvPA ST235 was reported by de Paula-Petroli et al. in Brazil [10]. These studies suggest that the reports on hvPA currently are mainly sporadic case reports, and relevant research on extensively drug-resistant P. aeruginosa (XDR-PA) is still lacking. Therefore, this study intended to (i) collect and screen extensively drug-resistant hypervirulent P. aeruginosa (XDR-hvPA) from clinical isolates, (ii) analyze the possible risk factors leading to its infection, and (iii) investigate its molecular epidemiological characteristics.
Methods
Bacterial strains and species identification
All non-repetitive clinically isolated XDR-PA were collected from January 2018 to January 2023 at Xiangya Hospital of Central South University, China; this is a large hospital with 3500 beds and more than 3 million outpatients every year. Matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI–TOF MS; Bruker Daltonics GmbH, Bremen, Germany) was used to identify all isolates, with Escherichia coli ATCC 25922 (National Center for Clinical Laboratories, Beijing, China) as the quality control strain.
Antimicrobial susceptibility testing
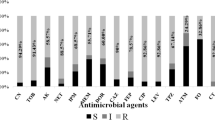
The classic broth microdilution test was used to determine the minimum inhibitory concentrations (MICs) of antimicrobial agents (Hangzhou Kangtai Biotechnology Co.), including piperacillin (PRL), ceftazidime (CAZ), cefepime (FEP), aztreonam (ATM), meropenem (MEM), imipenem (IPM), amikacin (AK), gentamicin (GEN), tobramycin (TOB), ciprofloxacin (CIP), levofloxacin (LEV), piperacillin/tazobactam (TZP), nitrofurantoin (F), ceftazidime/avibactam (CZA), and polymyxin B (PB). P. aeruginosa ATCC 27853 (National Center for Clinical Laboratories, Beijing, China) was considered the quality control strain. The susceptibility breakpoints were interpreted using the guidelines of the Clinical and Laboratory Standards Institute (2022) [11].
The extensively drug-resistant (XDR) isolate was defined to be non-susceptible to ≥ 1 agent in all but ≤ 2 categories (i.e., bacterial isolates remain susceptible to only one or two categories) [12].
Galleria mellonella infection model
The virulence of all collected clinical isolates was evaluated using the G. mellonella infection model (Tianjin Huiyude Biotech Company, Tianjin, China) [13]. In brief, 10 µL of P. aeruginosa overnight cultures adjusted to 1 × 106 CFU/mL in physiological saline were injected into G. mellonella larvae, followed by 5 days of incubation in the dark at 37 °C. The survival rate was measured using PBS as the negative group, and all experiments were done in triplicate [13].
Whole-genome sequencing and analysis
Genomic DNA was extracted from XDR-hvPA strains using TIANamp Bacteria DNA Kit (Tiangen Biochemical Technology Co., Ltd, Beijing) for NovaSeq 6000 sequencing. After passing through fastp filters and FastQC, clean reads were assembled and corrected using Unicycler to obtain the final genome sequence. Sequence typing (ST) and O serotype of XDR-hvPA strains were confirmed by multilocus sequence typing and PAst 1.0 (https://cge.cbs.dtu.dk/services/PAst/), respectively. To detect and characterize antimicrobial-resistant genes, the basic local alignment search tool (BLAST) alignments were conducted using the comprehensive antibiotic resistance database (https://card.mcmaster.ca/). All sequenced genomes were aligned to PAO1 (GenBank accession: NC_002516.2) to determine single-nucleotide polymorphisms (SNPs). Additionally, core genomes, single-nucleotide polymorphisms (cg-SNPs), and the phylogenetic tree were analyzed using Snippy (https://github.com/tseemann/snippy) and visualized using iTOL (https://itol.embl.de/) and ChiPlot (https://www.chiplot.online/). Notably, the sequence data have been deposited in NCBI with the accession number PRJNA1018421.
Statistical analyses
SPSS 26.0 was used for statistical analysis. The underlying clinical characteristics, underlying diseases, invasive procedure (e.g., urinary catheter, gastric tube, and peripherally inserted central catheter), and previous antibiotic exposure of XDR-hvPA and XDR-non-hvPA strains were compared. The Fisher’s exact test or χ2 test was used for categorical variables, and the Student’s t-test was used for continuous variables. A P value of < 0.05 indicated statistical significance.
Results
Collection of XDR-PA strains and screening of XDR-hvPA
A total of 77 non-repetitive P. aeruginosa strains were isolated from various types of clinical specimens taken from 77 patients; five of these patients were emergency patients, and the remainder were hospitalized patients. In terms of the distribution of patients in departments, most of the patients were in the intensive care unit (ICU, 40.3%, 31/77), followed by integrated Chinese and Western medicine departments (ICWM, 22.0%, 17/77), rehabilitation departments [11.7% (9/77)], emergency departments [6.5% (5/77)], respiratory departments [3.9% (3/77)], and other departments [15.6% (12/77)]. Among the sample sources, 85.7% (66/77) were respiratory specimens, followed by urine specimens [5.2% (4/77)], fecal specimens [3.9% (3/77)], wound secretion specimens [3.9% (3/77)], and tissue specimen [1.3% (1/77)].
According to the results of antimicrobial susceptibility tests, all 77 strains were susceptible only to polymyxin B, indicating XDR-PA. Notably, the drug sensitivity results of 14 ST235 strains carrying the blaGES gene to CZA showed that the MIC value was 2/4∼8/4 mg/L, and five strains (P54, P62, P68, P71, and P88) showed the MIC value of CZA at 8/4 mg/L, which was close to the resistance cutoff point (MIC break point for sensitivity: ≤8/4 mg/L).
In our study, the G. mellonella infection model and the important virulence-related genes (exoU and exoS) were used to evaluate the virulence of all collected strains, and the results suggested that 47 strains were XDR-hvPA. (Fig. 1).

The virulence of 77 XDR-PA isolates. The virulence characterization in the G. mellonella infection model. PBS was used as the negative group. After infection with XDR-PA isolates carrying exoE/exoS virulence-related genes, the mortality rate of the G. mellonella was significantly higher
Clinical characterizations of XDR-hvPA and XDR-non-hvPA
To clarify the clinical characteristics of XDR-hvPA and XDR-non-hvPA, a comparative analysis was conducted on these indicators, including demographic information, ICU admission, underlying diseases, invasive procedures, and patients’ history of antibiotic exposure. Patients were divided into three age groups: ≤18 years, 18–59 years, and ≥ 60 years. In patients aged ≥ 60 years, the detection rate of XDR-hvPA was significantly higher than that of XDR-non-hvPA (72.3% vs. 40.0%, P = 0.005); however, the rate did not differ significantly in the other two age groups. There was no significant difference in sex, length of hospitalization < 30 days, ICU admission, underlying diseases, invasive procedures, and previous antibiotic exposure. It was suggested that elderly patients were more likely to contract XDR-hvPA infection (Table 1).
Resistance and virulence genes of XDR-hvPA strains
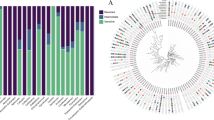
Among 47 XDR-hvPA strains, 51.1% (24/47) strains carried the carbapenemase gene, suggesting that carbapenem-producing played a major role in drug resistance of P. aeruginosa. Six types of carbapenem-resistant genes were detected, namely blaGES-1, blaVIM-2, blaGES-14, blaIMP-45, blaKPC-2, and blaNDM-14, with positivity rates of 21.3% (10/47), 12.8% (6/47), 8.5% (4/47), 4.3% (2/47), 2.1% (1/47), and 2.1% (1/47), respectively. Notably, some resistance genes are rare types of genes in XDR-hvPA, such as blaIMP-45, blaKPC-2, and blaNDM-14. (Fig. 2). In addition, these strains also co-carried other drug resistance-related genes, as shown in Figure S1.

Molecular characteristics of 47 XDR-hvPA strains. PAO1 (GenBank accession: NC_002516.2) was used as the reference strain
The detection rates of the virulence-associated genes exoT, exoY, exoU, and exoS, which are the important effectors of the type III secretion system of P. aeruginosa, were 100.0% (47/47), 97.9% (46/47), 66.0% (31/47), and 38.3% (18/47), respectively. Interestingly, two strains (P59 and P69) co-carried exoU and exoS simultaneously (Fig. 2). Genes encoding flagella, type IV pili and non-pilus adhesins, and extracellular virulence factors were also detected (Figure S2).
Homology analysis of XDR-hvPA
Notably, 47 XDR-hvPA strains belonged to 14 ST types, with ST235 (n = 14, 29.8%) being the most prevalent, followed by ST1158 (n = 13, 27.7%) and ST1800 (n = 7, 14.9%). All strains of ST235 carried the blaGES gene; ST1158 did not carry the carbapenemase gene, and three strains of ST1800 carried the blaVIM-2 gene. The strain carrying the blaNDM-14 gene belonged to ST4; the strain carrying blaIMP-45 belonged to ST2068 and ST3755, and the strain carrying the blaKPC-2 gene belonged to ST463. The 47 XDR-hvPA strains were divided into 17 clusters. Interestingly, the isolate carrying the blaGES gene (ST235) was identified to belong to the same cluster with the difference in SNP from 2 to 51 (mainly from 2021 to 2022), indicating the existence of clonal transmission (Fig. 2).
We downloaded the genetic data of global epidemic ST235 P. aeruginosa from the Pseudomonas genome database (https://www.pseudomonas.com/) and analyzed the evolutionary relationship between ST235 P. aeruginosa globally and XDR-hvPA strains in this study. Thus far, 206 strains of ST235 P. aeruginosa have been uploaded to the system globally. In addition, we conducted homology analysis on these 206 strains of ST235 P. aeruginosa, 15 strains of ST235 P. aeruginosa from Hangzhou, Zhejiang, which were previously reported by Li et al. [14], and 14 strains of XDR-hvPA reported in this study. The results showed that these 235 strains could be divided into 11 clusters, with clusters A, B, C, and D being the most prevalent, accounting for 98.3% of all strains, indicating clonal transmission. Fourteen XDR-hvPA strains were classified into A clone cluster. These had the closest similarity to JAPZLY01, which were obtained from Hangzhou, Zhejiang, in a previous report by Li et al. [14], with an SNP difference of 1–5 (except for P51 and P87), indicating that ST235 P. aeruginosa may have clonal transmission in China and should be highly valued. (Fig. 3).

The phylogenetic analyses of ST235 PA from the global region. The first circle represents the cluster; the second circle shows the countries or regions where the strains were distributed; the third circle indicates the O serotype. P54 in our study was used as the reference strain
Evolution relationship of serotype in the past six years
This study analyzed the change of O serotype during the evolution of the strain. A total of six O serotypes of 47 XDR-hvPA strains were detected, namely O1, O4, O5, O6, O7, and O11. Among them, O11 (40.4%, 19/47), O7 (27.6%, 13/47), and 04 (19.1%, 9/47) accounted for the highest proportion. From 2018 to 2020, O4 and O7 were the prevalent serotypes, whereas in the last three years, O11 was the most prevalent serotype, accounting for 61.5% of all serotypes (16/26). (Fig. 2). Notably, 235 strains of ST235 P. aeruginosa isolated globally were O11, except one strain (SAMN12127367) was O10. (Fig. 3).
Discussion
P. aeruginosa has long been recognized as a significant opportunistic pathogen for hospital infections, and in recent years, the number of resistant isolates, particularly MDR and XDR isolates, has increased. These isolates can cause treatment failure and higher mortality [3,4,5]. Moreover, hvPA has been an emergency in clinical in recent years [8,9,10]. However, it is currently unclear whether this hypervirulent strain also exists in XDR-PA. This study aimed to investigate the clinical and molecular characteristics of XDR-hvPA to establish a laboratory foundation for the efficient management of these isolates.
Owing to its ease of operation and preservation benefits, the G. mellonella infection model has been extensively used to evaluate bacterial virulence [15,16,17,18]. In this study, the virulence of all collected strains was evaluated using this model and the important virulence-related genes (exoU and exoS), and 61.0% of strains were found to be XDR-hvPA, suggesting that the detection rate of highly virulent strains in XDR-PA was much higher than expected. In addition, patients aged ≥ 60 years old were more vulnerable to XDR-hvPA infection, and this age is also considered an independent risk factor for healthcare-associated infections (HAIs) [19]. Therefore, such infections in elderly patients require more attention. Previous studies suggested that prior ICU hospitalization, history of invasive operation(s), and previous antibiotic exposure (e.g., carbapenems and cephalosporin) may be independent risk factors for P. aeruginosa infection [5, 20, 21]. However, no significant differences were noted between XDR-hvPA and XDR-non-hvPA in this study, which may be related to the serious condition of these patients, the use of multiple antibiotics, and a history of invasive procedures.
Carbapenem-resistant P. aeruginosa usually shows resistance to the vast majority of clinical antibiotics and even XDR due to the production of carbapenemase, high efflux pump expression, and deletion of outer membrane protein. In our study, most XDR-hvPA strains were carbapenemase producers, suggesting that the production of this enzyme is the main mechanism leading to their drug resistance, which is inconsistent with previous research [22,23,24]; this inconsistency can be attributed to all strains in this study being XDR and plasmids carrying multidrug resistance genes being obtained from outside. It is worth noting that among these carbapenemases in XDR-PA in our study, blaGES was the most prevalent, which is mainly detected in P. aeruginosa [25, 26]. Recent research has shown that increased blaGES-1 expression caused by the class 1 integron’s potent promoter reduces CRPA susceptibility to ceftazidime–avibactam [14, 27]. Although all strains carrying blaGES-1 in this study were not resistant to CZA, the MIC value of CZA in five strains had reached the susceptibility breakpoint (8/4 mg/L). This study also found that in addition to blaVIM-2 commonly seen in P. aeruginosa, some rare carbapenemase genotypes, such as blaKPC-2 and blaIMP-45, were also detected. The blaKPC first detected in Klebsiella pneumoniae has been worldwide dissemination in this strain [28, 29]. In recent years, it has come to be increasingly found in P. aeruginosa, and some reports have shown that the KPC gene is also found in hvPA, which was mainly in the southeastern coastal areas of China and predominantly in a potentially high-risk clone of P. aeruginosa ST463 [8, 9, 30, 31]. De Paula-Petroli et al. also reported a KPC-2-producing hvPA ST235 in Brazil [10]. This study is the first to detect hvPA ST463 carrying KPC in the central southern region, indicating that it may have spread from coastal areas to the central region, which is a noteworthy finding. In addition, a previously rare drug resistance gene, blaNDM-14, was also detected in XDR-hvPA. Certainly, these strains also carried multiple other drug resistance-related genes, as shown in Figure S1.
The virulence of P. aeruginosa may be related to its many flagella, type IV fimbriae, and non-fimbriae adhesins [32]. ExoU, exoT, exoY, and exoS, the four key effectors in the type III secretion system, can help inject toxic proteins into host cells and are also closely related to the virulence of P. aeruginosa [18, 30]. In our study, only one strain did not carry exoY, whereas all others carried exoT and exoY. Previous studies have shown mutual exclusion of the exoU and exoS genes [33, 34], and exoU-positive strains presented multiple resistance mechanisms and stronger virulence in G. mellonella [13]. The exoU and/or exoS genes were positive in XDR-hvPA, and two strains co-carried exoU and exoS genes simultaneously, which may increase drug resistance and virulence [35].
Although P. aeruginosa has a nonclonal epidemic nature, some genotypes are found to be linked to global outbreaks, including ST111, ST175, ST235, ST244, and ST395 [36]. ST235, which is the most common of these high-risk clones, has been linked to poor clinical outcomes, partly due to high levels of antibiotic resistance [4, 36], which was also detected in our XDR-hvPA carrying blaGES with clonal distribution. In addition, phylogenetic analyses of global ST235 P. aeruginosa were performed, and the results showed that ST235 P. aeruginosa was mainly divided into four large clusters and had clonal transmission. The 14 XDR-hvPA strains in this study were highly correlated with JAPZLY01 from the report in Zhejiang Province by Li et al. [14], with minimal SNP differences (ranging from 1 to 5), suggesting that ST235 P. aeruginosa also has clonal transmission in China and deserves clinical attention. This study also identified the existence of clone propagation in ST1158 and ST1800 XDR-hvPA.
Our study found six O serotypes in XDR-hvPA strains, and O11, O7, and O4 accounted for the highest proportion. Notably, O4 and O7 were the prevalent serotypes from 2018 to 2020, whereas O11 was the prevalent serotype in recent years, accounting for 61.5% of all serotypes (16/26), indicating that the main serotype may have changed from O4 and O7 to O11. It is worth noting that 235 strains of ST235 P. aeruginosa isolated globally are O11, except one strain (SAMN12127367), which is O10. This suggests that the O antigen serotype may be closely related to high-risk clones and virulence. According to Del Barrio-Tofio et al., there is a close connection among the P. aeruginosa O antigen serotypes, resistance profiles, and high-risk clones. For example, O4 is associated with ST175’s MDR/XDR profile [37]. However, we found that O4 mainly belonged to ST1800 XDR-hvPA and a blaKPC-2-carried ST463 isolate, which were also found in other regions in China [8, 9, 30, 31].
Conclusions
In summary, the clinical detection rate of XDR-hvPA may have exceeded our expectations, particularly for hospitalized patients aged over 60 years, and this age may be an important risk factor for increased vulnerability to this infection. There is a clonal transmission of XDR-hvPA carrying the GES-type carbapenemase, which belongs to the global epidemic ST235. To effectively prevent such transmission, it is necessary to strengthen the monitoring of XDR-hvPA in hospitalized patients, particularly the elderly.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- PA:
-
Pseudomonas aeruginosa
- XDR:
-
extensively drug-resistant
- MDR:
-
multidrug-resistant
- WHO:
-
World Health Organization
- hvPA:
-
hypervirulent P. aeruginosa
- XDR-PA:
-
extensively drug-resistant P. aeruginosa
- XDR-hvPA:
-
extensively drug-resistant hypervirulent P. aeruginosa
- MALDI–TOF MS:
-
matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry
- MICs:
-
minimum inhibitory concentrations
- ST:
-
sequence typing
- PRL:
-
piperacillin
- CAZ:
-
ceftazidime
- FEP:
-
cefepime
- ATM:
-
aztreonam
- MEM:
-
meropenem
- IPM:
-
imipenem
- AK:
-
amikacin
- GEN:
-
gentamicin
- TOB:
-
tobramycin
- CIP:
-
ciprofloxacin
- LEV:
-
levofloxacin
- TZP:
-
piperacillin/tazobactam
- F:
-
nitrofurantoin
- CZA:
-
ceftazidime/avibactam
- PB:
-
polymyxin B
- BLAST:
-
basic local alignment search tool
- SNPs:
-
single-nucleotide polymorphisms
- ICU:
-
intensive care unit
- PICC:
-
peripherally inserted central catheter
- HAIs:
-
healthcare-associated infections
References
Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med. 2014;370:1198–208.
Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, et al. Antimicrobial-resistant pathogens Associated with Healthcare-Associated infections: Summary of Data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect Control Hosp Epidemiol. 2016;37:1288–301.
Zhang Y, Chen XL, Huang AW, Liu SL, Liu WJ, Zhang N, et al. Mortality attributable to carbapenem-resistant Pseudomonas aeruginosa bacteremia: a meta-analysis of cohort studies. Emerg Microbes Infect. 2016;5:e27.
Horcajada JP, Montero M, Oliver A, Sorlí L, Luque S, Gómez-Zorrilla S, et al. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin Microbiol Rev. 2019;32.
Yuan Q, Guo L, Li B, Zhang S, Feng H, Zhang Y, et al. Risk factors and outcomes of inpatients with carbapenem-resistant Pseudomonas aeruginosa bloodstream infections in China: a 9-year trend and multicenter cohort study. Front Microbiol. 2023;14:1137811.
Hu F, Guo Y, Yang Y, Zheng Y, Wu S, Jiang X, et al. Resistance reported from China antimicrobial surveillance network (CHINET) in 2018. Eur J Clin Microbiol Infect Dis. 2019;38:2275–81.
WHO. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. https://www.infobioquimica.com/new/wp-content/uploads/2017/02/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf.
Zhang B, Xu X, Song X, Wen Y, Zhu Z, Lv J, et al. Emerging and re-emerging KPC-producing hypervirulent Pseudomonas aeruginosa ST697 and ST463 between 2010 and 2021. Emerg Microbes Infect. 2022;11:2735–45.
Zhang P, Wang J, Shi W, Wang N, Jiang Y, Chen H, et al. In vivo acquisition of bla(KPC-2) with low biological cost in bla(AFM-1)-harboring ST463 hypervirulent Pseudomonas aeruginosa from a patient with hematologic malignancy. J Glob Antimicrob Resist. 2022;31:189–95.
de Paula-Petroli SB, Campana EH, Bocchi M, Bordinhão T, Picão RC, Yamada-Ogatta SF, et al. Early detection of a hypervirulent KPC-2-producing Pseudomonas aeruginosa ST235 in Brazil. J Glob Antimicrob Resist. 2018;12:153–4.
CLSI. Performance stands for antimicrobial susceptibility testing. 32nd ed. CLSI supplement M100. Clinical and Laboratory Standards Institute. 2022.
Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–81.
Zhu Y, Kang Y, Zhang H, Yu W, Zhang G, Zhang J, et al. Emergence of ST463 exoU-positive, imipenem-nonsusceptible Pseudomonas aeruginosa isolates in China. Microbiol Spectr. 2023:e0010523.
Li X, Zhang X, Cai H, Zhu Y, Ji J, Qu T, et al. Overexpression of bla(GES-1) due to a strong promoter in the class 1 integron contributes to decreased ceftazidime-avibactam susceptibility in carbapenem-resistant Pseudomonas aeruginosa ST235. Drug Resist Updat. 2023;69:100973.
Zhou K, Tang X, Wang L, Guo Z, Xiao S, Wang Q, et al. An emerging clone (ST457) of Acinetobacter baumannii Clonal Complex 92 with enhanced virulence and increasing endemicity in South China. Clin Infect Dis. 2018;67:179–s88.
Ten KE, Muzahid NH, Rahman S, Tan HS. Use of the waxworm Galleria mellonella larvae as an infection model to study Acinetobacter baumannii. PLoS ONE. 2023;18:e0283960.
Zhou Y, Wu C, Wang B, Xu Y, Zhao H, Guo Y, et al. Characterization difference of typical KL1, KL2 and ST11-KL64 hypervirulent and carbapenem-resistant Klebsiella pneumoniae. Drug Resist Updat. 2023;67:100918.
Zhu Y, Jia P, Yu W, Chu X, Liu X, Yang Q. The epidemiology and virulence of carbapenem-resistant Pseudomonas aeruginosa in China. Lancet Microbe. 2023.
Zhao X, Wang L, Wei N, Zhang J, Ma W, Zhao H, et al. Epidemiological and clinical characteristics of healthcare-associated infection in elderly patients in a large Chinese tertiary hospital: a 3-year surveillance study. BMC Infect Dis. 2020;20:121.
Tsao LH, Hsin CY, Liu HY, Chuang HC, Chen LY, Lee YJ. Risk factors for healthcare-associated infection caused by carbapenem-resistant Pseudomonas aeruginosa. J Microbiol Immunol Infect. 2018;51:359–66.
Huang W, Wei X, Xu G, Zhang X, Wang X. Carbapenem-resistant Pseudomonas aeruginosa infections in critically ill children: prevalence, risk factors, and impact on outcome in a large tertiary pediatric hospital of China. Front Public Health. 2023;11:1088262.
Khalili Y, Omidnia P, Goli HR, Zamanlou S, Babaie F, Zahedi Bialvaei A, et al. Molecular characterization of carbapenem-resistant Pseudomonas aeruginosa isolated from four medical centres in Iran. Mol Biol Rep. 2022;49:8281–9.
Stanton RA, Campbell D, McAllister GA, Breaker E, Adamczyk M, Daniels JB, et al. Whole-genome sequencing reveals diversity of Carbapenem-Resistant Pseudomonas aeruginosa Collected through CDC’s Emerging infections Program, United States, 2016–2018. Antimicrob Agents Chemother. 2022;66:e0049622.
Zhao Y, Chen D, Chen K, Xie M, Guo J, Chan EWC, et al. Epidemiological and genetic characteristics of clinical carbapenem-resistant Pseudomonas aeruginosa strains in Guangdong Province, China. Microbiol Spectr. 2023;11:e0426122.
Hishinuma T, Tada T, Kuwahara-Arai K, Yamamoto N, Shimojima M, Kirikae T. Spread of GES-5 carbapenemase-producing Pseudomonas aeruginosa clinical isolates in Japan due to clonal expansion of ST235. PLoS ONE. 2018;13:e0207134.
McCracken MG, Adam HJ, Blondeau JM, Walkty AJ, Karlowsky JA, Hoban DJ, et al. Characterization of carbapenem-resistant and XDR Pseudomonas aeruginosa in Canada: results of the CANWARD 2007-16 study. J Antimicrob Chemother. 2019;74:iv32–iv8.
Recio R, Villa J, González-Bodí S, Brañas P, Orellana M, Mancheño-Losa M, et al. Genomic analysis of ceftazidime/avibactam-resistant GES-producing sequence type 235 Pseudomonas aeruginosa isolates. Antibiot (Basel). 2022;11.
Yigit H, Queenan AM, Anderson GJ, Domenech-Sanchez A, Biddle JW, Steward CD, et al. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother. 2001;45:1151–61.
Estabrook M, Muyldermans A, Sahm D, Pierard D, Stone G, Utt E. Epidemiology of resistance determinants identified in Meropenem-Nonsusceptible Enterobacterales Collected as Part of a global Surveillance Study, 2018 to 2019. Antimicrob Agents Chemother. 2023;67:e0140622.
Hu H, Zhang Y, Zhang P, Wang J, Yuan Q, Shi W, et al. Bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing P. aeruginosa sequence type 463, associated with high mortality rates in China: a retrospective cohort study. Front Cell Infect Microbiol. 2021;11:756782.
Hu Y, Peng W, Wu Y, Li H, Wang Q, Yi H, et al. A potential high-risk clone of Pseudomonas aeruginosa ST463. Front Microbiol. 2021;12:670202.
Veetilvalappil VV, Manuel A, Aranjani JM, Tawale R, Koteshwara A. Pathogenic arsenal of Pseudomonas aeruginosa: an update on virulence factors. Future Microbiol. 2022;17:465–81.
Bradbury RS, Roddam LF, Merritt A, Reid DW, Champion AC. Virulence gene distribution in clinical, nosocomial and environmental isolates of Pseudomonas aeruginosa. J Med Microbiol. 2010;59:881–90.
Juan C, Peña C, Oliver A. Host and Pathogen biomarkers for severe Pseudomonas aeruginosa infections. J Infect Dis. 2017;215:44–s51.
Zhao Y, Chen D, Ji B, Zhang X, Anbo M, Jelsbak L. Whole-genome sequencing reveals high-risk clones of Pseudomonas aeruginosa in Guangdong, China. Front Microbiol. 2023;14:1117017.
Treepong P, Kos VN, Guyeux C, Blanc DS, Bertrand X, Valot B, et al. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin Microbiol Infect. 2018;24:258–66.
Del Barrio-Tofiño E, Sánchez-Diener I, Zamorano L, Cortes-Lara S, López-Causapé C, Cabot G, et al. Association between Pseudomonas aeruginosa O-antigen serotypes, resistance profiles and high-risk clones: results from a Spanish nationwide survey. J Antimicrob Chemother. 2019;74:3217–20.
Acknowledgements
We would like to express our gratitude to the staff of the Department of Clinical Laboratory at Central South University’s Xiangya Hospital for their assistance in collecting and identifying the P. aeruginosa isolates. We also thank Medjaden Inc. for scientific editing of this manuscript.
Funding
The Natural Science Foundation of Hunan Province (No. 2022JJ70084, No. 2023JJ30900) and the Science Foundation of Hunan Health Commission in Hunan province (No. 202111000066) supported this study.
Author information
Authors and Affiliations
Contributions
Study design: Jun Li and Mingxiang Zou. Study conduct: Fengjun Xia, Zhaojun Liu, and Mengli Tang. Data collection: Mengli Tang, Yuhan Wei, Fengjun Xia and Yubing Xia. Data analysis: Jun Li and Mingxiang Zou. Data interpretation: Yongmei Hu and Haichen Wang. Drafting manuscript: Jun Li. Revising manuscript content: Mingxiang Zou. Approving the final version of the manuscript: Jun Li and Mingxiang Zou. The final manuscript was read and approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12941_2024_674_MOESM1_ESM.docx
Supplementary Material 1: Figure S1. Heatmap of XDR-hvPA carrying resistance genes; Figure S2. Heatmap of XDR-hvPA carrying virulence genes
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, J., Tang, M., Liu, Z. et al. Molecular characterization of extensively drug-resistant hypervirulent Pseudomonas aeruginosa isolates in China. Ann Clin Microbiol Antimicrob 23, 13 (2024). https://doi.org/10.1186/s12941-024-00674-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-024-00674-7