
Abstract
Objectives
Pseudomonas aeruginosa has intrinsic antibiotic resistance and the strong ability to acquire additional resistance genes. However, a limited number of investigations provide detailed modular structure dissection and evolutionary analysis of accessory genetic elements (AGEs) and associated resistance genes (ARGs) in P. aeruginosa isolates. The objective of this study is to reveal the prevalence and transmission characteristics of ARGs by epidemiological investigation and bioinformatics analysis of AGEs of P. aeruginosa isolates taken from a Chinese hospital.
Methods
Draft-genome sequencing was conducted for P. aeruginosa clinical isolates (n = 48) collected from a single Chinese hospital between 2019 and 2021. The clones of P. aeruginosa isolates, type 3 secretion system (T3SS)-related virulotypes, and the resistance spectrum were identified using multilocus sequence typing (MLST), polymerase chain reaction (PCR), and antimicrobial susceptibility tests. In addition, 17 of the 48 isolates were fully sequenced. An extensive modular structure dissection and genetic comparison was applied to AGEs of the 17 sequenced P. aeruginosa isolates.
Results
From the draft-genome sequencing, 13 STs were identified, showing high genetic diversity. BLAST search and PCR detection of T3SS genes (exoT, exoY, exoS, and exoU) revealed that the exoS+/exoU- virulotype dominated. At least 69 kinds of acquired ARGs, involved in resistance to 10 different categories of antimicrobials, were identified in the 48 P. aeruginosa isolates. Detailed genetic dissection and sequence comparisons were applied to 25 AGEs from the 17 isolates, together with five additional prototype AGEs from GenBank. These 30 AGEs were classified into five groups -- integrative and conjugative elements (ICEs), unit transposons, IncpPBL16 plasmids, Incp60512−IMP plasmids, and IncpPA7790 plasmids.
Conclusion
This study provides a broad-scale and deeper genomics understanding of P. aeruginosa isolates taken from a single Chinese hospital. The isolates collected are characterized by high genetic diversity, high virulence, and multiple drug resistance. The AGEs in P. aeruginosa chromosomes and plasmids, as important genetic platforms for the spread of ARGs, contribute to enhancing the adaptability of P. aeruginosa in hospital settings.
Similar content being viewed by others
Background
Pseudomonas aeruginosa is a common nosocomial pathogen [1] responsible for approximately 10% of all nosocomial infections [2]. Its extensive virulence and strong ability to evade antimicrobial therapeutic activity makes P. aeruginosa one of the most dangerous bacterial pathogens and is a major reason for its morbidity and mortality in immunocompromised individuals [3].
P. aeruginosa has a non-clonal epidemic population structure with 1392 sequence types (STs) defined worldwide (last accessed 5 May, 2022). The top 10 P. aeruginosa high-risk clones (ST235, ST111, ST233, ST244, ST357, ST308, ST175, ST277, ST654, and ST298) [4] are widespread in hospitals [5,6,7]. The success of P. aeruginosa high-risk clones likely reflects the acquisition and accumulation of antimicrobial resistance genes (ARGs) carried on accessory genetic elements (AGEs), given the large number s of distinct ARGs and AGEs detected in P. aeruginosa high-risk clones. AGEs, such as integrative and conjugative elements (ICEs), plasmids, and integrons, play a critical role in the accumulation and spread of ARGs in P. aeruginosa [10,11,12,13,14,15]. ICEs encode self-integration and self-conjugation modules, typically composed of an attL (attachment site at the left end), int (integrase), xis (excisionase), rlx (relaxase), oriT (origin of conjugative replication), cpl (coupling protein), a P (TivB)- or F (TivF)- type T4SS machinery (mating pair formation), and an attR (attachment site at the right end). Plasmid-mediated transmission of ARGs among P. aeruginosa has been widely reported, such as the IncP-2 plasmid carrying blaIMP−45 [16], the IncP-6 plasmid carrying blaKPC−2 [17], and the IncU plasmid carrying blaKPC−2 [18]. These plasmids harbor diverse resistance genes that can be horizontally transmitted in hospital settings. AGEs are often inserted within the genomes of P. aeruginosa by site-specific recombination events, facilitating the rapid spread of ARGs within communities and hospitals. Genetic analyses of AGEs from in-hospital P. aeruginosa contribute to our understanding of the transmission and infection dynamics of highly resistant bacteria isolates [7]. While many reports exist on AGEs and their associated ARGs among P. aeruginosa, few provide detailed modular structure dissection and evolutionary analysis of resistance genes in P. aeruginosa isolates from a single hospital.
In this study, we collected 48 P. aeruginosa isolates from a single Chinese hospital and conducted draft-genome sequencing to analyze the prevalence of STs and T3SS virulotypes. Moreover, we fully sequenced 17 of the 48 isolates to measure the prevalence of ARG-associated AGEs. From these fully sequenced isolates, we genetically dissected the modular structures of 25 AGEs and performed a detailed sequence comparison of these AGEs together with five prototype AGEs from GenBank. Data presented here provided a deeper understanding of the bioinformatics and epidemiology of ARGs and AGEs in P. aeruginosa from a single hospital.
Methods
Bacterial strains and identification
A total of 48 P. aeruginosa isolates (Table S2) were recovered from patients with nosocomial infections in a Chinese public hospital from 2019 to 2021. Bacterial antimicrobial susceptibility was tested by VITEK 2 Compact (BioMerieux, NC, USA), and interpreted as per the 2020 Clinical and Laboratory Standards Institute (CLSI) guidelines [19].
PCR identification
Primers of the exoT, exoY, exoS, and exoU genes were designed (data not shown, https://www.ncbi.nlm.nih.gov/tools/primer-blast). PCR was performed in a 30 µl volume using a ProFlex PCR System (Applied Biosystems, CA, USA). The reaction mixture (30 µl) consisted of 15 µl 2x Taq PCR Master Mix (MT201, Biomed, China), 0.3 µl each forward and reverse primers, 3 µl template DNA, and 11.4 µl ddH2O. The reaction mixture with no template DNA was included as a negative control. The cycling programs consisted of 1 × 94 °C for 3 min, 30 × 94 °C for 40 s, 60 °C (exoT, exoY, and exoU), 55°C (exoS) for 40 s and 72 °C for 1 min, and 1 × 72 °C for 5 min. After completion of all cycles, the PCR products were examined in 1.5% agarose (TSJ001, Tsingke, China) gel electrophoresis in the presence of TS-gelred (TSJ002, Tsingke, China) (7 µl/100ml gel). The stained gels were then visualized under UV light and photographed using iBringht 750 (Thermo Fisher, MA, USA).
Sequencing, and sequence assembly
All these 48 isolates were subjected to draft-genome sequencing using a paired-end library with an average insert size of 350 bp (range 150–600 bp) on a HiSeq sequencer (Illumina, CA, USA). In addition, 17 of them were subjected to complete-genome sequencing with a sheared DNA library with an average size of 15 kb (range 10–20 kb) on a PacBio RSII sequencer (Pacific Biosciences, CA, USA) (Table S1). The quality control analysis of sequencing data was conducted using NanoPack [20] and FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Sequence assembly and annotation were performed as described previously [21, 22].
Sequence annotation and comparison
Open reading frames (ORFs) and pseudogenes were predicted using RAST 2.0 [23] combined with BLASTP/BLASTN searches [24] against the UniProtKB/Swiss-Prot database [25] and the RefSeq database [26]. Annotation of resistance genes, AGEs, and other genome features was carried out using online databases including CARD [27], ResFinder [28], ISfinder [29], INTEGRALL [30], and Tn Number Registry [31]. Multiple and pairwise sequence comparisons were performed using MUSCLE 3.8.31 [32] and BLASTN, respectively. Gene organization diagrams were drawn in Inkscape 1.0 (https://inkscape.org/en/). Heatmaps were plotted with MeV 4.9.0 [33].
Multi-Locus sequence typing
The sequence types (STs) of P. aeruginosa isolates were identified according to the online P. aeruginosa MLST scheme (https://pubmlst.org/paeruginosa/). New STs discovered in our study were submitted to the curator of the database. GoeBURST was used for the MLST analysis, demonstrating the allelic relationship and prevalence of various STs. In this study, isolates were classified into the same clonal complex (CC) if six of the seven alleles were homologous. The PHYLOViZ 2.0 platform was used for data management and analysis of CCs, which were defined by related ST clusters exhibiting variation in a single locus (single locus variants-SLV) or in two loci (double locus variants-DLV) [34].
Conjugal transfer
Conjugal transfer experiments were carried out with rifampin-resistant P. aeruginosa ATCC 27,853 as a recipient, and the wild-type P. aeruginosa isolate as a donor. Three milliliters of overnight cultures of each of donor and recipient bacteria was mixed together, harvested, and resuspended in 80 mL of Brain Heart Infusion (BHI) broth (BD Biosciences). The mixture was spotted on a 1 cm2 hydrophilic nylon membrane filter with a 0.45 μm pore size (Millipore) that was placed on BHI agar (BD Biosciences) plate and then incubated for mating at 37 °C for 12–18 h. Bacteria were then washed from the filter membrane and spotted on Muller–Hinton (MH) agar (BD Biosciences) plates to select aadB-carrying ATCC 27,853 transconjugants. Transconjugant selection was done using 1500 mg/mL rifampin (for ATCC 27,853) together with 200 mg/L gentamicin (for aadB).
Results
Sample source and antimicrobial resistance profile of 48 clinical isolates
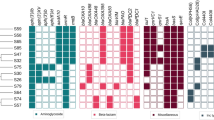
We collected 48 P. aeruginosa isolates from a hospital in the Henan Province of China. These isolates were mostly recovered from lung infection samples, such as sputum, bronchoalveolar lavage fluid, tracheal secretion, and airway lavage fluid (Fig. 1). Antimicrobial susceptibility/resistance profiles of these 48 isolates were described using nine different antimicrobials (Table S1). A high prevalence of multi-drug resistance (MDR) was observed, with 91.67% (n = 44) of isolates resistant to three classes of antibiotics.

Heatmap of specimens, virulence genes, and STs of the 48 isolates
Identification of 13 STs, T3SS virulotypes, and acquired ARGs from the 48 clinical P. aeruginosa isolates
We performed multi-locus sequence typing (MLST) analysis on 48 P. aeruginosa isolates and identified 13 STs presented in descending order by the number of isolates in which the ST was found: ST1976 (n = 13), ST244 (n = 11), ST235 (n = 9), ST270 (n = 3), ST274 (n = 2), ST1182 (n = 2), ST1710 (n = 2), ST277 (n = 1), ST292 (n = 1), ST357 (n = 1), ST782 (n = 1), ST3871 (n = 1), and ST1950 (n = 1) (Fig. 1 and Table S1).
ST3871 is a novel ST of P. aeruginosa. ST244, ST235, ST277, and ST357 are included in the worldwide top 10 P. aeruginosa high-risk clones [4] and were present in 45.8% of the 48 isolates. Thus, the P. aeruginosa isolates collected had high genetic diversity and many high-risk clones were prevalent in the hospital.
Based on BLAST search and PCR for the exoT, exoY, exoS, and exoU genes, we found exoT+/exoY+/exoS+/exoU- (34/48, 70.8%) was the predominant T3SS virulotype, (Fig. 1 and Table S1). The exoT and exoY genes were common in P. aeruginosa. All isolates that lacked the exoU gene carried the exoS gene. The widespread presence and diverse virulotypes of the T3SS in clinical isolates of P. aeruginosa suggest that the bacterial clones circulating and spreading were mostly those with high virulence.
At least 69 acquired ARGs involved in resistance to 10 different categories of antimicrobials were identified in the 48 P. aeruginosa isolates (Fig. S2). β-lactam-resistance genes, aminoglycoside-resistance genes, phenicol-resistance genes, and sulfonamide-resistance genes were distributed across all isolates.
Collection of 30 AGEs for sequence comparison
The 25 AGEs sequenced in this study and five additional reference/prototype AGEs (Tn6417, Tn1403, pRBL16, p60512-IMP, and pPA7790) from GenBank were compared in a detailed sequence comparison (Table 1). The 30 AGEs were classified into five groups: (i) integrative and conjugative elements (ICEs), (ii) unit transposons, (iii) IncpPBL16 plasmids, (iv) Incp60512−IMP plasmids, and (v) IncpPA7790 plasmids. Each group is discussed in detail below. At least 54 ARGs, involved in resistance to 11 different categories of antimicrobials and heavy metals were identified in these 30 AGEs (Fig. 2 and Table S2).

Heatmap of prevalence of resistance genes in the 30 AGEs. All data are provided in Table S2
Comparison of 11 Tn6417-related ICEs
Tn6417 (108.2 kb in length) was used as the reference ICE [35]. It was initially described in P. aeruginosa DHS01 [36]. The backbones of Tn6586, Tn7458, Tn7459, Tn7461, Tn6417, Tn7462, Tn7463, Tn7464, Tn7465, Tn7466, and Tn7482 varied in size from approximately 71.5 kb to nearly 95.1 kb, but all contained attL/R, int, cpl (coupling protein), rlx (relaxase), and an F-type T4SS gene set (Fig. S3). In addition, their backbones had at least three major modular differences: (i) the presence of a unique xerC–to–orf1068 region only in Tn6417; (ii) the presence of orf672, orf306, piL1–to–orf381, and orf3336–to–orf2514 regions in Tn7462, Tn7463, Tn7464, Tn7465, Tn7466, and Tn7482; and (iii) orf384–to–orf765 and orf1419–to–rlx regions from Tn7462, Tn7463, Tn7464, Tn7465, Tn7466, and Tn7482 displayed < 90% nucleotide identity with their counterparts in Tn6586, Tn7458, Tn7459, Tn7461, and Tn6417.
Each of the 11 ICEs carried a single accessory module, as follows for ICE (accessory module): Tn6586 (Tn6809), Tn7458 (Tn7404), Tn7459 (Tn7405), Tn7461 (Tn7460), Tn6417 (Tn6532), Tn7462 (In1815), Tn7463 (In2044), Tn7464 (In1836), Tn7465 (In1818), Tn7466 (In1979), and Tn7482 (In995) (Fig. S3).
In the first five ICEs, transposons were integrated at a site upstream of the ICE backbone gene orf582 and identified as derivatives of Tn6346. Tn6346, a prototype Tn3-family unit transposon originally identified in Achromobacter spp. AO22 [37], manifested as a hybrid of the core transposition module tnpAR–res from Tn5051 and the mer region from Tn501 (Fig. 3). The five Tn6346 derivatives differed from Tn6346 in two major aspects: (i) insertion of IS1071 at the same position within tnpA in all five Tn6346 derivatives and (ii) insertion of five different concise class 1 integrons (In159, In36, In1820, In717Tn7404, and In717Tn7405) into urf2.

Comparison of six Tn6346-related transposons. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). Numbers in brackets indicate nucleotide positions within the chromosomes of strains DHS01, NY5532, NY11254, F291007, and NY5523, respectively. The accession number of Tn6346 used as reference is EU696790.
In159 harbored a gene cassette array (GCA) with aadB as its only gene. In36 had the GCA dfrA16–aadA2. In1820, In717Tn7404, and In2044 in In717Tn7405 had the GCAs blaGES–aacA4’–gcuE15–aphA15 with different blaGES subtypes: blaGES−15, blaGES−5, and blaGES−1, respectively. (Fig. 3).
Each of the remaining six Tn6417-related ICEs had integrons integrated, including In1815, In2044, In1836, In1818, In1979, and In995, respectively (Fig. 4).These six integrons had the same insertion:Tn6758, a prototype Tn3-family unit transposon initially found in Achromobacter xylosoxidans X02736 [38]. Five of the Tn6758 interrupted the tnpA of IS6100, but in Tn7482, the insertion of Tn6758 plus a 4.0-kb region from p60512-IMP resulted in the truncation of IS6100.

Organization of In1815, In2044, In1836, In1818, In1979, and In995. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). Numbers in brackets indicate nucleotide positions within the chromosomes of strains NY5530, NY5507, NY5520, NY5510, NY11210, and NY5511, respectively
The GCAs blaOXA−10–aadA1e, blaOXA−10–aadA1e–qnrVC1–aacA4´, blaOXA−10–aadA1e–qnrVC1–aacA4´–aadB in In1815, In2044, and In1836 were incremental. This phenomenon demonstrates the process of accumulation and evolution of ARGs through integrons. In addition, the organization of In1836 and In1818 were highly consistent, with one difference in the position of aadB. In1979 had the GCA aadB–aadA2. In995 had the shortest GCA, which contained only blaIMP−10 (Fig. 4).
Comparison of seven Tn1403-related elements
Tn1403, a Tn3-family prototype unit transposon, was originally found in P. aeruginosa plasmid RPL11 [39] and displayed a backbone structure IRL–tnpAR–res–sup–uspA–dksA–yjiK–IRR, with the integration of two accessory modules In28 and Tn5393c into res and uspA, respectively [40]. We identified six chromosome-borne Tn1403 derivatives: Tn7483, Tn6846, Tn7484, Tn7485, T1403REcNY5525, and T1403REcNY5532 (Fig. 5). None of these T1403RE elements could be recognized as intact transposons due to the truncation of relevant core transposition modules. These six Tn1403 derivatives differed from Tn1403 in three major aspects: (i) integration of different class 1 integrons In1791, In1079, In458, In51, In1829, In44, and In167, instead of In28; (ii) absence of Tn5393c in Tn7485 and T1403REcNY5532; and (iii) insertion of the ISCR3–tetA(G)–cmlA9 unit into T1403REcNY5525 and T1403REcNY5532.

Comparison of Tn1403 and six related elements. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). Numbers in brackets indicate nucleotide positions within the chromosomes of strains NY13936, NY5524, NY5525, NY5523, NY5525, and NY5532, respectively. The accession number of Tn1403 used as reference is AF313472.
In1791 and In1079, two complex class 1 integrons, differed in variable region 1 (VR1) but were the same in VR2. The five concise class1 integrons In458, In51, In1829, In44, and In167 exhibited three additional major modular differences: (i) insertion of ISPa17 only in In51; (ii) the presence of different GCAs; and (iii) insertion of ΔtniTn402 only in In51.
Comparison of five IncpRBL16 plasmids
IncpRBL16 was originally identified and designated in P. citronellolis plasmid pRBL16 [41]. pRBL16 represented the IncpRBL16 reference plasmid as it was the complete IncpRBL16 backbone without any exogenous insertions. The four plasmids pNY5506-SIM, pNY11173-DIM, pNY5532-OXA, and pNY13932-PER were assigned to the IncpRBL16 group (Table 1), because each harbored a repA (replication initiation) gene sharing > 96% nucleotide identity to repAIncpRBL16 and contained a conserved IncpRBL16 backbone (> 98% nucleotide identity with > 90% coverage and highly similar gene organization).
The IncpRBL16 backbone genes or gene loci repAIncpRBL16 together with its iterons (replication), parB2–parAB (partition), cpl and several tivF genes (conjugal transfer), che (chemotaxis), pil (pilus assembly) and ter (tellurium resistance), were conserved among all five plasmids. Moreover, 2–20 kb deletions of backbone regions occurred in the four plasmids because of the insertion of exogenous genetic material.
Compared to pRBL16, each of the other four IncpRBL16 plasmids acquired 3–5 accessory modules integrated at various sites across the IncpRBL16 backbone (Fig. S4). Four transposon-like elements carried 5´-terminal regions (IRL–tnpAR–Δres) of Tn1403 and were named T1403REpNY5506 − SIM, T1403REpNY11173 − DIM, T1403REpNY5532 − OXA, and T1403REpNY13932 − PER. These elements displayed considerable modular diversification including: (i) the presence of diverse class 1 integrons with different collections of resistance genes; and (ii) the presence of various transposons with different collections of resistance genes (Fig. 6).

Comparison of Tn1403 and its four related derivatives. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). The numbers in brackets indicate nucleotide positions within the plasmids of strains NY5506, NY11173, NY5532, and NY13932 respectively. The accession numbers of Tn1403, Tn1548, Tn5053, Tn6855, and Tn6489a used as reference are AF313472, AF550415, L40585, MK347425, and CP017969, respectively
Comparison of five Incp60512−IMP plasmids
Four plasmids (pNY5535-IMP, pNY5530-IMP, pNY5520-IMP, and pNY5511-OXA) could not be assigned to any known Inc groups. A novel Incp60512−IMP group is thus proposed. p60512-IMP was used as the Incp60512−IMP reference plasmid. It was originally identified and described in P. aeruginosa 60,512 in our previous studies [42]. The four plasmids pNY5535-IMP, pNY5530-IMP, pNY5520-IMP, and pNY5511-OXA harbored not only homologous repA genes (100% nucleotide identity to repAIncp60512−IMP) together with its iterons but also similar backbone gene organization with p60512-IMP (> 99% nucleotide identity with > 98% coverage).
Seven accessory regions were inserted at three different sites within the backbones of the five Incp60512−IMP plasmids: (i) Tn7486, Tn7487, Tn7488, Tn6394, and Tn7494 plus adjacent Tn6758 were inserted at the site upstream of pine (DNA specific recombinase) in pNY5535-IMP, pNY5530-IMP, pNY5520-IMP, p60512-IMP, and pNY5511-OXA, respectively; (ii) Tn6758 was inserted into orf222 in pNY5535-IMP; and (iii) Tn5403 was inserted at the site upstream of Δorf498 in pNY5520-IMP (Fig. S5).
Tn7486, Tn7487, Tn7488, Tn6394, and Tn7494 were composed of an ISPa17 element (encoding a transposase, a recombinase, and a toxin/anti-toxin system) [43] together with In995, In1814, In1835, In992, or In1819, respectively, at its downstream end (Fig. 7). The connection of ISPa17 with each of these integrons might create the high levels of nucleotide identity between the IRL/IRR of ISPa17 and the IRi/IRt of these Tn402-associated class 1 integrons. In995, In1814, In1835, In992, and In1819 harbored GCAs blaIMP−10, blaIMP−10–aacA7, aacA7–blaIMP−10, aacA7–blaIMP−1, and blaOXA−10–aadA1–aadB–qacED1, respectively. The truncated tni modules of In995, In1814, In1835, and In992 are expected to impair the mobility of their respective plasmids.

Comparison of five ISPa17-based transposition units Tn6394, Tn7486. Tn7487, Tn7488, and Tn7494. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). Numbers in brackets indicate nucleotide positions within the plasmids of strains NY5535, NY5530, NY5520, and NY5511, respectively. The accession number of Tn6394 used as reference is MF344578
Comparison of two IncpPA7790plasmids
Plasmid pNY13932-OXA could not be assigned to any known Inc groups. A novel IncpPA7790 group was thus designated. Plasmid pPA7790, initially extracted from P. aeruginosa PA7790 [44], was used as the reference because it was the first sequenced plasmid carrying repAIncpPA7790 without any exogenous insertions. pNY13932-OXA was assigned to IncpPA7790 due to the repA sharing > 98% nucleotide identity to repAIncpPA7790 and conserved IncpPA7790 backbones (> 96% nucleotide identity with > 93% coverage; highly similar gene organization).
A comparative genomics analysis was applied to the two IncpPA7790 plasmids. The IncpPA7790 backbone genes included repABIncpPA7790 together with its iterons (replication), parAB (partition), rlx–traM–tivB–pilL genes (conjugal transfer), topA (DNA topoisomerase), radC (DNA repair protein), and ssb (single-stranded DNA-binding protein) (Fig. S6).
Compared with pPA7790, an 8.7-kb deletion in pNY13932-OXA was replaced by an 11.54-kb MDR region inserted at the site downstream of orf285. It was composed of a 3.7-kb Tn1722 remnant (IRL–tnpAR–res) plus a concise class 1 integron In2151 carrying GCA aadB–catB3–blaOXA−1–aadA1a, and one IS6100. The blaOXA−1, catB3, aadB, and aadA1a genes mediate resistance to β-lactam, phenicol, and aminoglycosides (Fig. 8).

Organization of 11.54-kb MDR region in pNY13932-OXA. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). Numbers in brackets indicate nucleotide positions within the plasmids of strains NY13932. The accession number of Tn1722 used as reference is X61367
Summary of newly identified or designated AGEs
We identified 33 new AGEs. Of these, 16 were directly integrated into chromosomes or plasmids, including nine ICEs: Tn7458, Tn7459, Tn7461, Tn7462, Tn7463, Tn7464, Tn7465, Tn7466, and Tn7482; four IS-based transposition units: Tn7486, Tn7487, Tn7488, and Tn7494; three unit transposons: Tn7483, Tn7484, and Tn7485. The remaining 17 AGEs were inner components of the above 16 and included three unit transposons (Tn7404, Tn7405, Tn7460) and 14 integrons (In2144, In1815, In2044, In1836, In1818, In1979, In1829, In1809, In1810, In1980, In1814, In1835, In1819, and In2151). Moreover, two novel Inc groups Incp60512−IMP and IncpPA7790 were designated in this study.
Transferability and antimicrobial susceptibility. pNY13932-OXA, selected to represent IncpPA7790, transferred from the wild-type isolate NY13932 into ATCC 27,853 through conjugation, generating the transconjugant ATCC 27,853/pNY13932-OXA. Both NY13932 and ATCC 27,853/pNY13932-OXA showed high resistance to gentamicin with a minimum inhibitory concentration (MIC) value ≥ 128 µg/mL owing to the production of aminoglycoside-2’-adenylyltransferase.
Discussion
The P. aeruginosa isolates collected are genetically diverse and belong to mostly high-risk clones, including ST235, ST244, ST277, and ST357. All ST235 isolates contained the exoU gene, as has been found in previous studies, which leads to poor clinical outcomes and early mortality [45, 46]. All ST244 isolates contained the exoS gene, which is also consistent with previous literature [4, 47, 48]. These observations provide evidence that clonal lineages are linked to specific T3SS virulotypes, and this linkage may play a major role in their intrinsic virulence levels.
In addition, all 13 exoU + isolates are resistant to β-lactams and 12 are resistant to fluoroquinolones. Nearly all (97.1%; 34/35,) exoS + isolates are resistant to β-lactams. Many studies have reported that high virulence isolates often also show multidrug resistance, especially fluoroquinolone resistance [49,50,51]. This correlation between T3SS virulotypes and antimicrobial resistance suggests a coevolutionary response leading to the formation of high-risk clones [8]. Thus, the identification of virulence genes facilitates the prediction of resistance patterns and guides antibiotic treatment.
Among the nine ST235 isolates in this study, class A β-lactamases, including multiple GES and PER variants, were detected most frequently. This observation is consistent with previously reported results [9, 52, 53]. OXA variants are the most widespread β-lactamases among the ST244 isolates of the current study, followed by PER-1 and TEM-116, which matches previous data indicating ST244 is associated with multiple different β-lactamases [54, 55]. The ST277 and ST357 isolates encode DIM and OXA enzymes, respectively. This suggests that a widely successful clone has more opportunities to encounter and acquire ARGs, which may explain the high prevalence and diversity of ARGs in P. aeruginosa high-risk clones.
The combination of T3SS-related virulence and antimicrobial resistance facilitates the emergence and spread of high-risk clones in hospitals to some extent, leading P. aeruginosa to become a leading cause of morbidity and mortality in cystic fibrosis patients and immunocompromised individuals. Our study illustrates the emerging trends and threats of P. aeruginosa high-risk clones in a hospital setting in the era of antimicrobial resistance.
Of the 54 ARGs found in the 30 AGEs in the current study, 49 are found in class 1 integrons. These integrons, which have captured various ARGs, are further integrated into ICEs and plasmids with intercellular mobility and relevant unit transposons with intracellular mobility. The remaining five ARGs were found in other various subregions: the gene aphA7 in the IS26-aphA7-IS26 unit, the genes armA, mph(E), and msr(E) in the Tn1548 related region and ter in the IncpRBL16 plasmid backbone region. Therefore, dissecting the genome structure of P. aeruginosa reveals that ARGs are always dependent on a variety of vectors for expression and spread.
Two groups of AGEs provide intuitive insight into the evolution of integrons: (i) the gradual accumulation of GCAs observed in In1815, In2044, In1836, and In1818 from Tn6417-related ICEs that show up as blaOXA−10–aadA1e, blaOXA−10–aadA1e–qnrVC1–aacA4´, blaOXA−10–aadA1e–qnrVC1–aacA4´–aadB, and aadB–blaOXA−10–aadA1e–qnrVC1–aacA4´ (Fig. 5); and (ii) the accumulation, transversion, and substitution of GCAs seen in In995, In1814, In1835, and In992 from Incp60512−IMP plasmids show up as GCAs blaIMP−10, blaIMP−10–aacA7, aacA7–blaIMP−10, and aacA7–blaIMP−1,respectively (Fig. 7). GCA sharing can generate diversity within members of a bacterial clone or species [56, 57], and a diverse collection of GCAs plays an important role in enabling P. aeruginosa to overcome the deleterious effects of antibiotics [58] and provides an added survival advantage in specific or challenging environments, such as hospitals.
Up to 150 subregions were identified in the 30 AGEs in this study, including 32 unit transposons, 39 integrons, 11 putative resistance units, 66 insertion sequences (ISs), a Tn1548 associated region, and a 4.5-kb truncated Tn2-rmtB region. These intact or residue subregions together constitute the diverse mosaic structure of AGEs that are created by complex transposition and homologous recombination, facilitate horizontal genetic exchange and promote the acquisition and spread of resistance genes.
Our conjugal transfer experiment confirms the IncpPA7790 plasmid pNY13932-OXA can to transfer between cells. Furthermore, the horizontal transferability of Tn6417-related ICEs and Incp60512−IMP plasmids was demonstrated in our previous studies [42, 59, 60]. Of note, Incp60512−IMP plasmids are transferred through electroporation rather than conjugation because they contain only four conjugal transfer genes (traA, traC, traD and cpl), which is insufficient for plasmid conjugation [43, 61].
All identified AGEs were initially described in P. aeruginosa, except the IncpRBL16 plasmid, which was originally identified in P. citronellolis. Tn6417-related ICEs have spread worldwide in numerous bacterial species, including P. aeruginosa [59, 60, 62], and, less frequently, in Klebsiella pneumoniae (accession number CP085740), Bordetella (accession number CP049957), Alcaligenes (accession number CP032153), Achromobacter xylosoxidans (accession number LN890477), Morganella morganii (accession number CP061513), Aeromonas caviae (accession number AP024402), Casimicrobium huifangae (accession number CP041352), and Delftia acidovorans (accession number CP000884). Tn1403-related regions have a wide range of hosts and are frequently identified in Pseudomonadaceae [40, 63,64,65], including P. putida (accession number CP045551), P. asiatica (accession number CP101701), P. juntendi (accession number CP091088), P. soli (accession number CP009365), P. fulva (accession number CP064945), P. migulae (accession number CP043572), P. shirazica (accession number CP063457), and also sporadically in Aeromonas media (accession number CP047963). The Incp60512−IMP plasmids have been most often identified in P. aeruginosa [42, 66] and occasionally in Achromobacter (accession number KM659090). By contrast, the IncpRBL16 plasmids and the IncpPA7790 plasmids have been identified only in P. aeruginosa to date (last accessed 9 September, 2022). In conclusion, the high prevalence of these five groups of AGEs in Pseudomonas isolates is surprising, suggesting that they might already be widespread in Chinese hospital and play a dominant role in the spread of ARGs.
Conclusion
The five groups of AGEs display a high-level diversification in modular structures, have complex mosaic natures, and carry many ARGs. Integration of these ARG-containing AGEs into P. aeruginosa likely contributes to the accumulation and dissemination of ARGs in P. aeruginosa, enhancing the adaptability of P. aeruginosa in hospital settings and its survivability under drug selection pressure. To better understand the prevalence of STs, T3SS virulotypes, and AGEs in hospital settings and to assess the clinical implications for humans, a comprehensive survey of P. aeruginosa is necessary.
Data availability
The complete chromosome sequences of NY5507,NY5510, NY5511, NY5530, NY11210, NY5520, NY5524, NY5532, NY5523, F291007, NY11254, NY5535, NY5506, NY13936, NY13932, NY11173, and NY5525 and those plasmids of pNY5507-IMP, pNY5511-OXA, pNY5530-IMP, pNY5520-IMP, pNY5520-KPC, pNY5532-OXA, pNY5535-IMP, pNY5506-SIM, pNY13932-OXA, pNY13932-PER, and pNY11173-DIM were submitted to GenBank under accession numbers CP096929, CP096932, CP096934, CP096946, CP096958, CP096937, CP096942, CP096950, CP096941, CP081345, CP096960, CP096953, CP096927, CP096964, CP096961, CP096956, CP096945, CP096930, CP096935, CP096949, CP096938, CP096939, CP096951, CP096955, CP096928, CP096962, CP096963, and CP096957, respectively.
Data Availability
The data used and analyzed during this study are available from the corresponding author on reasonable request.
Abbreviations
- ST:
-
Sequence type
- T3SS:
-
Type III secretion system
- ARG:
-
Antimicrobial resistance gene
- AGE:
-
Accessory genetic element
- ICE:
-
Integrative and conjugative element
- GCA:
-
Gene cassette array
- MDR:
-
Multi-drug resistance
- MLST:
-
Multi-locus sequence typing
- CLSI:
-
Clinical and Laboratory Standards Institute
References
Breidenstein EBM, de la Fuente-Núñez C, Hancock REW. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 2011;19(8):419–26.
De Oliveira DMP, Forde BM, Kidd TJ, Harris PNA, Schembri MA, Beatson SA, Paterson DL, Walker MJ. Antimicrobial resistance in ESKAPE pathogens. Clin Microbiol Rev 2020, 33(3).
Alvarez-Ortega C, Wiegand I, Olivares J, Hancock REW, Martínez JL. The intrinsic resistome of Pseudomonas aeruginosa to β-lactams. Virulence. 2011;2(2):144–6.
Del Barrio-Tofiño E, López-Causapé C, Oliver A. Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int J Antimicrob Agents. 2020;56(6):106196.
Papagiannitsis CC, Verra A, Galani V, Xitsas S, Bitar I, Hrabak J, Petinaki E. Unravelling the features of success of VIM-producing ST111 and ST235 in a greek hospital. Microorganisms 2020, 8(12).
Feng W, Sun F, Wang Q, Xiong W, Qiu X, Dai X, Xia P. Epidemiology and resistance characteristics of Pseudomonas aeruginosa isolates from the respiratory department of a hospital in China. J Glob Antimicrob Resist. 2017;8:142–7.
Rodrigues YC, Furlaneto IP, Maciel AHP, Quaresma AJPG, de Matos ECO, Conceição ML, Vieira MCdS, Brabo GLdC, Sarges EdSNF, Lima LNGC et al. High prevalence of atypical virulotype and genetically diverse background among Pseudomonas aeruginosa isolates from a referral hospital in the Brazilian Amazon. PLoS One 2020, 15(9):e0238741.
Subedi D, Vijay AK, Kohli GS, Rice SA, Willcox M. Association between possession of ExoU and antibiotic resistance in Pseudomonas aeruginosa. PLoS ONE. 2018;13(9):e0204936.
Ranellou K, Kadlec K, Poulou A, Voulgari E, Vrioni G, Schwarz S, Tsakris A. Detection of Pseudomonas aeruginosa isolates of the international clonal complex 11 carrying the blaPER-1 extended-spectrum β-lactamase gene in Greece. J Antimicrob Chemother. 2012;67(2):357–61.
Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, Koehrsen M, Rokas A, Yandava CN, Engels R, Zeng E, et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc Natl Acad Sci U S A. 2008;105(8):3100–5.
Roy Chowdhury P, Scott M, Worden P, Huntington P, Hudson B, Karagiannis T, Charles IG, Djordjevic SP. Genomic islands 1 and 2 play key roles in the evolution of extensively drug-resistant ST235 isolates of Pseudomonas aeruginosa. Open Biology 2016, 6(3).
Botelho J, Grosso F, Peixe L. Unravelling the genome of a Pseudomonas aeruginosa isolate belonging to the high-risk clone ST235 reveals an integrative conjugative element housing a blaGES-6 carbapenemase. J Antimicrob Chemother. 2018;73(1):77–83.
Botelho J, Grosso F, Quinteira S, Brilhante M, Ramos H, Peixe L. Two decades of blaVIM-2-producing Pseudomonas aeruginosa dissemination: an interplay between mobile genetic elements and successful clones. J Antimicrob Chemother. 2018;73(4):873–82.
van der Zee A, Kraak WB, Burggraaf A, Goessens WHF, Pirovano W, Ossewaarde JM, Tommassen J. Spread of carbapenem resistance by transposition and conjugation among Pseudomonas aeruginosa. Front Microbiol. 2018;9:2057.
Chew KL, Octavia S, Ng OT, Marimuthu K, Venkatachalam I, Cheng B, Lin RTP, Teo JWP. Challenge of drug resistance in Pseudomonas aeruginosa: clonal spread of NDM-1-positive ST-308 within a tertiary hospital. J Antimicrob Chemother. 2019;74(8):2220–4.
Zhang X, Wang L, Li D, Li P, Yuan L, Yang F, Guo Q, Wang M. An IncP-2 plasmid sublineage associated with dissemination of blaIMP-45 among carbapenem-resistant Pseudomonas aeruginosa. Emerg Microbes Infections. 2021;10(1):442–9.
Wang L-J, Chen E-Z, Yang L, Feng D-H, Xu Z, Chen D-Q. Emergence of clinical isolate Guangzhou-PaeC79 carrying crpP, blaGES-5, and blaKPC-2 in Guangzhou of China. Microb Drug Resist (Larchmont NY). 2021;27(7):965–70.
Tartari DC, Zamparette CP, Martini G, Christakis S, Costa LH, Silveira ACdO, Sincero TCM. Genomic analysis of an extensively drug-resistant Pseudomonas aeruginosa ST312 harbouring IncU plasmid-mediated blaKPC-2 isolated from ascitic fluid. J Global Antimicrob Resist. 2021;25:151–3.
WPCaLS I. CLSI: Performance standards for antimicrobial susceptibility testing. 30th ed. CLSI supplement M100. Clinical and Laboratory Stantardeds Institute 2020.
De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinf (Oxford England). 2018;34(15):2666–9.
Fu J, Zhang J, Yang L, Ding N, Yue L, Zhang X, Lu D, Jia X, Li C, Guo C et al. Precision methylome and in vivo methylation kinetics characterization of Klebsiella Pneumoniae. Genomics, Proteomics & Bioinformatics 2021.
Li C, Jiang X, Yang T, Ju Y, Yin Z, Yue L, Ma G, Wang X, Jing Y, Luo X et al. Genomic epidemiology of carbapenemase-producing Klebsiella pneumoniae in china. Genomics Proteom Bioinf 2022.
Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:8365.
Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, Madden TL, Matten WT, McGinnis SD, Merezhuk Y et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 2013, 41(Web Server issue):W29–W33.
Boutet E, Lieberherr D, Tognolli M, Schneider M, Bansal P, Bridge AJ, Poux S, Bougueleret L, Xenarios I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods In Molecular Biology (Clifton, NJ) 2016, 1374:23–54.
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–45.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–73.
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–4.
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34(Database issue):D32–6.
Moura A, Soares M, Pereira C, Leitão N, Henriques I, Correia A. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinf (Oxford England). 2009;25(8):1096–8.
Roberts AP, Chandler M, Courvalin P, Guédon G, Mullany P, Pembroke T, Rood JI, Smith CJ, Summers AO, Tsuda M, et al. Revised nomenclature for transposable genetic elements. Plasmid. 2008;60(3):167–73.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–8.
Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carriço JA. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics. 2012;13:87.
Zeng L, Zhan Z, Hu L, Jiang X, Zhang Y, Feng J, Gao B, Zhao Y, Yang W, Yang H, et al. Genetic characterization of a blaVIM-24-carrying IncP-7β plasmid p1160-VIM and a blaVIM-4-harboring integrative and conjugative element Tn6413 from clinical Pseudomonas aeruginosa. Front Microbiol. 2019;10:213.
Valot B, Rohmer L, Jacobs MA, Miller SI, Bertrand X, Hocquet D. Comparative genomic analysis of two multidrug-resistant clinical isolates of ST395 epidemic strain of Pseudomonas aeruginosa obtained 12 years apart. Genome Announcements 2014, 2(3).
Ng SP, Davis B, Palombo EA, Bhave M. A Tn5051-like mer-containing transposon identified in a heavy metal tolerant strain Achromobacter sp. AO22. BMC Res Notes. 2009;2:38.
Hu Y, Zhu Y, Ma Y, Liu F, Lu N, Yang X, Luan C, Yi Y, Zhu B. Genomic insights into intrinsic and acquired drug resistance mechanisms in Achromobacter xylosoxidans. Antimicrob Agents Chemother. 2015;59(2):1152–61.
Levesque RC, Jacoby GA. Molecular structure and interrelationships of multiresistance beta-lactamase transposons. Plasmid. 1988;19(1):21–9.
Stokes HW, Elbourne LDH, Hall RM. Tn1403, a multiple-antibiotic resistance transposon made up of three distinct transposons. Antimicrob Agents Chemother. 2007;51(5):1827–9.
Jiang X, Yin Z, Yuan M, Cheng Q, Hu L, Xu Y, Yang W, Yang H, Zhao Y, Zhao X, et al. Plasmids of novel incompatibility group IncpRBL16 from Pseudomonas species. J Antimicrob Chemother. 2020;75(8):2093–100.
Zhan Z, Hu L, Jiang X, Zeng L, Feng J, Wu W, Chen W, Yang H, Yang W, Gao B, et al. Plasmid and chromosomal integration of four novel blaIMP-carrying transposons from Pseudomonas aeruginosa, Klebsiella pneumoniae and an Enterobacter sp. J Antimicrob Chemother. 2018;73(11):3005–15.
Di Pilato V, Pollini S, Rossolini GM. Characterization of plasmid pAX22, encoding VIM-1 metallo-β-lactamase, reveals a new putative mechanism of In70 integron mobilization. J Antimicrob Chemother. 2014;69(1):67–71.
Nascimento APB, Ortiz MF, Martins WMBS, Morais GL, Fehlberg LCC, Almeida LGP, Ciapina LP, Gales AC, Vasconcelos ATR. Intraclonal genome stability of the metallo-β-lactamase SPM-1-producing ST277, an endemic clone disseminated in Brazilian hospitals. Front Microbiol 2016, 7:1946.
Peña C, Cabot G, Gómez-Zorrilla S, Zamorano L, Ocampo-Sosa A, Murillas J, Almirante B, Pomar V, Aguilar M, Granados A, et al. Influence of virulence genotype and resistance profile in the mortality of Pseudomonas aeruginosa bloodstream infections. Clin Infect Diseases: Official Publication Infect Dis Soc Am. 2015;60(4):539–48.
Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, Miyata S, Diggins LT, He J, Saucier M, Déziel E, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006;7(10):R90.
Kiyaga S, Kyany’a C, Muraya AW, Smith HJ, Mills EG, Kibet C, Mboowa G, Musila L. Genetic diversity, distribution, and genomic characterization of antibiotic resistance and virulence of clinical strains in Kenya. Front Microbiol. 2022;13:835403.
Weber C, Schultze T, Göttig S, Kessel J, Schröder A, Tietgen M, Besier S, Burbach T, Häussler S, Wichelhaus TA, et al. Antimicrobial activity of ceftolozane-tazobactam, ceftazidime-avibactam, and cefiderocol against multidrug-resistant Pseudomonas aeruginosa recovered at a german university hospital. Microbiol Spectr. 2022;10(5):e0169722.
Takata I, Yamagishi Y, Mikamo H. Association of the exoU genotype with a multidrug non-susceptible phenotype and mRNA expressions of resistance genes in Pseudomonas aeruginosa. J Infect Chemother. 2018;24(1):45–52.
Ali FA. Association between biofilm formation gene bla exoU and metallo and extend spectrum beta-lactamase production of multidrug resistance Pseudomonas aeruginosa in clinical samples. Comb Chem High Throughput Screen. 2022;25(7):1207–18.
Zarei O, Mahmoudi H, Bardbari AM, Karami P, Alikhani MY. Detection of virulence factors and antibiotic resistance pattern of clinical and intensive care unit environmental isolates of Pseudomonas aeruginosa. Infect Disord Drug Targ. 2020;20(5):758–62.
Larché J, Pouillot F, Essoh C, Libisch B, Straut M, Lee JC, Soler C, Lamarca R, Gleize E, Gabard J, et al. Rapid identification of international multidrug-resistant Pseudomonas aeruginosa clones by multiple-locus variable number of tandem repeats analysis and investigation of their susceptibility to lytic bacteriophages. Antimicrob Agents Chemother. 2012;56(12):6175–80.
Avetisian LR, Voronina OL, Chernukha MI, Kunda MS, Gabrielian NI, Lunin VG, Shaginian IA. Persistence of Pseudomonas aeruginosa strains in patients of Federal Scientific Center of Transplantology and Artificial Organs. Zhurnal Mikrobiologii, Epidemiologii I Immunobiologii 2012(4).
Mano Y, Saga T, Ishii Y, Yoshizumi A, Bonomo RA, Yamaguchi K, Tateda K. Molecular analysis of the integrons of metallo-β-lactamase-producing Pseudomonas aeruginosa isolates collected by nationwide surveillance programs across Japan. BMC Microbiol. 2015;15:41.
Vatcheva-Dobrevska R, Mulet X, Ivanov I, Zamorano L, Dobreva E, Velinov T, Kantardjiev T, Oliver A. Molecular epidemiology and multidrug resistance mechanisms of Pseudomonas aeruginosa isolates from bulgarian hospitals. Microb Drug Resist (Larchmont NY). 2013;19(5):355–61.
Hall RM, Collis CM, Kim MJ, Partridge SR, Recchia GD, Stokes HW. Mobile gene cassettes and integrons in evolution. Ann N Y Acad Sci. 1999;870:68–80.
Boucher Y, Labbate M, Koenig JE, Stokes HW. Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. 2007;15(7):301–9.
Partridge SR, Tsafnat G, Coiera E, Iredell JR. Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol Rev. 2009;33(4):757–84.
Yu T, Yang H, Li J, Chen F, Hu L, Jing Y, Luo X, Yin Z, Zou M, Zhou D. Novel chromosome-borne accessory genetic elements carrying multiple antibiotic resistance genes in Pseudomonas aeruginosa. Front Cell Infect Microbiol. 2021;11:638087.
Chen F, Wang P, Yin Z, Yang H, Hu L, Yu T, Jing Y, Guan J, Wu J, Zhou D. VIM-encoding Inc plasmids and chromosome-borne integrative and mobilizable elements (IMEs) and integrative and conjugative elements (ICEs) in Pseudomonas. Ann Clin Microbiol Antimicrob. 2022;21(1):10.
Bonnin RA, Poirel L, Nordmann P, Eikmeyer FG, Wibberg D, Pühler A, Schlüter A. Complete sequence of broad-host-range plasmid pNOR-2000 harbouring the metallo-β-lactamase gene blaVIM-2 from Pseudomonas aeruginosa. J Antimicrob Chemother. 2013;68(5):1060–5.
Zeng L, Zhan Z, Hu L, Jiang X, Zhang Y, Feng J, Gao B, Zhao Y, Yang W, Yang H, et al. Genetic characterization of a blaVIM-24-carrying IncP-7β plasmid p1160-VIM and a blaVIM-24-harboring integrative and conjugative element Tn from clinical Pseudomonas aeruginosa. Front Microbiol. 2019;10:213.
Vézina G, Levesque RC. Molecular characterization of the class II multiresistance transposable element Tn1403 from Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1991;35(2):313–21.
Zhang X, Wang L, Li D, Wang C, Guo Q, Wang M. Characterization of the novel plasmid-encoded MBL gene blaAFM-1, integrated into a blaIMP-45-bearing transposon Tn6485e in a carbapenem-resistant Pseudomonas aeruginosa clinical isolate. J Antimicrob Chemother. 2021;77(1):83–8.
Roy Chowdhury P, Merlino J, Labbate M, Cheong EYL, Gottlieb T, Stokes HW. Tn6060, a transposon from a genomic island in a Pseudomonas aeruginosa clinical isolate that includes two class 1 integrons. Antimicrob Agents Chemother. 2009;53(12):5294–6.
Feng W, Lv J, Wang H, Yao P, Xiong L, Xia P, Yuan Q, Sun F. The first report of the blaIMP-10 gene and complete sequence of the IMP-10-encoding plasmid p12NE515 from Pseudomonas aeruginosa in China. Acta Trop. 2022;228:106326.
Acknowledgements
All experiments and data analyses were done in Dr. Dongsheng Zhou’s laboratory.
Funding
This work was supported by the National Key Research and Development Program of China (Grant no: 2022YFC2303900) and the Key Project Plan of Medical Science Research in Hebei Province under Grant 20181072.
Author information
Authors and Affiliations
Contributions
Xiaofei Mu and Xinyue Li contributed equally to this work. Data analysis was performed by Xiaofei Mu and Xinyue Li. The first draft of the manuscript was written by Xiaofei Mu and all authors commented on earlier versions of the manuscript. Xiaofei Mu, Xinyue Li, Zhe Yin, Ying Jing, Fangzhou Chen, Huixia Gao, Zhi Zhang, Yueyang Tian, Huiqian Guo, Xiuhui Lu, Jiaqi He, Yali Zheng, Dongsheng Zhou, Peng Wang, and Erhei Dai all contributed to the study concept and design. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12941_2023_600_MOESM1_ESM.jpg
Supplementary Material 1: Figure S1. A minimum spanning tree of MLST among P. aeruginosa isolates. This analysis is displayed by the goeBURST diagram. The MLST database (http://pubmlst.org/paeruginosa, last accessed May 5th, 2022) contained 8251 isolates producing a total of 3912 sequence types (STs). A cluster of linked isolates corresponds to a CC (clonal complex). 2635 STs are divided into 492 CCs and 1277 STs The red boxes show the STs of the 48 isolates in this study. Blue rots are the others in the P. aeruginosa MLST database.
12941_2023_600_MOESM2_ESM.jpg
Supplementary Material 2: Figure S2. Heatmap of prevalence of resistance genes in the 48 clinical isolates.
12941_2023_600_MOESM3_ESM.jpg
Supplementary Material 3: Figure S3. Comparison of 11 Tn6417->related ICEs. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading in light blue denotes regions of homology (nucleotide identity ≥ 95%), light orange (nucleotide identity < 90%). The accession number of Tn6417 used as reference is EU696790.
12941_2023_600_MOESM4_ESM.jpg
Supplementary Material 4: Figure S4. Comparison of five IncpRBL16 plasmids pRBL16, pNY5506-SIM, pNY11173-DIM, pNY5532-OXA, and pNY13932-PER. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). The accession number of pRBL16 used as reference is CP015879.
12941_2023_600_MOESM5_ESM.jpg
Supplementary Material 5: Figure S5. Comparison of five Incp60512−IMP plasmids p60512-IMP, pNY5535-IMP, pNY5530-IMP, pNY5520-IMP, and pNY5511-OXA. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). The accession number of p60512-IMP used as reference is MF344578.
12941_2023_600_MOESM6_ESM.jpg
Supplementary Material 6: Figure S6. Comparison of two IncpPA7790 plasmids pPA7790 and pNY13932-OXA. Genes are denoted by arrows. Genes, AGEs, and other features are colored based on their functional classification. Shading denotes regions of homology (nucleotide identity ≥ 95%). The accession number of pPA7790 used as reference is CP015000.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Mu, X., Li, X., Yin, Z. et al. Abundant diversity of accessory genetic elements and associated antimicrobial resistance genes in pseudomonas aeruginosa isolates from a single Chinese hospital. Ann Clin Microbiol Antimicrob 22, 51 (2023). https://doi.org/10.1186/s12941-023-00600-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-023-00600-3