
Abstract
Objective
This study aims to identify the most effective diagnostic method for distinguishing pathogenic and non-pathogenic Gram-negative bacteria (GNB) in suspected pneumonia cases using metagenomic next-generation sequencing (mNGS) on bronchoalveolar lavage fluid (BALF) samples.
Methods
The effectiveness of mNGS was assessed on BALF samples collected from 583 patients, and the results were compared with those from microbiological culture and final clinical diagnosis. Three interpretational approaches were evaluated for diagnostic accuracy.
Results
mNGS outperformed culture significantly. Among the interpretational approaches, Clinical Interpretation (CI) demonstrated the best diagnostic performance with a sensitivity of 87.3%, specificity of 100%, positive predictive value of 100%, and negative predictive value of 98.3%. CI’s specificity was significantly higher than Simple Interpretation (SI) at 37.9%. Additionally, CI excluded some microorganisms identified as putative pathogens by SI, including Haemophilus parainfluenzae, Haemophilus parahaemolyticus, and Klebsiella aerogenes.
Conclusion
Proper interpretation of mNGS data is crucial for accurately diagnosing respiratory infections caused by GNB. CI is recommended for this purpose.
Similar content being viewed by others
Introduction
Lower respiratory tract infections (LRTIs) are the most common type of infectious disease and the leading infectious cause of mortality worldwide [1]. Early recognition and clearance of pathogens are crucial in the management of LRTIs. Microbial culture, a traditional method for bacterial recognition, suffers from low sensitivity and requires a time-consuming process. It yields negative results in approximately 40–60% of patients with acute infections and sepsis [2, 3]. The failure to recognize pathogens with traditional approaches may lead to extended hospitalizations, readmissions, and increased mortality and morbidity [4]. Clearly, conventional methods are no longer sufficient to meet current clinical needs.
A variety of molecular diagnostic techniques have been employed for the rapid and precise identification of pathogens. These techniques encompass Polymerase Chain Reaction (PCR) and metagenomic Next-Generation Sequencing (mNGS), which are utilized either as stand-alone methods or in conjunction with traditional culture-based approaches. mNGS offers a significant advancement over PCR in pathogen identification. Unlike PCR, which requires prior knowledge of the pathogen and is limited by potential false negatives, mNGS enables broad-spectrum, non-targeted detection of multiple pathogens simultaneously [5,6,7,8]. This method is not only more sensitive and faster but also provides comprehensive pathogen characterization [9]. Its ability to identify a wide range of pathogens, including low-abundance ones, without prior genomic information, makes mNGS particularly effective in complex diagnostic scenarios, surpassing the capabilities of PCR in both efficiency and scope. Many clinical reports demonstrate the diagnostic utility of mNGS in detecting pathogens, especially in respiratory tract infections [10, 11]. Despite widespread acceptance of mNGS in clinical settings, a standardized method for interpreting its results is lacking, complicating the interpretation of mNGS results.
The clinical applications of mNGS are more common in patients with high severity. Gram-negative bacteria (GNB) are critical pathogens and are increasingly significant in LRTIs for these patients [12]. Furthermore, GNB are the most commonly identified pathogens in mNGS studies [13,14,15,16,17]. However, the respiratory tract often experiences high levels of GNB colonization, and there is no widely accepted method for interpreting these results. Distinguishing between colonization and actual infection can be challenging for clinicians. Given this situation, we sought to study the distribution of Gram-negative bacteria in LRTIs using mNGS and evaluate different approaches for interpreting mNGS results.
Materials and methods
Study population and ethical considerations
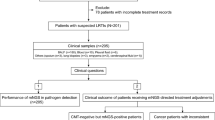
Between June 1, 2020, and November 1, 2022, a total of 583 patients with suspected pulmonary infection were enrolled in this study, originating from four medical institutions located in China. All enrolled patients were required to meet the following criteria: Firstly, they had to present with symptoms such as fever, cough, expectoration, shortness of breath, dyspnea, and abnormal imaging findings. Secondly, bronchoalveolar lavage (BALF) samples were collected concurrently for both metagenomic next-generation sequencing (mNGS) and culture to identify pathogens. Thirdly, the quality inspection and BALF sample testing process met the standards of mNGS. Patients with confirmed Gram-positive bacterial infections, fungal infections, or infections caused by other pathogens were excluded from the analysis. Demographic and baseline characteristics, clinical presentation, radiography and laboratory findings, treatment, and outcomes of the 583 patients were investigated for clinical diagnosis. The study was approved by the Institutional Review Board of the First Affiliated Hospital of Nanjing Medical University (approval no. 2022-SR-014).
Clinical groups of patients
Patients were stratified into three groups based on the presence of co-existing diseases. The Simple Pulmonary Infection Group: This group included patients without underlying diseases. The Immunosuppressed Group: This group comprised patients diagnosed with autoimmune diseases, post-splenectomy individuals, and/or those on long-term treatment with glucocorticoids, immunosuppressive agents, cytotoxic drugs, hematological malignancies, or those who had undergone chemotherapy in the last 6 months or solid organ transplantation. The Chronic Airway Disease Group: This group included patients with chronic bronchitis, bronchiectasis, or chronic obstructive pulmonary disease (COPD), but without immunosuppression.
Specimen collection and processing
Bronchoalveolar lavage fluid (BALF) for metagenomic next-generation sequencing (mNGS) and traditional culture was collected by an experienced clinician using standard procedures from patients with suspected pulmonary infection. Before the procedure, patients’ nasal or oral cavities were cleansed with normal saline. Dexmedetomidine was administered for sedation before the bronchoscopy, and local anesthesia with 2% lidocaine was applied during the examination. The electronic bronchoscope was used to examine all bronchi in detail, and any lesions found on a chest CT scan were brush-examined before BALF collection. The area for lavage was determined based on the lesion area found on the chest CT scan, with BALF collected from the right middle lobe or the subsegment of the left lingual lobe if scattered lesions were present.
Approximately 100 mL of sterile normal saline was injected into the target bronchus in batches at 37 °C, with the first 20 mL discarded to avoid contamination, and approximately 5 mL collected into sterile tubes. The BALF samples were then divided into aliquots for pathogen detection, with one aliquot inactivated (56 °C, 30 min) before nucleic acid extraction.
mNGS assay
(i) Nucleic Acid Extraction: Bronchoalveolar lavage fluid (BALF) samples were procured following standard protocols. DNA extraction employed the TIANamp Micro DNA Kit (Tiangen Biotech, Beijing, China), adhering to the manufacturer’s guidelines. Each batch included a no-template control (NTC) alongside clinical specimens. DNA quantification and quality assessment utilized Qubit and NanoDrop devices (Thermo Fisher Scientific). (ii) Library Preparation and Sequencing: The Hieff NGS C130P2 OnePot II DNA Library Prep Kit for MGI (Yeasen Biotechnology) facilitated DNA library construction, in line with manufacturer instructions. Post-preparation, libraries underwent Agilent 2100 qualification and were sequenced as 50 bp single-ends on DNBSEQ-200 (MGI Tech, China). Quality control (QC) criteria required over 18 ng of DNA post-library construction and a minimum of eight million raw reads. (iii) Bioinformatics Analysis: An in-house bioinformatics pipeline was employed for microorganism identification. Quality sequencing data were refined by removing low-quality reads, adapter contaminants, duplicates, and reads shorter than 36 bp. Human sequences were filtered out using bowtie2 software against the hs37d5 human reference genome. The residual data was aligned with the NCBI microorganism genome database using Kraken2, enabling the determination of the samples’ microbial composition.
Culture method
The BLAF was inoculated onto bacteriological media, including blood agar, chocolate agar, and blue agar plates, using sterile wire loops. Incubation was carried out at 35 °C for 48 h in a 5% CO2 atmosphere within a thermostatic incubator. Dominant colonies were then selected for bacterial identification using the VITEK2-Compact, an automated system from BioMerieux, France. Bacterial strains identified in the BALF at concentrations of ≥ 10^3 colony-forming units per milliliter were deemed causative pathogens.
Three interpretational approaches of mNGS
Simple Interpretation (SI): Gram-negative bacteria were identified by mNGS. Laboratory Interpretation (LI) [18, 19]: the parameter of gram-negative bacteria reached one of the following criteria: (i) relative abundance of pathogens detected by mNGS at the genus level was greater than or equal to 30%, regardless of the culture results, or (ii) the coverage rate scored 10-fold greater than that of any other microbes according to Langelier’s study. Laboratory interpretation serves as the positive standard for mNGS. Clinical Interpretation (CI): the bacteria identified in LI were further screened based on additional criteria. This microbe must have unambiguous literature evidence of its pulmonary pathogenicity, and the matched patient must have had risk factors for its infection.
Clinical diagnosis
Two physicians with expertise in managing infections (WKS and XSC) conducted an independent review of all patient medical records, as well as the results of culture and mNGS. The physicians initially determined whether patients had an infectious or noninfectious etiology. Following this, they identified the causative pathogens by evaluating a combination of clinical manifestations, laboratory tests, chest radiology, and microbiological tests (including culture and mNGS). Any disagreements between the two intensivists were resolved through in-depth discussion, and another senior physician (SLD) was consulted if consensus could not be reached.
Statistical analysis
Following the extracted data, 2 × 2 contingency tables were derived to determine sensitivity, and the McNemar test was used for discrete variables when appropriate. Differences between qualitative variables were assessed using the Fisher exact test, and the chi-square test was used to compare differences in positivity rates. Concordance was assessed using kappa statistics (kappa ≤ 0.4 low, kappa 0.41–0.6 fair, kappa > 0.6 good). Data analyses were performed using SPSS18 and GraphPad Prism7 software. P values smaller than 0.05 were considered statistically significant, and all tests were two-tailed.
Results
Patient characteristics
The study included a total of 583 patients (Fig. 1A), with a median age of 57 years (Table 1). Among these patients, 247 had at least one comorbidity. Chest CT scans revealed abnormalities in all patients, with solitary lesions being the most common radiologic finding upon admission (262, 45.4%). Bronchoscopy revealed abnormal secretions, mucosal abnormalities (such as erythema and edema), ulceration, and plague.

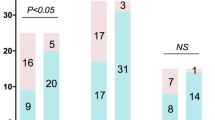
Detection performance of mNGS and culture. (A) The enrolled patients and research findings. (B) The comparison between mNGS and culture results in patients with positive findings. (C) The numbers and spectrum of bacteria detected with diffenent tools. Abbreviations: mNGS, metagenomic next-generation sequencing. H. parainfluenzae, Haemophilus parainfluenzae. P. aeruginosa, Pseudomonas aeruginosa. H. influenzae, Haemophilus influenzae. K. pneumoniae, Klebsiella pneumoniae. A. baumannii, Acinetobacter baumannii. H. parahaemolyticus, Haemophilus parahaemolyticus. K. aerogenes, Klebsiella aerogenes. S. maltophilia, Stenotrophomonas maltophilia
Comparison of gram-negative bacteria detection by mNGS and culture
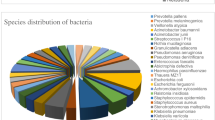
Figure 1B illustrates the positive findings in both mNGS and culture. mNGS identified GNB in 389 patients (66.7%), while conventional culture-based methods detected GNB in only 54 patients (9.2%). Moreover, mNGS detected a broader spectrum of Gram-negative bacteria (11 types) compared to culture (3 types). The top five GNB identified by mNGS were Haemophilus parainfluenzae (H. parainfluenzae, 45.1%), Pseudomonas aeruginosa (P. aeruginosa, 11.6%), Haemophilus influenzae (H. influenzae, 10.6%), Klebsiella pneumoniae (K. pneumoniae, 8.2%), Acinetobacter baumannii (A. baumannii, 7.5%). In contrast, culture identified P. aeruginosa (5.3%), K. pneumoniae (1.8%), and A. baumannii (2.0%), all of which were also recognized by mNGS (Fig. 1C).
Distribution of gram-negative bacteria across different groups
We utilized mNGS to investigate the bacterial distribution among patients with varying underlying diseases and immune backgrounds. As depicted in Fig. 2A, H. parainfluenzae was the most frequently identified species across all groups, with the highest prevalence in the simple pulmonary infection group. Additionally, P. aeruginosa was more commonly found in the chronic airway disease group. No specific bacterial distribution was observed in the immunosuppressed group.

The distribution and identification of putative pathogen. (A) The distribution of identified bacteria in different clinical population. (B) The identification of putative pathogen in different interpretational approaches. Abbreviations: h. parainfluenzae, Haemophilus parainfluenzae. P. aeruginosa, Pseudomonas aeruginosa. H. influenzae, Haemophilus influenzae. K. pneumoniae, Klebsiella pneumoniae. A. baumannii, Acinetobacter baumannii. H. parahaemolyticus, Haemophilus parahaemolyticus. K. aerogenes, Klebsiella aerogenes. S. maltophilia, Stenotrophomonas maltophilia
Evaluation of putative pathogens via three interpretational approaches
Putative pathogens were analyzed using three distinct methods: simple Interpretation (SI), laboratory Interpretation (LI), and clinical Interpretation (CI). SI identified 588 pathogens, significantly more than LI and CI, which identified 110 and 65 pathogens respectively (Fig. 2B). Notably, bacterial species differed between SI and CI, with H. parainfluenzae, Haemophilus parahaemolyticus (H. parahaemolyticus), and Klebsiella aerogenes (K. aerogenes) excluded by CI.
Diagnostic performance of MNGS across interpretational approaches
A total of 389 (SI), 99 (LI), and 62 (CI) subjects were diagnosed with Gram-negative bacterial infections. Through comprehensive assessment by two clinical physicians, the final count of pulmonary GNB infection was 71. Sensitivity and specificity of mNGS testing compared to culture were as follows: SI, 100% (95% CI 91.5–100%) and 36.6% (95% CI 32.5–40.8%); LI, 83.0% (95% CI 69.7–91.4%) and 89.6% (95% CI 86.6–92.0%); CI, 83.0% (95% CI 69.7–91.4%) and 96.6% (95% CI 94.7–97.9%) (Fig. 3A). A consensus analysis revealed significant agreement (p < 0.001) but weak concordance (Kappa = 0.095) between SI and culture. When comparing to the final result by clinical physicians, the Positive Predictive Value (PPV) and Negative Predictive Value (NPV) were 18.3% (95% CI 14.6–22.5%) and 100% (95% CI 97.5–100%) for SI, 62.6% (95% CI 52.3–71.9%) and 98.1% (95% CI 96.4–99.1%) for LI, and 100% (95% CI 92.7–100%) and 98.3% (95% CI 96.6–99.2%) for CI (Fig. 3B). Among the approaches, CI demonstrated the highest PPV.

Diagnostic performance of mNGS with different interpretation. (A) 2 × 2 Contingency tables for culture. (B) 2 × 2 Contingency tables for final clinical diagnosis. Abbreviations: mNGS, metagenomic next-generation sequencing. Sens, sensitivity. Spec, specificity. NPV, negative predictive value. PPV, positive predictive value
Impact of antibiotic exposure on pathogen detection
In our study, 474 of 583 patients (81.3%) were administered antibiotics prior to mNGS and culture testing, while the remaining patients were not exposed to any antibiotic treatment. Empirical antibiotic regimens covered 59.5% of the gram-negative bacteria (Fig. 4A). The detection rate of putative pathogens by mNGS was analyzed between the Uncovered and Covered groups across three interpretational approaches (Fig. 4B). The detection rates in all three approaches were unaffected by antibiotic coverage. However, antibiotic coverage significantly reduced the culture detection rate to 7.1%, compared to 11.9% in the uncovered group (p = 0.044).

Impact of Antibiotic exposure on pathogen detection. (A) The pathogen coverage of EAT. (B) The effect of antibiotic exposure on detection rate for putative pathogens. Abbreviations: EAT, empirical antibiotic therapy. mNGS, metagenomic next-generation sequencing. SI, simple interpretation. LI, laboratory interpretation. CI, clinical interpretation. No-Uncovered, no eat and uncovered
Discussion
Lower respiratory tract infections are a significant source of morbidity and mortality in hospitalized patients [20]. GNB are considered canonical pathogens and are becoming increasingly important [21, 22]. In this study, we investigated the diagnostic accuracy of mNGS in detecting pulmonary infections caused by GNB. Our results demonstrated that mNGS was more effective than culture, but its effectiveness depended on the correct interpretation method.
In our study, mNGS identified a greater variety and number of GNB, including fastidious bacteria that are difficult to grow in culture. Fastidious GNB such as H. influenzae, H. parainfluenzae, H. parahemolyticus, Eikenella corrodens, and Moraxella catarrhalis are often found in the human respiratory tract and can cause disease under certain conditions [23, 24]. The presence of these bacteria in mNGS data underscores the need for careful analysis to avoid incorrect diagnoses. To address this, we established three different interpretive approaches.
The diagnostic accuracy of mNGS varied depending on the method used. While the sensitivity of mNGS testing was comparable to that of culture testing across all three approaches, SI had very low specificity and weak concordance with culture. In contrast, CI demonstrated the highest specificity and best concordance with culture. When compared to the confirmed cases, all three approaches had high Negative Predictive Values (NPV). The Positive Predictive Value (PPV) varied, with SI having the lowest at 18.3%, LI in line with other studies at 62.6%, and CI the highest at 100% [25, 26]16. This variation was attributed to the fact that BALF from the respiratory tract often mixes with oral flora and colonizers, leading to false-positive mNGS results [27]. Only CI effectively eliminated all false positives.
Some microorganisms may be excluded by CI. For example, H. parainfluenzae was ranked first in detection in our study, consistent with previous reports [28, 29], but was identified as a putative pathogen only by SI and LI, not CI. Similar situations occurred with other bacteria, such as H. parahemolyticus [30]. Stenotrophomonas maltophilia (S. maltophilia), Eikenella corrodens and Moraxella catarrhalis were partially included by CI, reflecting their status as opportunistic pathogens that primarily infect specific populations with immunosuppression or structural lung disease [31,32,33]. Klebsiella aerogenes (K. aerogenes), an opportunistic pathogen often linked to severe infections in mechanically ventilated patients, was evaluated in this study [34,35,36,37]. The patient under investigation lacked pertinent risk factors and the bacterial sequence count fell short of our assessment criteria, leading to the exclusion of K. aerogenes from our considerations.
Core components in the WHO priority list [31, 38], such as P. aeruginosa, K. pneumoniae, A. baumannii, H. influenzae, and Escherichia coli, were identified as putative pathogens as long as they met the criterion of LI. These bacteria are responsible for a significant global clinical and epidemiological burden.
Unlike culture tests, mNGS is less affected by prior antibiotic usage, and it is widely accepted that antibiotics reduce the sensitivity of culture [13, 39]. In our study, the detection rate across all three approaches was unaffected by antibiotic coverage, although antibiotic coverage significantly decreased the detection rate in culture (11.9% vs. 7.1%, p = 0.044).
In our study, mNGS demonstrates efficacy in detecting respiratory GNB, markedly enhancing clinical pathogen diagnosis. Yet, the treatment of these pathogens encounters difficulties in drug selection, exacerbated by the rise of multifaceted drug-resistant strains. Our forthcoming research focuses on assessing the precision and dependability of mNGS in identifying resistance genes, thereby bolstering its theoretical foundation for clinical application in the detection of respiratory pathogens.
This study has limitations, including the need for a larger sample size, the lack of universally accepted criteria for mNGS, and the potential for subjective bias in the final diagnosis by clinical experts.
Overall, mNGS data require meticulous analysis to distinguish true pathogens from colonization. With proper interpretation, mNGS could become a valuable tool for precision diagnosis and tailored therapy for Gram-negative bacteria.
Data availability
The datasets used during the current study are available from the corresponding author on reasonable request.
References
Collaborators GL. Age-sex differences in the global burden of lower respiratory infections and risk factors, 1990–2019: results from the global burden of Disease Study 2019. Lancet Infect Dis. 2022;22(11):1626–47.
Fenn D, et al. Composition and diversity analysis of the lung microbiome in patients with suspected ventilator-associated pneumonia. Crit Care. 2022;26(1):203.
Liao WQ, et al. A molecular perovskite solid solution with piezoelectricity stronger than lead zirconate titanate. Science. 2019;363(6432):1206–10.
Glimaker M, et al. Adult bacterial meningitis: earlier treatment and improved outcome following guideline revision promoting prompt lumbar puncture. Clin Infect Dis. 2015;60(8):1162–9.
Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol. 2007;45(9):2761–4.
Reuwer AQ, et al. Added diagnostic value of broad-range 16S PCR on periprosthetic tissue and clinical specimens from other normally sterile body sites. J Appl Microbiol. 2019;126(2):661–6.
Diao Z, et al. Metagenomics next-generation sequencing tests take the stage in the diagnosis of lower respiratory tract infections. J Adv Res. 2022;38:201–12.
Gu W, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 2021;27(1):115–24.
Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20(6):341–55.
Peng JM, et al. Metagenomic next-generation sequencing for the diagnosis of suspected pneumonia in immunocompromised patients. J Infect. 2021;82(4):22–7.
Serpa PH, et al. Metagenomic prediction of antimicrobial resistance in critically ill patients with lower respiratory tract infections. Genome Med. 2022;14(1):74.
Cilloniz C, Dominedo C, Torres A. Multidrug resistant gram-negative Bacteria in Community-Acquired Pneumonia. Crit Care. 2019;23(1):79.
Miao Q, et al. Microbiological Diagnostic performance of Metagenomic Next-generation sequencing when Applied to Clinical Practice. Clin Infect Dis. 2018;67(suppl2):S231–40.
Jin X, et al. Improving suspected pulmonary infection diagnosis by Bronchoalveolar Lavage Fluid Metagenomic Next-Generation sequencing: a Multicenter Retrospective Study. Microbiol Spectr. 2022;10(4):e0247321.
Zhao Z, et al. Comparison of quality/quantity mNGS and usual mNGS for pathogen detection in suspected pulmonary infections. Front Cell Infect Microbiol. 2023;13:1184245.
Chen Y, et al. Application of Metagenomic Next-Generation sequencing in the diagnosis of pulmonary infectious pathogens from Bronchoalveolar Lavage Samples. Front Cell Infect Microbiol. 2021;11:541092.
Qu J, et al. Aetiology of severe community acquired pneumonia in adults identified by combined detection methods: a multi-centre prospective study in China. Emerg Microbes Infect. 2022;11(1):556–66.
Maxted AE, et al. Infant colic and maternal depression. Infant Ment Health J. 2005;26(1):56–68.
Li Y, et al. Application of metagenomic next-generation sequencing for bronchoalveolar lavage diagnostics in critically ill patients. Eur J Clin Microbiol Infect Dis. 2020;39(2):369–74.
Wang Y, et al. Association between Medicare expenditures and adverse events for patients with Acute Myocardial Infarction, Heart failure, or Pneumonia in the United States. JAMA Netw Open. 2020;3(4):e202142.
Brereton CJ, et al. Is gentamicin safe and effective for severe community-acquired pneumonia? An 8-year retrospective cohort study. Int J Antimicrob Agents. 2018;51(6):862–6.
Peto L, et al. The bacterial aetiology of adult community-acquired pneumonia in Asia: a systematic review. Trans R Soc Trop Med Hyg. 2014;108(6):326–37.
Jorgensen JH, Hindler JF. New consensus guidelines from the Clinical and Laboratory Standards Institute for antimicrobial susceptibility testing of infrequently isolated or fastidious bacteria. Clin Infect Dis. 2007;44(2):280–6.
Doern GV. Detection of selected fastidious bacteria. Clin Infect Dis. 2000;30(1):166–73.
Parize P, et al. Untargeted next-generation sequencing-based first-line diagnosis of infection in immunocompromised adults: a multicentre, blinded, prospective study. Clin Microbiol Infect. 2017;23(8):574e1–6.
Brown JR, Bharucha T, Breuer J. Encephalitis diagnosis using metagenomics: application of next generation sequencing for undiagnosed cases. J Infect. 2018;76(3):225–40.
Dickson RP, et al. The Microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504.
Musher DM, et al. Can an etiologic agent be identified in adults who are hospitalized for community-acquired pneumonia: results of a one-year study. J Infect. 2013;67(1):11–8.
Jain S, et al. Community-Acquired Pneumonia requiring hospitalization among U.S. adults. N Engl J Med. 2015;373(5):415–27.
Le Floch AS, et al. Haemophilus parahaemolyticus septic shock after aspiration pneumonia, France. Emerg Infect Dis. 2013;19(10):1694–5.
Tacconelli E, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18(3):318–27.
Hoyler SL, Antony S. Eikenella corrodens: an unusual cause of severe parapneumonic infection and empyema in immunocompetent patients. J Natl Med Assoc. 2001;93(6):224–9.
Murphy TF, Parameswaran GI. Moraxella catarrhalis, a human respiratory tract pathogen. Clin Infect Dis. 2009;49(1):124–31.
Alvarez-Marin R, et al. A prospective, multicenter case control study of risk factors for acquisition and mortality in Enterobacter species bacteremia. J Infect. 2020;80(2):174–81.
Davin-Regli A, Pages JM. Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front Microbiol. 2015;6:392.
Laupland KB, et al. Significant clinical differences but not outcomes between Klebsiella aerogenes and Enterobacter cloacae bloodstream infections: a comparative cohort study. Infection. 2023;51(5):1445–51.
Cui X, et al. A novel phage carrying capsule depolymerase effectively relieves pneumonia caused by multidrug-resistant Klebsiella aerogenes. J Biomed Sci. 2023;30(1):75.
Resistance EA. The burden of bacterial antimicrobial resistance in the WHO European region in 2019: a cross-country systematic analysis. Lancet Public Health. 2022;7(11):e897–e913.
Cheng MP, et al. Blood Culture results before and after Antimicrobial Administration in patients with severe manifestations of Sepsis: a diagnostic study. Ann Intern Med. 2019;171(8):547–54.
Funding
This study was supported by High-end Training Program for Teacher Professional Leaders in Higher Vocational Colleges in Jiangsu Province [grant number 2022-GRFX-026], Major project of Jiangsu Health Vocational College [grant number JKA-2021-002].
Author information
Authors and Affiliations
Contributions
W.Y.L. and Q.Z. designed the study and wrote the main manuscript text, Q.Q. also participated in writing the manuscript. M.Y.W. and Y.C.D. were responsible for preparing the figures. J.Z., Y.Z., Y.J., X.S.C., H.K., W.S., X.L., X.D.W., X.Y.X., and S.L.D. contributed to collecting and providing relevant clinical data. W.K.S. oversaw the overall planning and revised the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
The study was approved by the Institutional Review Board of the First Affiliated Hospital of Nanjing Medical University (approval no. 2022-SR-014). Before undergoing mNGS, patients consented to the anonymized use of their specimens, test results, and sequencing data for subsequent research, negating the need for additional consent in this study as no further personal data were collected.
Competing interests
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, W., Zhang, Q., Qian, Q. et al. Diagnostic strategy of metagenomic next-generation sequencing for gram negative bacteria in respiratory infections. Ann Clin Microbiol Antimicrob 23, 10 (2024). https://doi.org/10.1186/s12941-024-00670-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-024-00670-x