
Abstract
Background
The SIRT1 gene was associated with the lifespan in several organisms through inflammatory and oxidative stress pathways. Long-term air particulate matter (PM) is detrimental to health through the same pathways.
Methods
We used the Chinese Longitudinal Healthy Longevity Survey (CLHLS) to investigate whether there is a gene-environment (G × E) interaction of SIRT1 and air pollution on mortality in an older cohort in China. Among 7083 participants with a mean age of 81.1 years, we genotyped nine SIRT1 alleles for each participant and assessed PM2.5 concentration using 3-year average concentrations around each participant’s residence. We used Cox-proportional hazards models to estimate the independent and joint effects of SIRT1 polymorphisms and PM2.5 exposure on all-cause mortality, adjusting for a set of confounders.
Results
There were 2843 deaths over 42,852 person-years. The mortality hazard ratio (HR) and 95% confidence interval (CI) for each 10 μg/m3 increase in PM2·5 was 1.08 (1.05–1.11); for SIRT1_391 was 0.77 (0.61, 0.98) in the recessive model after adjustment. In stratified analyses, participants carrying two SIRT1_391 minor alleles had a significantly higher HR for each 10 μg/m3 increase in PM2.5 than those carrying zero minor alleles (1.323 (95% CI: 1.088, 1.610) vs. 1.062 (1.028, 1.096) p for interaction = 0.03). Moreover, the interaction of SIRT1 and air pollution on mortality is significant among women but not among men. We did not see significant relationships for SIRT1_366, SIRT1_773, and SIRT1_720.
Conclusion
We found a gene-environment interaction of SIRT1 and air pollution on mortality, future experimental studies are warranted to depict the mechanism observed in this study.
Similar content being viewed by others
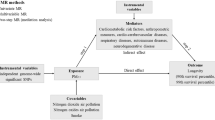


Introduction
Sirtuin 1 (SIRT1) is a gene well-documented to be associated with aging and longevity [1]. The SIRT1 gene encodes proteins related to mammalian nicotinamide adenine dinucleotide (NAD+) dependent histone deacetylase. The sirtuin pathway is implicated in regulating lifespan in many model organisms such as yeast, Caenorhabditis elegans, and rodents through several biopathways, including anti-inflammation, regulation of metabolism, hypoxic responses, and circadian rhythms [2, 3]. In humans, SIRT1 improves healthy aging and affects human life expectancy through its protective role in various biological processes related to age-related diseases ranging from metabolic disorders, cellular senescence, cardiac aging, oxidative stress, neurodegeneration, inflammatory signaling, and placental cell survival [1, 4]. A chronic, low-grade inflammation level characterizes human aging. Ambient air pollution is involved in developing degenerative diseases inclusive of aging arteries and brains through shared inflammatory pathways [5]. Biologically, SIRT1 can modulate inflammatory genes such as NF-κB and NLRP3 and further leads to delayed onset of age-related symptoms and pathologies [6, 7]. To date, quite a few observational studies linked SIRT1 (i.e., SNP rs7896005, rs12778366, rs4746720) to long-term survival and longevity in the human populations, though these findings were restricted by either small population size or a cross-sectional cohort study design [8,9,10,11].
Air pollution is a significant detrimental environmental risk to human health, attributing to one in every nine deaths annually, and pollution mitigation is a top priority in the UN sustainable development goal (SDG) agenda [12]. The disease burden due to air pollution in many places is likely to continue to exacerbate, with many epidemiological studies continuing to document morbidities and mortalities [13, 14]. According to an analysis of the trend of air pollution in Global Burden of Disease Study, exposure to ambient fine particulate matter (PM2.5: particulate matter (PM) with aerodynamic diameters< 2.5 μm) is the fifth leading risk factor for death, accounting for 7.6% of total global deaths and 4.2% of global DALYs, with China accounting for a large share of these burdens [15]. China’s coal-based energy-intensive development path has led to a steep increase in emissions of PM2.5 and other pollutants [16], estimated to have led to 1.6 million deaths from heart and lung diseases or stroke, approximately accounting for one in six premature deaths in China [17]. Thus, the comprehensive health impact of air pollutions in Low- and Middle-income countries such as China remains one of the major health issues, and further attention, as well as joint approaches, are needed.
Increasingly, we have seen that air pollution does not affect everyone equally, with some populations more susceptible to its detrimental effects. Genetic susceptibility is likely to play a vital role in response to air pollution [18]. Inflammation and oxidative stress are documented to play a role in the mechanistic pathways, including nuclear factor kappa B cells (NF-kB) signaling, Krüppel-like Factor 2 (Klf2) mediated immune response, nuclear factor E2-realest factor 2 (Nrf2)-mediated oxidative stress response, NLR family pyrin domain containing 3 (NLRP3) inflammasome activation, glutathione metabolism, coagulation system, endogenous reactive oxygen species (ROS) production, and other cytokines signaling, between air pollution exposures and adverse health outcome including mortality [19]. Notably, biological studies indicated that SIRT1 can be modulated through most of those pathways, including NF-kB [20]. Klf2 [21], Nrf2 [22], NLRP3 [7], and ROS [23]. Additionally, previous experimental studies observed that air pollution and SIRT1 have an interactive effect on pulmonary diseases [24, 25], cardiovascular diseases [26]. Although the associations between exposure to air pollutants and the SIRT1 gene were observed in in vitro studies, less effort has been put forth in the investigation on a population level, especially for the vulnerable older adults. Additionally, epidemiologic evidence and rodent model showed the effect of SIRT1 on disease and longevity vary by inflammatory levels [11, 27] and can be a double-edged sword: lower levels of SIRT1 (short term exposure of toxicants) accentuate acute inflammation-related autotoxicity by increasing NFκB RelA/p65 activity, but prolonged upgrading in SIRT1 in later inflammation are associated with immunosuppression and increased mortality [28]. Given the SIRT1 and PM2.5 shared several common biological pathways containing inflammation and oxidative stress on mortality and are tended to interplay with each other [26, 29], we hypothesized a potential synergistic effect between PM2.5 exposure and SIRT1 polymorphisms on mortality.
To test our hypotheses, we used a nationally representative cohort of individuals aged 65 and older from the Chinese Longitudinal Healthy Longevity Study (CLHLS). First, we aim to estimate the independent and joint effects of SIRT1 polymorphisms and PM2.5 exposure on all-cause mortality. Second, we take advantage of the sample size to study the interaction effect of SIRT1 and air pollution on mortality. Considering the “male-female health-survival paradox” in longevity research, there are marked sex differences in genetic associations with longevity; thus, we also assessed the three-way interaction by sex.
Method
Study population
We used data from the Chinese Longitudinal Healthy Longevity Study (CLHLS), which are publicly available from Peking University Open Research Data (https://opendata.pku.edu.cn/dataverse/CHADS). The baseline and follow-up surveys were conducted in 1998, 2000, 2002, 2005, 2008–2009, 2011–2012, and 2014 in a randomly selected half of the counties and cities in 23 out of 31 provinces in China. The study was the first national longitudinal survey on the determinants of healthy aging among the oldest old individuals in China. Details of descriptions of the CLHLS including the rationale and design have been described previously [30]. With 631 cities and counties randomly selected as the sample sites, the population in 23 selected provinces represents 85% of the total population in China (Fig. 1). CLHLS was approved by the Institutional Review Board, Duke University (Pro00062871), and the Biomedical Ethics Committee, Peking University (IRB00001052–13074). All participants or their legal representatives signed written consent forms to participate in the baseline and follow-up surveys. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guidelines.

Distribution of the study population from participants of the Chinese Longitudinal Healthy Longevity Survey (CLHLS)
There was a total of 34,394 participants in 5 waves of CLHLS, recruited from 2000 to 2011. We excluded 25,220 participants without available SIRT1 genotypic data or did not meet the selection criteria of sample filtering, 545 participants aged 64 or younger, 212 participants with missing PM2.5 values, 567 participants were not of Han ethnicity (according to the ID card or household registry) or having a missing value in covariates; 767 participants lost to follow-up at the first follow-up survey. The samples were required to meet 3 selection criteria: (1) a genotype calling rate > 90%; (2) no existing population stratification according to multidimensional scaling (MDS) procedure implemented in PLINK v1.07, based on which individuals deviating from the main population cluster were removed; and (3) no inclusion of duplicates or first-degree relatives when evaluating pairwise through identity by descent (IBD). Accordingly, the final sample that met inclusion criteria for this study was 7083 participants (Fig. S1). The sample consisted of 3677 women and 3406 men; 3272 participants were 65 to 79 years of age, 1840 were 80–89 years of age, 1305 were 90–99 years of age, and 667 were 100 years of age or older. To test the possibility of potential selection bias, gender, age, and residence were compared between participants who lost to follow-up (767 participants) or not (7083 participants) at the first follow-up survey; the significant difference for ages (86.0 vs 81.1) and residence (rural: 53.8% vs 66.7%) between two groups, while no significant difference for sex (female: 52.7% vs 51.9%).
Assessment of PM2.5 exposure
We estimated ground-level concentrations of PM2.5 from the Atmospheric Composition Analysis Group based on participants’ residential addresses [31]. It combines remote sensing from National Aeronautics and Space Administration’s Moderate Resolution Imaging Spectroradiometer, Multiangle Imaging Spectroradiometers, and Sea-viewing Wide field-of-view Sensor satellite instruments; vertical profiles derived from the GEOS-Chem chemical transport model; and calibration to ground-based observations of PM2.5 using geographically weighted regression. Annual PM2.5 estimates were calculated from 2000 to 2014, at 1 km × 1 km spatial resolution, which was the longest and the highest resolution exposure dataset available [32, 33]. Additionally, our estimations were highly consistent with out-of-sample cross-validated concentrations from monitors (R2 = 0·81) and another exposure dataset in China (R2 = 0·79) [31]. A previous study found that the three-year average PM2.5 before death or the end of the study had the strongest association with mortality among old adults in China [32]. Therefore, we used a three-year average PM2.5 to reflect ambient air pollution in this study.
The SIRT1 genotypes
We customize an SNP chip containing 27,656 selected longevity and disease-related SNPs for targeted genotyping [34]. We selected candidate SNPs from the public database of the National Center for Biotechnology Information (NCBI; http://www-ncbi-nlm-nih-gov/gene/23411) to cover the SIRT1 gene region in equally spaced intervals [35]. The minor allele frequencies (MAF) of the polymorphisms are required to be > 10% [36]. The selected and genotyped SIRT1 SNPs were rs12778366 (promoter), rs3758391 (promoter), rs2273773 (exon), rs2236319 (intron), rs1885472 (intron), rs7069102 (intron), rs10823112 (intron), rs3818291 (intron), and rs4746720 (intron), which have covered major functional SNPs in SIRT1 gene. Among the 9 SNPs, SIRT1_366 (rs12778366), SIRT1_391 (rs3758391), SIRT1_773 (rs2273773), and SIRT1_720 (rs4746720) were annotated and regarded as tagging SNPs due to the high linkage disequilibrium, which were used as proxies for the rest of 5 SNPs in the analysis (Table S1). The Hardy-Weinberg equilibrium of the 9 SIRT1 SNPs was tested with the GENEPOP package (version 1.2). To determine the SIRT1 carrier, we coded the genotype based on the minor allele number [37]. For each SNP, three different inheritance models (additive, dominant, and recessive models) were tested. In the additive model, the genotype that contains zero, one, or two copies of minor allele was coded as 0, 1 or 2; In the dominant model, the genotype that contains at least one copy of minor allele was coded as 1, otherwise, it was coded as 0; In the recessive model, the genotype that contains two copies of minor allele was coded as 1 and otherwise it was coded as 0.
All-cause mortality
The primary outcome was all-cause mortality. Mortality information was obtained from the follow-up survey done in 2011 and 2014. The date of death would be validated by death certificates when available - otherwise, the close family member’s report was used.
Covariates
The investigator chose covariates that may be potential confounders between exposures and outcomes or predictors of outcomes based on a prior knowledge and a causal framework. All self-reported information was collected through face-to-face home interviews by trained research staff members. Interviewees were encouraged to answer as many questions as possible. If they were unable to answer questions, a close family member or another proxy, such as a primary caregiver, provided answers. We included baseline age, gender, marital status, residence, education, occupation, smoking status, drinking status, physical activity, and the wave of the first interview as covariates. We classified marital status into two categories: currently married and living with spouse as married, and widowed/separated/divorced/never married/married but not living with spouse as not married. We divided residences into urban and rural areas based on governmental administrative categories. We used the schooling year to evaluate education level. We categorized the occupation into two groups: professional and technical personnel, governmental, institutional, or managerial personnel as non-manual, and agriculture, forest, animal husbandry, fishery worker, industrial worker, and others as manual. We divided the regular exercise, smoking, and alcohol drinking status into three categories: “Current”, “Former”, and “Never”. For example, participants were asked, “do you do exercise regularly at present (planned exercise like walking, playing balls, running and so on)?” and/or “did you do exercise regularly in the past?”. We defined the regular exercise status as “Current” for participants who answered “Yes” to the first question, “Former” for those who answered “No” to the first question and “Yes” to the second question, and “Never” for those who answered “No” to both two questions. We categorized the participants into two geographical regions: South China (Guangdong, Guangxi, Hainan, Chongqing, Sichuan, Anhui, Hubei, Fujian, Jiangxi, Jiangsu, Shanghai, and Zhejiang) and North China (Beijing, Shandong, Heilongjiang, Jilin, and Liaoning, Hebei, Henan, Shanxi, Tianjin, and Shaanxi).
Statistical analysis
We used the cox proportional hazard model for every SIRT1 SNP and three-year average PM2.5 (as continuous variables or category variables by quartiles) separately to evaluate their single effect on mortality. We added the interaction term of SNP and three-year average PM2.5 in the cox model to investigate the interaction of SIRT1 and three-year average PM2.5. The genotype can be defined as low-risk genotype (G0) and high-risk genotype (G1) based on the genotypes’ hazard ratio for mortality risk. The three-year average PM2.5 was discretized into two categories as low PM2.5 exposure (E0) versus high PM2.5 exposure (E1) using the median of three-year average PM2.5 as the cut-off point. The gene effect in different environment exposure and environmental effect among participants with different genotypes can be evaluated by comparing the difference among the four categories (G0 × E0 stands for low-risk genotype under low PM2.5 exposure, G0 × E1 stands for low-risk genotype under high PM2.5 exposure, G1 × E0 stands for high-risk genotype under low PM2.5 exposure, and G1 × E1 stands for high-risk genotype under high PM2.5 exposure) of the combined term. To investigate the gender-specific G × E effect, we adopt an integrated statistical model of three-way interaction to assess varied effect magnitude in the hazard ratio of mortality between those who have different combinations of sex, SIRT1 genotypes, and exposure to PM2.5 [38].
We measured the survival time in months from the first interview date to the recorded death date or last interview date. Our statistician adjusted all models for age, gender, marital status, residence area, education, occupation, smoking status, drinking status, and physical activity. We calculated hazard ratios (HRs) and 95% CIs to indicate the effect magnitude of SIRT1 and PM2.5 on mortality. We analyzed for effect modification by potential modifier variables, then did stratified analyses by sex, urban or rural residence, financial status, smoking status, and North or South geographical regions.
We did additional analyses to verify the results. We compared baseline characteristics between the included participants and all participants in the five waves of CLHLS to assess the sample’s representativeness. We adjusted the regression models by using more informative covariates and by excluding covariates with missing values. We also plotted the geographical distribution of the included participants.
We used R 3.6.1 (R Foundation for Statistical Computing) and SAS university edition to perform all the analyses. All p values were from 2-sided tests, and results were deemed statistically significant at p < .05 for all analyses.
Results
Among the 7083 participants, 48.1% were men, and 51.9% were women, with a mean (SD) age of 81.1 (11.5) years. More than half of the participants were illiterate (57.1%), residents in rural (66.7%), responded to no engage in regular exercise (64.5%), never smoked (64·0%), and never drank alcohol (68.5%) (Table S2). The mean 3-year PM2.5 was 51.0 μg/m3 (13.5), with populations in rural regions and southern regions experiencing lower ambient air pollution. Four tag SNPs (SIRT1_366, SIRT1_391, SIRT1_773, and SIRT1_720) were used to represent the 9 SIRT1 SNPs as they had high Linkage Disequilibrium (r2 ≥ 0.975; Fig. S2). The distributions of the tag SNPs of SIRT1 are similar across varied baseline characteristics, including sex, age, and education (Table S2), indicating SIRT1 SNPs were randomly distributed across population characteristics. While participants who were older, illiterate, responded of no regular exercise, reside in northern China tended to live in areas with higher PM2.5 exposure (Table S2). The final sample has a lower average of age, more likely to be men and rural residents, had a higher proportion of currently married status, than those who were excluded; no significant difference has been detected regarding the education level, exercise habit, smoking and drinking status between the two groups (Table S3).
The mean follow-up time (SD) was 6.1 (3.5) years (range: 0 to 14 years). During the 42,852 person-years of follow-up, we saw 2843 mortality events (40.1%) between 2002 and 2014. The mortality rate was 6.6 per 100 person-years for our entire study population. In the additive model, SIRT1_391, SIRT1_472, and SIRT1_102 minor allele carriers had lower mortality than their counterparts. The recessive model showed similar results to the addictive model, yet no statistical significance was observed in the dominant model (Table 1). We also did not see a significant association of SIRT1_366, SIRT1_773, and SIRT1_720 with mortality.
In the fully adjusted model, the all-cause mortality hazard ratio (HR) and 95% CI for each 10 μg/m3 increase in PM2.5 was 1.082 (1.053, 1.111). We also conducted our analysis by quartiles to account for a possible nonlinear dose-response relationship between PM2.5 exposure and risk of longevity. Compared with the participants who resided in areas with the lowest quartile of PM2.5, the HR of mortality for the 2nd quartile, 3rd quartile, and 4th quartile were 1.203 (1.078, 1.343), 1.431 (1.287, 1.593), 1.297 (1.167, 1.442), respectively (Table 2). In the stratified analyses by tags of SIRT1 genotypes, participants who carry zero or one SIRT1_391 minor allele had an HR of 1.078 (95% CI: 1.049,1.107) on mortality for each 10 μg/m3 increase in PM2.5, while the HR was 1.336 (95% CI: 1.079, 1.653) in those who carry two SIRT1_391 minor alleles (data not shown).
According to an addictive model with interaction terms of SIRT1 SNPs and PM2.5, the detrimental effect of PM2.5 exposure on mortality for the participants carrying two SIRT1_391 minor alleles was stronger than those carrying zero alleles. We saw similar results in the SIRT1_472 and SIRT1_102, of which were in LD with SIRT1_391. For instance, the test for the interaction between SIRT1_391 and PM2.5 in additive models revealed a significant gene-environment interaction (p for interaction = 0.03). The HR of 10-μg/m3 3-year average PM2.5 on mortality for the participants carrying two SIRT1_391 minor alleles was e(0.06 + 0.220) = 1.323 (95% CI: 1.088, 1.610) and for those carrying zero minor allele would be e0.06 = 1.062 (1.028, 1.096) (Table 3). We also tested the interaction effect of SIRT1 SNPs and PM2.5 in the recessive model, and the results are not meaningfully different from findings based on the additive model (Table S4). As for the dominant model, we found there was a difference of PM2.5 in mortality between participants carrying one or two alleles and those carrying zero alleles (Table S5).
Further, we conducted a three-way interaction analysis to examine whether there is a sex difference in the effect of G × E interaction. Figure 2 demonstrates exposure to higher PM2.5 does not affect mortality significantly in males and females who carry zero or one SIRT1_391 allele, but PM2.5 exposure substantially increases the mortality risk among male and female carriers of two SIRT1_391 alleles. It is noted that among the participants with exposure to the high concentration of PM2.5, females carry two SIRT1_391 alleles have a significant excess risk of mortality than those with zero or one SIRT1_391 allele; but the interaction is not significant among male participants. Specifically, in females who were carrying two SIRT1_391, exposure to a higher level of PM2.5 is associated with a 47.7% higher risk of mortality, while the effect size is 16.8% in men.

The sex-specific interaction analysis on PM2.5, SIRT1_391 and mortality. Note: The male participants without two SIRT1_391 minor allele copies and with low exposure of PM2.5 was regarded as the reference group. The model adjusted for age at baseline, sex, education, marriage, occupation, residence, exercise, smoking, and alcohol consumption
Discussion
Our study indicated/replicated the detrimental effect of air pollution on all-cause mortality. We found a protective effect of some alleles of SIRT1 on all-cause mortality, in concurrence with several prior animal studies and population studies. Our study’s added evidence is finding an interaction between PM2.5 induced premature mortality and carriers of SIRT1_391, SIRT1_472, and SIRT1_102 genotypes, which carriers of 2 alleles of SIRT1_391 can counteract the detrimental effect of PM2.5 and reduce 26.1% risk of premature mortality. Although none of the associations between SNPs and mortality would be statistically significant after the most stringent Bonferroni correction (α = 0.05/4 ≈ 0.0125), we used candidate SNPs tested in previous studies instead of random SNPs so that the need for stringent correction may not be necessitated. Our study used a large old cohort to elucidate better evidence of several cellular and molecular pathways studies on the protective role of SIRT1. Prior studies indicate SIRT1 is involved in the pathway of PM-induced airway inflammation with the activation of SIRT1 to prevent airway disorders [24, 25]. On the cellular and molecular side, using in vivo models of airway inflammation and in vitro culture of human bronchial epithelial (HBE) cells exposed to PM2.5 and resveratrol (SIRT1 activator), SIRT1 expression was decreased in HBE cells and lung tissues after PM2.5 exposure, suggesting that SIRT1 is involved in the pathogenesis of PM-induced airway inflammation [29]. Second, a plethora of animal studies investigating PM2.5 has documented pathways of inducing oxidative stress to trigger inflammation and thrombosis [39]. It was hypothesized that SIRT1 as a member of class III histone deacetylase, controls lung inflammation and coagulation after PM2.5 exposure. SIRT1 knock-out mice exhibited aggravated lung vascular leakage and inflammation after PM2.5 exposure, correlated with increased NF-κB acetylation and activation. This indicates that SIRT1 functions as a suppressor of coagulation after particulate matter exposure [29]. Further molecular evidence also implicates the role of SIRT1 in protein/histone deacetylase and as a protective factor of the development of pulmonary emphysema [40]. PM2.5 exposure in animal models was also demonstrated to introduce the repression activity of SIRT1 in the mice liver [41].
Our study builds on these animal models and is the most extensive population-based study on the role of SIRT1 in air pollution-induced mortality. Previously, a series of genes involved in oxidative stress and inflammatory pathways were studied for interaction with air pollutants, including GSTM1, GSTP1, NQO1, and TNF. There were both positive and null findings [42]. Studies indicate genetic susceptibility is likely to play a role in response to air pollution, especially on cardiovascular and respiratory outcomes [29]. A recent review indicated that SIRT1 plays in toxicological damage caused by environmental toxicants such as PM2.5, the PM-induced injury affect SIRT1 expression, which then affects the expression and activity of downstream proteins, resulting in toxic damage [43]. In addition, one molecular biology finding suggested that PM2.5 can upregulate MicroRNA-146a-3p and induces inflammatory macrophage polarization by targeting SIRT1 [44]. A recent population study also found that exposure to long-term air pollution, even at a low level, can alter gene expression and, in turn to impact an individual’s health [18]. Various studies have indicated that aging and lifespan are modulated by genetic and environmental factors as well as their interactions. Extant basic and translational studies have reported the interaction effects of air pollution and SIRT1 on the incidence or progression of pulmonary diseases [25, 29, 45] and cardiovascular diseases [26]. As we do not have a complete understanding of the synergistic health effects of air pollution and SIRT1 in humans, and prior studies are mainly focused on animal models, our finding of an interactive effect needs validation.
We also observe significant gender differentials in the interaction effect of PM2.5 and SIRT1 genotypes on mortality. The association of PM2.5 exposure with a reversal of the negative effects of carrying two SIRT1_391 alleles on mortality is much stronger in females than in males. In females who were carrying two SIRT1_391, exposure to a higher level of PM2.5 is associated with a 47.7% higher risk of mortality, while the effect size is 16.8% in men. Underlying mechanisms of gender difference may be due to sex hormones and distinct innate immune systems [46]. First, estrogen can induce downregulation of SIRT1, which in turn reduces the anti-oxidative anti-inflammatory effect on PM-induced toxicants [47]. Although production of estrogen declined dramatically in females after menopause status, the estrogen remained at moderately low level. SIRT1, possibly through the AKT and ERK signaling pathways, plays a crucial role in estrogen in protecting arteries from senescence and atherosclerosis [48]. Resveratrol also functions as a suppressor of PM-induced inflammatory signaling pathways by inhibiting COX-2/PGE2 expression [49]. Second, sex-specific analyses of longevity gene indicate “the innate immune system in men and of the tryptophan and PGC-1 pathways in the regulation of immune-related pathways in women suggests that women and men have different approaches for longevity, in which the SIRT1 deacetylates PGC-1α and enhances PGC-1α activity, insufficient NAD+ availability and SIRT1 enzymatic activity may be contributing factors [50]. Moreover, another sex-specific study on the human heart reveals a female sex-specific downregulation of SIRT1 and SIRT3 in aged hearts, as well as a decline in mitochondrial anti-oxidative defense and a pro-inflammatory shift in old female hearts but not in male hearts. The ex-specific downgrade of SIRT1 in females than in males might predispose females to a more susceptible condition when exposed to PM-derived toxicants [51].
Our study has several limitations. The biggest limitation is that our cohort did not ascertain reliable cause-specific mortality, which means we could not determine the possible pathways of airway-induced mortality. Second, we used satellite-derived residential area-level PM2.5 measurement, which has been shown to be reliable but may not be as precise as ground-level monitors and personal monitors. However, we do not expect differential misclassification that biases our effect estimates. In addition, we used a 3-year average PM2.5, so we can only study long-term air pollution difference. Third, our study did not have biomarkers or dietary determinants of nicotinamide mononucleotide (NMN), an NAD+ precursor, to assess SIRT1 activation activity; however, we do not expect these factors to varying with air pollution levels. Lastly, we also did not have biomarker data or clinical diagnosis of pulmonary function of the participants to ascertain differences based on SIRT1 genotype status. Thus, our study cannot distinguish the possible mechanism of the interaction between SIRT1 and air pollution on neuroprotection, metabolism, and cell survival. Lastly, we currently do not possess epigenetic markers in this cohort to measure gene expression.
Our study has many strengths. First, it is one of the largest population studies by samples size on the role of SIRT1 in human populations. Second, we had over one decade of follow-up on the survival status of participants. We had a highly diverse group of individuals from within China, including all climatic and geographical regions covering 23 provinces. Third, as our cohort design was initially intended to study socioeconomic determinants of health, we had a wide range of possible confounders to adjust for. Fourth, conducting this study in a developing country, we had high heterogeneity of a range of low and high ambient air pollution levels to see a dose-response relationship. Lastly, a relatively large sample size gave us statistical power for interaction variables and stratified analyses.
Conclusions
In conclusion, our study suggested a gene-environmental interaction of SIRT1 genotype and air pollution in a large population-based cohort, provided human epidemiological evidence to validate the many animal studies. If SIRT1 does protect against PM2.5 exposure, those living in areas with high levels of air pollution may benefit from induced SIRT1 activity through supplementation or interventions. Our findings indicate future clinical trials are needed to validate this hypothesis. If successful, this may be public health and clinical intervention through targeting mechanisms to protect against air pollution insult on the human body.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
References
Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1(1):3. https://doi.org/10.1038/ncomms1001.
Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120(4):473–82. https://doi.org/10.1016/j.cell.2005.01.029.
Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18(3):416–30. https://doi.org/10.1016/j.cmet.2013.07.013.
Bonda DJ, Lee HG, Camins A, Pallas M, Casadesus G, Smith MA, Zhu X. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol. 2011;10(3):275–9. https://doi.org/10.1016/S1474-4422(11)70013-8.
Finch CE: Chapter 3 - Air Pollution in Diseases of Aging. In: The Role of Global Air Pollution in Aging and Disease. Edited by Finch CE: Academic Press; 2018: 83–130.
Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, Oh DY, Lu M, Milne JC, Westphal C, Bandyopadhyay G, Olefsky JM. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298(3):E419–28. https://doi.org/10.1152/ajpendo.00417.2009.
Park S, Shin J, Bae J, Han D, Park SR, Shin J, Lee SK, Park HW. SIRT1 Alleviates LPS-Induced IL-1beta Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells. 2020:9(3).
Kim S, Bi X, Czarny-Ratajczak M, Dai J, Welsh DA, Myers L, Welsch MA, Cherry KE, Arnold J, Poon LW, Jazwinski SM. Telomere maintenance genes SIRT1 and XRCC6 impact age-related decline in telomere length but only SIRT1 is associated with human longevity. Biogerontology. 2012;13(2):119–31. https://doi.org/10.1007/s10522-011-9360-5.
Figarska SM, Vonk JM, Boezen HM. SIRT1 polymorphism, long-term survival and glucose tolerance in the general population. PLoS One. 2013;8(3):e58636. https://doi.org/10.1371/journal.pone.0058636.
Huang J, Sun L, Liu M, Zhou L, Lv ZP, Hu CY, Huang ZZ, Zheng CG, Zhou L, Yang Z. Association between SIRT1 gene polymorphisms and longevity of populations from Yongfu region of Guangxi. Chin J Med Genet. 2013;30(1):55–9.
Albani D, Mazzuco S, Chierchia A, Fusco F, Boeri L, Martines R, Giorgi ED, Frigato A, Durante E, Caberlotto L, Zanardo A, Siculi M, Gallucci M, Forloni G. The SIRT1 promoter polymorphic site rs12778366 increases IL-6 related human mortality in the prospective study “Treviso Longeva (TRELONG)”. Int J Mol Epidemiol Genet. 2015;6(1):20–6.
WHO WHO: Ambient air pollution: a global assessment of exposure and burden of disease. 2016.
Liu C, Chen R, Sera F, Vicedo-Cabrera AM, Guo Y, Tong S, Coelho M, Saldiva PHN, Lavigne E, Matus P, et al. Ambient particulate air pollution and daily mortality in 652 cities. N Engl J Med. 2019;381(8):705–15. https://doi.org/10.1056/NEJMoa1817364.
Wang Q, Wang J, He MZ, Kinney PL, Li T. A county-level estimate of PM2.5 related chronic mortality risk in China based on multi-model exposure data. Environ Int. 2018;110:105–12. https://doi.org/10.1016/j.envint.2017.10.015.
Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, Balakrishnan K, Brunekreef B, Dandona L, Dandona R, Feigin V, Freedman G, Hubbell B, Jobling A, Kan H, Knibbs L, Liu Y, Martin R, Morawska L, Pope CA III, Shin H, Straif K, Shaddick G, Thomas M, van Dingenen R, van Donkelaar A, Vos T, Murray CJL, Forouzanfar MH. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the global burden of diseases study 2015. Lancet. 2017;389(10082):1907–18. https://doi.org/10.1016/S0140-6736(17)30505-6.
Huang ZY, Bozzetti C, Ho KF, Cao JJ, Han Y, Daellenbach KR, Slowik JG, Platt SM, Canonaco F, et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature. 2014;514(7521):218–22. https://doi.org/10.1038/nature13774.
Song C, He J, Wu L, Jin T, Chen X, Li R, Ren P, Zhang L, Mao H. Health burden attributable to ambient PM2.5 in China. Environ Pollut. 2017;223:575–86. https://doi.org/10.1016/j.envpol.2017.01.060.
Madaniyazi L, Li S, Li S, Guo Y. Candidate gene expression in response to low-level air pollution. Environ Int. 2020:105610.
Huang YC, Karoly ED, Dailey LA, Schmitt MT, Silbajoris R, Graff DW, Devlin RB. Comparison of gene expression profiles induced by coarse, fine, and ultrafine particulate matter. J Toxicol Environ Health A. 2011;74(5):296–312. https://doi.org/10.1080/15287394.2010.516238.
Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25(10):1939–48. https://doi.org/10.1016/j.cellsig.2013.06.007.
Gracia-Sancho J, Villarreal G Jr, Zhang Y, Garcia-Cardena G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Res. 2010;85(3):514–9. https://doi.org/10.1093/cvr/cvp337.
Kawai Y, Garduno L, Theodore M, Yang J, Arinze IJ. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J Biol Chem. 2011;286(9):7629–40. https://doi.org/10.1074/jbc.M110.208173.
Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int J Mol Sci. 2013;14(2):3834–59. https://doi.org/10.3390/ijms14023834.
He B, Zhang W, Qiao J, Peng Z, Chai X. Melatonin protects against COPD by attenuating apoptosis and endoplasmic reticulum stress via upregulating SIRT1 expression in rats. Can J Physiol Pharmacol. 2019;97(5):386–91. https://doi.org/10.1139/cjpp-2018-0529.
Tien CP, Chen CH, Lin WY, Liu CS, Liu KJ, Hsiao M, Chang YC, Hung SC. Ambient particulate matter attenuates Sirtuin1 and augments SREBP1-PIR axis to induce human pulmonary fibroblast inflammation: molecular mechanism of microenvironment associated with COPD. Aging (Albany NY). 2019;11(13):4654–71. https://doi.org/10.18632/aging.102077.
Wittkopp S, Staimer N, Tjoa T, Stinchcombe T, Daher N, Schauer JJ, Shafer MM, Sioutas C, Gillen DL, Delfino RJ. Nrf2-related gene expression and exposure to traffic-related air pollution in elderly subjects with cardiovascular disease: an exploratory panel study. J Expo Sci Environ Epidemiol. 2016;26(2):141–9. https://doi.org/10.1038/jes.2014.84.
Braidy N, Jayasena T, Poljak A, Sachdev PS. Sirtuins in cognitive ageing and Alzheimer’s disease. Curr Opin Psychiatry. 2012;25(3):226–30. https://doi.org/10.1097/YCO.0b013e32835112c1.
Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, Vachharajani VT, Yoza BK, McCall CE. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012;92(3):499–507. https://doi.org/10.1189/jlb.0212078.
Lai T, Wen X, Wu D, Su G, Gao Y, Chen C, Wu W, Lv Y, Chen Z, Lv Q, Li W, Li D, Chen M, Wu B. SIRT1 protects against urban particulate matter-induced airway inflammation. Int J Chron Obstruct Pulmon Dis. 2019;14:1741–52. https://doi.org/10.2147/COPD.S202904.
Zeng, Poston DL, Vlosky DA, Gu D: Healthy longevity in China: demographic, socioeconomic, and psychological dimensions, vol. 20: Springer Science & Business Media; 2008.
van Donkelaar A, Martin RV, Brauer M, Hsu NC, Kahn RA, Levy RC, Lyapustin A, Sayer AM, Winker DM. Global estimates of fine particulate matter using a combined geophysical-statistical method with information from satellites, models, and monitors. Environ Sci Technol. 2016;50(7):3762–72. https://doi.org/10.1021/acs.est.5b05833.
Li T, Zhang Y, Wang J, Xu D, Yin Z, Chen H, Lv Y, Luo J, Zeng Y, Liu Y, Kinney PL, Shi X. All-cause mortality risk associated with long-term exposure to ambient PM2.5 in China: a cohort study. Lancet Public Health. 2018;3(10):e470–7. https://doi.org/10.1016/S2468-2667(18)30144-0.
Ji JS, Zhu A, Lv Y, Shi X. Interaction between residential greenness and air pollution mortality: analysis of the Chinese longitudinal healthy longevity survey. Lancet Planet Health. 2020;4(3):e107–15. https://doi.org/10.1016/S2542-5196(20)30027-9.
Zeng Y, Nie C, Min J, Chen H, Liu X, Ye R, Chen Z, Bai C, Xie E, Yin Z, Lv Y, Lu J, Li J, Ni T, Bolund L, Land KC, Yashin A, O’Rand AM, Sun L, Yang Z, Tao W, Gurinovich A, Franceschi C, Xie J, Gu J, Hou Y, Liu X, Xu X, Robine JM, Deelen J, Sebastiani P, Slagboom E, Perls T, Hauser E, Gottschalk W, Tan Q, Christensen K, Shi X, Lutz M, Tian XL, Yang H, Vaupel J. Sex differences in genetic associations with longevity. JAMA Netw Open. 2018;1(4):e181670. https://doi.org/10.1001/jamanetworkopen.2018.1670.
Kuningas M, Putters M, Westendorp RG, Slagboom PE, van Heemst D. SIRT1 gene, age-related diseases, and mortality: the Leiden 85-plus study. J Gerontol A Biol Sci Med Sci. 2007;62(9):960–5. https://doi.org/10.1093/gerona/62.9.960.
Tabangin ME, Woo JG, Martin LJ: The effect of minor allele frequency on the likelihood of obtaining false positives. BMC Proc 2009, 3 Suppl 7:S41.
Zeng CH, Ni T, Ruan R, Feng L, Nie C, Cheng L, Li Y, Tao W, Gu J, et al. GxE interactions between FOXO genotypes and drinking tea are significantly associated with prevention of cognitive decline in advanced age in China. J Gerontol A Biol Sci Med Sci. 2015;70(4):426–33. https://doi.org/10.1093/gerona/glu060.
Aiken LS, West SG, Reno RR: Multiple regression: testing and interpreting interactions: sage; 1991.
Rao X, Zhong J, Brook RD, Rajagopalan S. Effect of particulate matter air pollution on cardiovascular oxidative stress pathways. Antioxid Redox Signal. 2018;28(9):797–818. https://doi.org/10.1089/ars.2017.7394.
Yao H, Hwang JW, Sundar IK, Friedman AE, McBurney MW, Guarente L, Gu W, Kinnula VL, Rahman I. SIRT1 redresses the imbalance of tissue inhibitor of matrix metalloproteinase-1 and matrix metalloproteinase-9 in the development of mouse emphysema and human COPD. Am J Physiol Lung Cell Mol Physiol. 2013;305(9):L615–24. https://doi.org/10.1152/ajplung.00249.2012.
Qiu Y, Zheng Z, Kim H, Yang Z, Zhang G, Shi X, Sun F, Peng C, Ding Y, Wang A et al: Inhalation Exposure to PM2.5 Counteracts Hepatic Steatosis in Mice Fed High-fat Diet by Stimulating Hepatic Autophagy. Sci Rep 2017, 7(1):16286.
Romieu I, Moreno-Macias H, London SJ. Gene by environment interaction and ambient air pollution. Proc Am Thorac Soc. 2010;7(2):116–22. https://doi.org/10.1513/pats.200909-097RM.
Ren Z, He H, Zuo Z, Xu Z, Wei Z, Deng J. The role of different SIRT1-mediated signaling pathways in toxic injury. Cell Mol Biol Lett. 2019;24(1):36. https://doi.org/10.1186/s11658-019-0158-9.
Zhong Y, Liao J, Hu Y, Wang Y, Sun C, Zhang C, Wang G. PM2.5 Upregulates MicroRNA-146a-3p and induces M1 polarization in RAW264.7 cells by targeting Sirtuin1. Int J Med Sci. 2019;16(3):384–93. https://doi.org/10.7150/ijms.30084.
Yanagisawa S, Baker JR, Vuppusetty C, Koga T, Colley T, Fenwick P, Donnelly LE, Barnes PJ, Ito K. The dynamic shuttling of SIRT1 between cytoplasm and nuclei in bronchial epithelial cells by single and repeated cigarette smoke exposure. PLoS One. 2018;13(3):e0193921. https://doi.org/10.1371/journal.pone.0193921.
Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16(10):626–38. https://doi.org/10.1038/nri.2016.90.
Shimabukuro M. SIRT1 and gender differences in atherosclerotic cardiovascular disease. J Atheroscler Thromb. 2020;27(1):8–10. https://doi.org/10.5551/jat.ED119.
Lee CH, Su SC, Chiang CF, Chien CY, Hsu CC, Yu TY, Huang SM, Shieh YS, Kao HW, Tsai CS, Hung YJ, Lin CY. Estrogen modulates vascular smooth muscle cell function through downregulation of SIRT1. Oncotarget. 2017;8(66):110039–51. https://doi.org/10.18632/oncotarget.22546.
Tsai MH, Hsu LF, Lee CW, Chiang YC, Lee MH, How JM, Wu CM, Huang CL, Lee IT. Resveratrol inhibits urban particulate matter-induced COX-2/PGE2 release in human fibroblast-like synoviocytes via the inhibition of activation of NADPH oxidase/ROS/NF-kappaB. Int J Biochem Cell Biol. 2017;88:113–23. https://doi.org/10.1016/j.biocel.2017.05.015.
Canto G-HZ, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–60. https://doi.org/10.1038/nature07813.
Barcena de Arellano ML, Pozdniakova S, Kuhl AA, Baczko I, Ladilov Y, Regitz-Zagrosek V: Sex differences in the aging human heart: decreased sirtuins, pro-inflammatory shift and reduced anti-oxidative defense. Aging (Albany NY) 2019, 11(7):1918–1933.
Acknowledgements
We are grateful to the CLHLS study, which provided the data in this research. The CLHLS was supported by funds from the US National Institute on Aging (NIA), the China Natural Science Foundation, the China Social Science Foundation, and the United Nations Fund for Population Activities (UNFPA) and was managed by the Center for Healthy Aging and Development Studies, Peking University.
Funding
This work was supported by Bill & Melinda Gates Foundation, National Institute of Health of the United States (2P01AG031719), National Key R&D Program of China (2018YFC2000400) and National Natural Sciences Foundation of China (71490732, 81903392, 81941021), China Postdoctoral Science Foundation funded project (2019 M650359).
Author information
Authors and Affiliations
Contributions
Yao Yao drafted the original manuscript. Linxin Liu cleaning the data, performed the analysis and aided in writing. Guang Guo and Yi Zeng made key revisions and edits. John S. Ji conceptualized the idea, designed the methodology, made revision and edits. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
CLHLS was approved by the Institutional Review Board, Duke University (Pro00062871), and the Biomedical Ethics Committee, Peking University (IRB00001052–13074). All participants or their legal representatives signed written consent forms to participate in the baseline and follow-up surveys. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guidelines.
Consent for publication
Written informed consent for publication was obtained from all participants.
Competing interests
None reported.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yao, Y., Liu, L., Guo, G. et al. Interaction of Sirtuin 1 (SIRT1) candidate longevity gene and particulate matter (PM2.5) on all-cause mortality: a longitudinal cohort study in China. Environ Health 20, 25 (2021). https://doi.org/10.1186/s12940-021-00718-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12940-021-00718-x