
Abstract
Aim
To explore the genetic effects of CYP2C8, CYP2C9, CYP2J2, and EPHX2, the key genes involved in epoxyeicosatrienoic acid processing and degradation pathways in gestational diabetes mellitus (GDM) and metabolic traits in Chinese pregnant women.
Methods
A total of 2548 unrelated pregnant women were included, of which 938 had GDM and 1610 were considered as controls. Common variants were genotyped using the Infinium Asian Screening Array. Association studies of single nucleotide polymorphisms (SNPs) with GDM and related traits were performed using logistic regression and multivariable linear regression analyses. A genetic risk score (GRS) model based on 12 independent target SNPs associated with GDM was constructed. Logistic regression was used to estimate odds ratios and 95% confidence intervals, adjusting for potential confounders including age, pre-pregnancy body mass index, history of polycystic ovarian syndrome, history of GDM, and family history of diabetes, with GRS entered both as a continuous variable and categorized groups. The relationship between GRS and quantitative traits was also evaluated.
Results
The 12 SNPs in CYP2C8, CYP2C9, CYP2J2, and EPHX2 were significantly associated with GDM after adjusting for covariates (all P < 0.05). The GRS generated from these SNPs significantly correlated with GDM. Furthermore, a significant interaction between CYP2J2 and CYP2C8 in GDM (PInteraction = 0.014, ORInteraction= 0.61, 95%CI 0.41–0.90) was observed.
Conclusion
We found significant associations between GDM susceptibility and 12 SNPs of the four genes involved in epoxyeicosatrienoic acid processing and degradation pathways in a Chinese population. Subjects with a higher GRS showed higher GDM susceptibility with higher fasting plasma glucose and area under the curve of glucose and poorer β-cell function.
Similar content being viewed by others


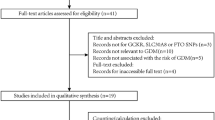
Introduction
Gestational diabetes mellitus (GDM) is defined as diabetes first diagnosed in the second or third trimester of pregnancy that was not clearly overt prior to gestation [1]. The prevalence of GDM varies in different populations and based on diagnostic criteria being used. The latest report from the International Diabetes Federation (IDF) Atlas indicated that the global standardised prevalence of GDM was 14% after adjusting for age [2]. It is estimated that 21.1 million (16.7%) live births to women in 2021 were affected by any type of hyperglycaemia in pregnancy (HIP), of which 80.3% were due to GDM [3]. Against the backdrop of the escalating obesity epidemic and advanced maternal age, the prevalence of GDM has rapidly increased and continues to surge. The importance of detecting GDM is exemplified by the fact that the hyperglycaemia status during pregnancy not only increases the maternal risk of subsequent progression to type 2 diabetes (T2D) by approximately 10 folds [4], compared with healthy controls but also predisposes the offspring to poor metabolic conditions in later life [5, 6]. This contributes to a vicious intergenerational cycle of diabetes and obesity that can impact the global health.
The well-documented risk factors contributing to GDM include maternal features (for example, advanced maternal age, weight, and high parity), previous GDM, and family history of diabetes [7]. Ethnicity has also been shown to be an independent determinant of GDM [8]. Additionally, polycystic ovarian syndrome (PCOS) has been frequently reported to be associated with an elevated risk of GDM [9,10,11]. Several pathogenic processes are involved in the development of GDM such as pancreatic islet β-cell dysfunction and chronic insulin resistance during pregnancy [12]. In addition, an imbalance between pro-inflammatory and anti-inflammatory processes leads to the progression of GDM. Various inflammatory factors, such as interleukin (IL)-1β, IL-6, IL-8, and tumour necrosis factor alpha, have been confirmed to have an independent positive correlation with GDM [13,14,15,16]. Accumulating evidence indicate that GDM is associated with strong genetic predisposition. Over the past few decades, gene loci responsible for insulin secretion and resistance and lipid and glucose metabolism have been found to be associated with GDM [17]. However, its genetics is complex and not fully defined.
Epoxyeicosatrienoic acids (EETs), metabolites of arachidonic acid produced by cytochrome P450 enzymes (CYP450), exhibit multiple biological activities, including anti-inflammatory, vasodilatory, and electrophysiological effects [18,19,20,21]. Several in vitro and animal studies have suggested that CYP450-derived EETs exert protective effects on insulin sensitivity and glucose metabolism, which are critical processes in GDM development [22,23,24]. In humans, the predominant epoxygenases involved in EET formation are CYP2J2, CYP2C8, and CYP2C9, which are encoded by the corresponding genes. Highly unstable EETs are hydrolysed to less active dihydroxyeicosatrienoic acids (DHETs) by soluble epoxide hydrolase (sEH) encoded by EPHX2 [25, 26]. Previous studies have shown that single nucleotide polymorphisms (SNPs) in CYP2J2, CYP2C8, CYP2C9, and EPHX2 are associated with diabetes and diabetic kidney disease (DKD); however, their genetic effects on GDM remain unclear.
Therefore, to address this knowledge gap, the present study was designed to investigate the association of common genetic variants of CYP2J2, CYP2C8, CYP2C9 and EPHX2 with GDM, with the aim of providing novel genetic basis for GDM susceptibility.
Materials and methods
Ethics statement
The present study was approved by the Institutional Review Board (IRB) of the University of Hong Kong-Shenzhen Hospital ([2017]13) and conducted according to the principles of the Declaration of Helsinki as revised in 2013. Written informed consent was obtained from each participant prior to enrolment.
Study design and participants
A total of 2548 Chinese women in early pregnancy were recruited between January 2016 and December 2018 at the University of Hong Kong-Shenzhen Hospital. All participants routinely underwent a standard 75-g oral glucose tolerance test (OGTT) at 24–28 weeks of gestation after an overnight fast of at least eight hours [28]. According to the criteria recommended by the International Association of Diabetes and Pregnancy Study Groups (IADPSG) [27], GDM was diagnosed if any of the following threshold values were equalled or exceeded: fasting plasma glucose: 5.1 mmol/L (92 mg/dL), one-hour plasma glucose (1 h-PG): 10.0 mmol/L (180 mg/dL), or two-hour plasma glucose (2 h-PG): 8.5 mmol/L (153 mg/dL). Participants with diabetes antedating pregnancy were excluded.
Clinical measurements
Information on demographics, family, and medical history of PCOS and GDM was collected using a standard questionnaire. Weight and standing height were measured with light clothes and no shoes, according to standard protocols by trained investigators [29, 30]. Pre-pregnancy weight was self-reported, and pre-pregnancy body mass index (BMI; kg/m2) was calculated as pre-pregnancy weight (kg) divided by the square of height (m). Blood pressure measurements were performed in both arms using a mercury sphygmomanometer after a rest period of at least 5 min, and the arm with the higher reading was tested twice at 3-min intervals to calculate the mean value [31]. Blood samples collected in the fasting status were used to measure the levels of fasting plasma glucose (FPG), fasting insulin (FINS), glycated haemoglobin A1c (HbA1c), total cholesterol, triglyceride, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C); details of these biochemical measurements have been described previously by Lu W et al. [32, 33]. Homeostasis model assessment of β-cell function (HOMA-β) and insulin resistance (HOMA-IR) were used to evaluate basal insulin secretion and insulin resistance, respectively, which were calculated using insulin and glucose concentrations as follows: HOMA-β = 20 × FINS (mU/L)/[FPG (mmol/L) − 3.5] and HOMA-IR = FINS (mU/L)/(22.5e− lnFPG (mmol/L)) [34, 35]. The area under curve of glucose (GAUC) from the 75-g OGTT was calculated as 1/2 × [FPG (mmol/L) + 1 h-PG (mmol/L)] × 1 h + 1/2 × [1 h-PG (mmol/L) + 2 h-PG (mmol/L)] × 1 h [36]. In addition, PCOS was diagnosed according to the revised 2003 consensus [37]. The birth of an infant before completion of 37 weeks of gestation was defined as preterm delivery. Macrosomia, preterm delivery, and caesarean delivery referred to current pregnancies.
SNP genotyping and quality control analysis
DNA was extracted from the peripheral blood using DNA Extraction Kit (Qiagen, Duesseldorf, Germany) according to the manufacturer’s instructions. The concentration of DNA in each sample was measured using NanoDrop2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). A 260/280 ratio of ~ 1.8 was generally accepted as “pure” for DNA. Genotyping was performed using Infinium Asian Screening Array-24 v1.0 BeadChip (Illumina, Inc., San Diego, CA, United States). Four critical genes related to EET processing and degradation pathways were selected. Genetic variants located within 3 kb upstream and 3 kb downstream of each gene region were extracted for further analysis [Fig. 1]. All single nucleotide polymorphisms (SNPs) were filtered based on the Hardy-Weinberg equilibrium in controls (HWE; P < 0.01), minor allele frequency (MAF > 0.001), and success rate (> 0.97) using the PLINK version 1.9 (https://www.cog-genomics.org/plink/1.9/) [38]. Pairwise linkage disequilibrium (LD) analysis was performed to obtain independent target SNPs using the Haploview version 4.2 (https://www.broadinstitute.org/haploview/downloads) [39]. To avoid the inflation of the estimates due to linkage disequilibrium, the threshold of r2 < 0.49 was used to select independent SNPs. Quality control procedures for individual samples required a call rate > 95%.

Regional association plot of SNPs in CYP2J2 (A),EPHX2 (B), CYP2C8 (C), and CYP2C9 (D) with GDM. The most signifcant SNP of each gene is highlighted in black triangle. The -log10P-value for the associations are given at the y-axis and the chromosomal positions (Genome Reference Consortium Human Build 37, GRCh37) of the SNPs are plotted
Statistical analysis
All data were collected on standard forms, checked for completeness, double-entered, compared, and corrected for inconsistencies. The normality of the distribution of continuous variables was tested using the Kolmogorov–Smirnov test. Continuous variables with normal distribution, that is, age, systolic blood pressure, and diastolic blood pressure were presented as the mean ± standard deviation (SD), and variables with non-normal distribution were reported as the median (interquartile range). Frequencies and proportions were used for categorical variables. The differences in mean, median, or frequency between groups were compared using independent samples Student’s t-test, Mann–Whitney U test, Kruskal–Wallis test, or Pearson χ2 test, as appropriate. The HWE in the control group was calculated using chi-square goodness of fit test. Genotype and allele distributions were compared using χ2 test. Data with skewed distribution were logarithmically transformed before analysis. Association tests were performed within a linear or logistic regression framework using the PLINK software. An additive genetic model was used to analyse allele dosage in which the genotypes AA, Aa, aa were coded as 0, 1, and 2, respectively (‘A’ represents the common allele and ‘a’ represents the rare allele). Age, pre-pregnancy BMI, history of PCOS, history of GDM and family history of diabetes were included as covariates and adjusted using a multivariable regression model; unadjusted and adjusted odds ratios (OR) and 95% confidence intervals (95% CI) were calculated. Gene-gene interaction (SNP-SNP epistasis) were performed by PLINK with a model based on allele dosage for each SNP, A and B, and fits in the form of ‘Y ~ b0 + b1.A + b2.B + b3.AB + e’. The test for interaction is based on the coefficient b3. All pairwise combinations of SNPs were tested. Multiple testing corrections were made using the Benjamini-Hochberg false discovery rate (FDR) method with a threshold of 0.05 for statistical significance [40].
The number of risk alleles (zero, one, or two) is summed for 12 independent variants, to generate unweighted genetic risk scores (GRS). Moreover, a weighted GRS was generated for each individual by taking the sum of the weighted number of observed risk alleles, each risk allele weighted by SNP-specific per allele effect size (loge[OR]) from the single SNP analysis, and dividing by the mean per allele effect size for the SNPs. Association between GRS and GDM susceptibility was evaluated [41]. To better interpret the results, the subjects were stratified into five GRS groups considering the distribution of GRS. We performed logistic regression analyses of the GRS groups against GDM case-control status and GRS group (1,2,3,4,5) was entered as a continuous variable. The association between GRS group and traits were analysed using linear regression. To better reflect the changing trend of metabolic traits across the GRS groups, trait levels were normalised using Z-scores, which were calculated by subtracting the mean from the raw score and then dividing the difference by the standard deviation [42]. Restricted cubic splines (RCS) were implemented to detect potential nonlinear association of GRS with GDM and metabolic traits, and the RCS models were adjusted for covariates. All statistical analyses were performed using the IBM SPSS Statistics for Windows (version 25.0; IBM Corp., Armonk, New York, USA) and R software v.4.1.3. A P-value of less than 0.05 (two-sided test of significance) was considered statistically significant.
Results
Subject characteristics
The clinical and demographic characteristics of the two groups are shown in Table 1. In total, 2548 participants were included in our study. Among them, 938 were diagnosed with GDM, and 1610 with normal glucose tolerance (NGT) were considered as controls. The mean age was 31.79 year (± 4.08), and the median gestational age at delivery was 39.29 weeks (38.57–40.00). Compared with the control group, pregnant women with GDM were older and more likely to report PCOS history, GDM history and family history of diabetes. Additionally, pre-pregnancy BMI, blood pressure, lipid parameters (total cholesterol, triglyceride, LDL-C, and HDL-C), and glycaemic parameters (FPG, 1 h-PG, 2 h-PG, GAUC, HbA1c, and HOMA-IR) were significantly higher, and BMI changes during pregnancy and HOMA-β were significantly lower in the GDM group than in the control group (all P < 0.001). There were no significant differences in the multiparous status, macrosomia, or primary caesarean delivery between the two groups (all P > 0.05).
Association between SNPs and GDM
We first examined the potential effects of these SNPs on GDM susceptibility in the study population. After the quality control procedure, 31 SNPs were included in the LD analysis [Suppl. Figure 1]. Among these, 12 SNPs were selected for further analysis (r2 < 0.49). Table 2 presents the genotype and allele distribution of the 12 independent SNPs and the corresponding odds ratios for GDM. The A allele of rs61790001 in CYP2J2 was the most significant SNP associated with GDM in the unadjusted model (OR 0.73 [95% CI 0.62–0.86], P = 0.0001). Considering the confounders, only 7 SNPs (rs61790001, rs76271683, rs57699806, rs11572177, rs9332092, rs4918758, and rs2860905) retained significance after multiple testing corrections (FDR < 0.05). The missense variant rs57699806 in EPHX2 was associated with GDM (adjusted OR 1.46 [95% CI 1.10–1.93], FDR = 0.044). Moreover, only rs61790001 in CYP2J2 presented a decresed risk of GDM (adjusted OR 0.73 [95% CI 0.61–0.86]). CYP2J2-rs76271683 and CYP2C8-rs11572177 were associated with an increased risk of GDM (adjusted OR 1.27 [95% CI 1.07–1.49], 1.34[95% CI 1.07–1.67]; FDR = 0.028, 0.046, respectively). The minor allele frequency(MAF) of these 12 SNPs in the current study and in other populations is shown in Suppl. Table 1.
Association between SNPs and metabolic traits
We subsequently analysed the association between SNPs and metabolic traits. As shown in Fig. 2, CYP2J2-rs76271683 was significantly associated with glucose indicators, including 1 h-PG (β = 0.011, SE = 0.003, FDR = 0.0125), 2 h-PG (β = 0.011, SE = 0.003, FDR = 0.0145), and GAUC (β = 0.010, SE = 0.003, FDR = 0.0047) [Suppl. Table 2]. Moreover, EPHX2-rs57699806 and CYP2C8-rs11572177 were associated with higher level of 1 h-PG and GAUC after multiple testing corrections (all FDR < 0.05), while CYP2C9-rs9332146 was in negative association with 1 h-PG and GAUC [Suppl. Figure 2 A-B]. Both rs9332092 and rs2860905 in CYP2C9 were associated with lower level of HOMA-β (β = -0.048, -0.034; SE = 0.018, 0.012, respectively) [Suppl. Table 3; Suppl. Figure 2D]. In addition, the A allele of CYP2J2-rs144619025 was associated with lower LDL-C level (β = -0.033, SE = 0.010, FDR = 0.0164) [Suppl. Table 4; Suppl. Figure 2C]. However, no association was observed between SNPs and fasting plasma glucose, HbA1c, fasting insulin, HOMA-IR, total cholesterol, triglyceride, or HDL-C levels.

Association between CYP2J2-rs76271683 and metabolic traits. Median values and 95% CI of blood glucose (A) and box plot of area under the curve of glucose (GAUC) (B) indicate that the glucose level was significantly higher in the minor allele carrier group. A: major allele; a: minor allele. Within each box, horizontal lines denote median values; boxes extend from the 25th to the 75th percentile of each group’s distribution of values; whiskers above and below the box indicate the 5th and 95th percentiles. Points above and below the whiskers indicate outliers
*P < 0.05 vs. AA group after adjusted for maternal age, pre-pregnancy BMI, history of PCOS, history of GDM and family history of diabetes
GRS of risk variants associated with GDM and metabolic traits
To evaluate population with higher susceptibility to GDM and determine whether it affected the assessment of GDM and metabolic traits in our study, genetic risk score (GRS) was generated based on the 12 independent SNPs. GRS (continuous) slightly increase the susceptibility to GDM (adjusted OR 1.07 [95% CI 1.02–1.13]). Then, to better interpret the results, the subjects were stratified into five risk groups considering the distribution of GRS [Fig. 3B]. Compared to GRS < 2 group, susceptibility to GDM increased in both unadjusted and adjusted models with the increase in the GRS[Figure 3 C]. The subjects harbouring GRS of 2, 3, 4 and > 4 all presented an increased risk of GDM (adjusted OR 1.57 [95% CI 1.09–2.26], 1.65 [95% CI 1.15–2.37], 1.81 [95% CI 1.24–2.65], and 1.80 [95% CI 1.24–2.63], respectively, all P < 0.05). Multivariable linear regression indicated that the median FPG and GAUC increased while the median HOMA-β decreased in higher GRS groups after adjusted for confounders (all P < 0.05). The heatmap shows the relative change trend of metabolic traits with the GRS [Fig. 3A] and the characteristics of the GRS groups are presented in Table 3. Overall, individuals with a higher GRS presented a higher susceptibility to GDM, higher FPG and GAUC and impaired insulin secretion than those with a lower GRS.

Association of genetic risk score (GRS) based on 12 SNPs with GDM and metabolic traits. Heatmap of metabolic traits z-scores computed for different GRS groups (A). Frequency distribution across the GRS groups (B). The histogram is plotted on the x-axis representing each GRS category as the sum of the number of risk alleles across the 12 loci, and the y-axis plots the number of individuals in each GRS category. Forest plot of the association between GRS and GDM (C). Adjusted OR and P-value refer to adjustment for maternal age, pre-pregnancy BMI, history of PCOS, history of GDM, and family history of diabetes
Gene-gene interaction
Gene-gene interaction (epistasis) analyses were carried out to avoid overlooking the heritability of GDM due to undiscovered interactions between them. After all pairwise combinations of the 12 SNPs were tested, only CYP2J2-rs76271683 and CYP2C8-rs11572177 exhibited significant epistatic effects on GDM susceptibility in all subjects (PInteraction = 0.014, ORInteraction = 0.61, 95%CI 0.41–0.90) [Suppl.Table 5]. The proportion of patients with GDM increased in subgroups with a greater number of risk alleles [Fig. 4].

Epistatic analysis between rs76271683 in CYP2J2 and rs11572177 in CYP2C8. The percentage of patients with GDM based on the genotypes is shown
Discussion
Several genome-wide association studies (GWAS) and candidate gene association studies have been performed to examine the association between genetic variants and the risk of GDM, which can be utilised to identify individuals at high risk for GDM or in the early stage of the disease. Notwithstanding, these studies have largely focused on common variants known to be associated with T2D and glycaemic traits outside of pregnancy based on the hypothesis that a shared genetic architecture exists between GDM and T2D [32, 43]. For example, TCF7L2, GCK, KCNJ11, CDKAL1, IGF2BP2, and MTNR1B are thought to modulate pancreatic islet β-cell function, all of which were associated with GDM (OR 1.15–1.46) [44, 45]. EETs are epoxygenase derivatives of arachidonic acid and closely related to pancreatic β-cell function, insulin resistance, glucose homeostasis, and other pathophysiological processes of glucose metabolism. Preclinical studies have consistently shown a protective role of EETs in the aetiology and progression of various metabolic diseases, such as diabetes and its complications [46]. Back in 1983, EETs were shown to stimulate insulin secretion in isolated rat islets and were relatively regioselective for EET formation [47], involving three critical genes (CYP2C8, CYP2C9, and CYP2J2). In addition to production of EETs and anti-inflammatory process, these genes are also involved in both insulin sensitivity in peripheral tissues and the capacity of the islets to respond to insulin resistance [48, 49]. CYP-derived EETs induce insulin secretion and protect pancreatic islet cells from apoptosis [50, 51]. Previous study indicated that CYP2J3 overexpression improved insulin resistance in rats treated with fructose and in db/db diabetic mice, improving insulin resistance by activating insulin receptor signaling and adiponectin-mediated AMPK signaling pathways [51, 52]. CYP2J3 gene delivery markedly reversed insulin resistance via upregulated AMPK signaling, which was associated with decreased ER stress response in adipose tissue [51]. The CYP2C gene family locus is highly polymorphic. Previous studies have indicated that CYP2J2 G-50T polymorphism (rs890293) was significantly associated with younger onset (less than 40 years old) T2D in a Chinese population. CYP2C8-rs10509681 was associated with an increased risk of DKD [53]. Our results demonstrated that both CYP2J2-rs76271683 and CYP2C8-rs11572177 were associated with an increased risk of GDM. In addition, the G allele of rs76271683 was associated with glucose metabolism indicators, including 1 h-PG, 2 h-PG, and GAUC. EPHX2 encodes the enzyme (sEH) responsible for the hydrolysis of EETs. EPHX2 rs751141 (Arg287Gln) polymorphism has been reported to be associated with insulin resistance in patients with T2D in Japanese population [54]. In our study, we found that the missense variant rs57699806 in EPHX2 was not only associated with an increased risk of GDM, but also with a higher level of 1 h-PG and GAUC.
GDM appears to influence the transfer of PUFAs from mothers to fetuses, which may affect the development of the fetal brain and impair the cognitive ability of the infant [55]. Ortega-Senovilla et al. proposed that there is a higher requirement for maternal fatty acids (both AA and DHA) in the fetuses of women with GDM than in those without GDM [56]. We found that individuals with certain variants in genes involved in epoxyeicosatrienoic acid (EET) processing and degradation pathways showed higher susceptibility to GDM. Considering variants may affect activity or yield of cytochrome P450 enzymes, and thus the substrate arachidonic acid content, individuals with certain variants may need to supplement more arachidonic acid, especially for pregnant women.
The discovery of multiple loci associated with GDM has demanded investigation of its clinical implications. The most common disease-associated genetic variants have a small effect size and are likely to explain only a limited fraction of heritability [43]. Thus, we attempted to aggregate the information of variants by constructing genetic risk scores. Weighted GRS is often based on the effect size reported in prior GWAS summary statistics. Therefore, a simple GRS was first used because for the risk alleles included there was inadequate information in the literature to assign a weight. An unweighted GRS was generated based on 12 risk variants by summing the number of risk alleles (zero, one, or two) for variants and subjects were stratified into five groups considering the distribution of GRS [41]. Correlation of GRS with GDM susceptibility was tested and we found that the GRS > 4 group had a nearly 1.80-fold increased susceptibility to GDM (adjusted OR 1.80 [95% CI 1.24–2.63]) than that of the lowest GRS group. After analysis of restricted cubic spline (RCS) regression between GRS and metabolic traits, only total cholesterol showed a significant nonlinear relationship between GRS (P for nonlinearity = 0.0038) [Suppl. Figure 3F]. Association between GRS groups and metabolic traits was evaluated and individuals with higher GRS had higher FPG and GAUC and poorer β-cell function. We further used the effect sizes from the single SNP analysis (Table 2) to construct a weighted GRS. Restricted cubic spline (RCS) analysis showed a significant nonlinear relationship between weighted GRS and GDM (P for nonlinearity = 0.0133) [Suppl. Figure 4], according to which 0 and 10 were selected as the cutoff values of weighted GRS. Subjects with weighted GRS > 10 had a nearly 2.22-fold increased susceptibility to GDM (adjusted OR 2.22 [95% CI 1.64–2.99]) than GRS < 0 group [Suppl. Figure 5]. Our results highlight the value of the GRS based on the EETs pathway in GDM risk assessment in the Chinese population.
Complementing simple additive main effects of individual loci, gene-gene interactions (epistasis) can explain some of the unexplained heritability of common diseases [57]. Fisher [58] defined epistasis in a statistical manner as an explanation for deviation from additivity in a linear model. In our study, a significant interaction (epistasis) was identified between CYP2J2-rs76271683 and CYP2C8-rs11572177, suggesting that individuals carrying variants of both genes might be more susceptible to GDM than those carrying variants in single genes. However, statistical interaction does not necessarily imply interaction on the biological or mechanistic level and it is a challenge to go from a population-level statistical gene-gene interaction to the biological interactions occurring at the cellular level [59]. But they do suggest directions for the discovery of biological interactions.
To the best of our knowledge, this is the first study to suggest a genetic association between polymorphisms in the EET pathway and GDM, which can provide novel insights into the genetic architecture and aetiology of GDM. This study has several limitations. First, it was conducted only in Chinese population, which could lead to inherent bias or ethnicity-specific observations. Thus, further studies in other ethnic populations are needed. Second, while the analysis of genetic polymorphism in EET metabolic pathways supports its association with GDM and indicates possible risk factors, the potential relationship between enzyme activity and EET levels and its roles in the pathogenesis of GDM remain to be investigated. Therefore, further functional assays are necessary to explore the underlying functions and mechanisms of these polymorphisms. Third, lifestyle factors, such as cigarette smoking and dietary pattern were not included in the genotype-disease analysis. Prudent dietary pattern which is characterised by a high intake of fruit, green leafy vegetables, poultry, and fish was significantly and inversely associated with GDM risk and smoking has been found to increase the risk of developing GDM [60,61,62]. However, whether interactions existed between lifestyle factors and genetic variants remains unknown. Finally, we adopted an unusual threshold (r2 < 0.49) for SNPs selection, which may have caused the inclusion of non-independent SNPs in the GRS model and may have inflated the estimates.
Conclusion
Our study suggests that common SNPs in key genes of EET processing and degradation pathways are associated with GDM in Chinese population. The GRS based on 12 independent SNPs was found to be positively associated with GDM. The mechanisms by which CYP2C8, CYP2C9, CYP2J2, and EPHX2 influence GDM susceptibility warrant further investigation.
Data Availability
The datasets used or analyzed during the current study are available from the corresponding author (Cheng Hu) on reasonable request.
References
American Diabetes A. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care. 2021;44:15–S33.
Wang H, Li N, Chivese T, Werfalli M, Sun H, Yuen L, Hoegfeldt CA, Elise Powe C, Immanuel J, Karuranga S, et al. IDF Diabetes Atlas: estimation of Global and Regional Gestational Diabetes Mellitus Prevalence for 2021 by International Association of diabetes in pregnancy Study Group’s Criteria. Diabetes Res Clin Pract. 2022;183:109050.
International Diabetes Federation. IDF Diabetes Atlas. 10th ed. Brussels, Belgium: International Diabetes Federation; 2021.
Vounzoulaki E, Khunti K, Abner SC, Tan BK, Davies MJ, Gillies CL. Progression to type 2 diabetes in women with a known history of gestational diabetes: systematic review and meta-analysis. BMJ. 2020;369:m1361.
Ruchat SM, Hivert MF, Bouchard L. Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr Rev. 2013;71(Suppl 1):88–94.
Abokaf H, Shoham-Vardi I, Sergienko R, Landau D, Sheiner E. In utero exposure to gestational diabetes mellitus and long term endocrine morbidity of the offspring. Diabetes Res Clin Pract. 2018;144:231–5.
Ben-Haroush A, Yogev Y, Hod M. Epidemiology of gestational diabetes mellitus and its association with type 2 diabetes. Diabet Med. 2004;21:103–13.
Weijers RN, Bekedam DJ, Smulders YM. Determinants of mild gestational hyperglycemia and gestational diabetes mellitus in a large dutch multiethnic cohort. Diabetes Care. 2002;25:72–7.
Bals-Pratsch M, Grosser B, Seifert B, Ortmann O, Seifarth C. Early onset and high prevalence of gestational diabetes in PCOS and insulin resistant women before and after assisted reproduction. Exp Clin Endocrinol Diabetes. 2011;119:338–42.
Boomsma CM, Eijkemans MJ, Hughes EG, Visser GH, Fauser BC, Macklon NS. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum Reprod Update. 2006;12:673–83.
Reyes-Muñoz E, Castellanos-Barroso G, Ramírez-Eugenio BY, Ortega-González C, Parra A, Castillo-Mora A. De la Jara-Díaz JF: the risk of gestational diabetes mellitus among mexican women with a history of infertility and polycystic ovary syndrome. Fertil Steril. 2012;97:1467–71.
Buchanan TA, Xiang AH. Gestational diabetes mellitus. J Clin Invest. 2005;115:485–91.
Pantham P, Aye IL, Powell TL. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta. 2015;36:709–15.
Vitoratos N, Valsamakis G, Mastorakos G, Boutsiadis A, Salakos N, Kouskouni E, Creatsas G. Pre- and early post-partum adiponectin and interleukin-1beta levels in women with and without gestational diabetes. Horm (Athens). 2008;7:230–6.
Wolf M, Sauk J, Shah A, Vossen Smirnakis K, Jimenez-Kimble R, Ecker JL, Thadhani R. Inflammation and glucose intolerance: a prospective study of gestational diabetes mellitus. Diabetes Care. 2004;27:21–7.
Zhang J, Chi H, Xiao H, Tian X, Wang Y, Yun X, Xu Y. Interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) single nucleotide polymorphisms (SNPs), inflammation and metabolism in gestational diabetes Mellitus in Inner Mongolia. Med Sci Monit. 2017;23:4149–57.
Zhang C, Bao W, Rong Y, Yang H, Bowers K, Yeung E, Kiely M. Genetic variants and the risk of gestational diabetes mellitus: a systematic review. Hum Reprod Update. 2013;19:376–90.
Ramirez CE, Shuey MM, Milne GL, Gilbert K, Hui N, Yu C, Luther JM, Brown NJ. Arg287Gln variant of EPHX2 and epoxyeicosatrienoic acids are associated with insulin sensitivity in humans. Prostaglandins Other Lipid Mediat. 2014;113–115:38–44.
Kaspera R, Totah RA. Epoxyeicosatrienoic acids: formation, metabolism and potential role in tissue physiology and pathophysiology. Expert Opin Drug Metab Toxicol. 2009;5:757–71.
Capdevila J, Wang W. Role of cytochrome P450 epoxygenase in regulating renal membrane transport and hypertension. Curr Opin Nephrol Hypertens. 2013;22:163–9.
Aliwarga T, Evangelista EA, Sotoodehnia N, Lemaitre RN, Totah RA. Regulation of CYP2J2 and EET levels in Cardiac Disease and Diabetes. Int J Mol Sci 2018, 19.
Ma B, Xiong X, Chen C, Li H, Xu X, Li X, Li R, Chen G, Dackor RT, Zeldin DC, Wang DW. Cardiac-specific overexpression of CYP2J2 attenuates diabetic cardiomyopathy in male streptozotocin-induced diabetic mice. Endocrinology. 2013;154:2843–56.
Li R, Xu X, Chen C, Wang Y, Gruzdev A, Zeldin DC, Wang DW. CYP2J2 attenuates metabolic dysfunction in diabetic mice by reducing hepatic inflammation via the PPARγ. Am J Physiol Endocrinol Metab. 2015;308:E270–282.
Gangadhariah MH, Dieckmann BW, Lantier L, Kang L, Wasserman DH, Chiusa M, Caskey CF, Dickerson J, Luo P, Gamboa JL, et al. Cytochrome P450 epoxygenase-derived epoxyeicosatrienoic acids contribute to insulin sensitivity in mice and in humans. Diabetologia. 2017;60:1066–75.
Deng Y, Theken KN, Lee CR. Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J Mol Cell Cardiol. 2010;48:331–41.
Beetham JK, Tian T, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305:197–201.
International Association of D, Pregnancy Study Groups, Consensus P, Metzger BE, Gabbe SG, Persson B, Buchanan TA, Catalano PA, Damm P, Dyer AR, Leiva A, et al. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33:676–82.
Sacks DA, Greenspoon JS, Abu-Fadil S, Henry HM, Wolde-Tsadik G, Yao JF. Toward universal criteria for gestational diabetes: the 75-gram glucose tolerance test in pregnancy. Am J Obstet Gynecol. 1995;172:607–14.
Lohman TG, Roche AF, Martorell R. Anthropometric standardization reference manual. Human kinetics books; 1988.
Rose GA, Blackburn H, Gillum R, Prineas R. Cardiovascular survey methods Geneva, Switzerland; WHO; 1982.
Panayiotou BN. Measuring blood pressure: which arm? JAMA. 1995;274:1343.
Fang X, Jin L, Tang M, Lu W, Lai S, Zhang R, Zhang H, Jiang F, Luo M, Hu C. Common single-nucleotide polymorphisms combined with a genetic risk score provide new insights regarding the etiology of gestational diabetes mellitus. Diabet Med. 2022;39:e14885.
Lu W, Luo M, Fang X, Zhang R, Li S, Tang M, Yu X, Hu C. Discovery of metabolic biomarkers for gestational diabetes mellitus in a chinese population. Nutr Metab (Lond). 2021;18:79.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Lee SH, Rhee M, Yang HK, Ha HS, Lee JH, Kwon HS, Park YM, Yim HW, Kang MI, Lee WC, et al. Serum preadipocyte factor 1 concentrations and risk of developing diabetes: a nested case-control study. Diabet Med. 2016;33:631–8.
Belfiore F, Iannello S, Volpicelli G. Insulin sensitivity indices calculated from basal and OGTT-induced insulin, glucose, and FFA levels. Mol Genet Metab. 1998;63:134–41.
Rotterdam EA-SPcwg. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod. 2004;19:41–7.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5.
Benjamini Y, Hochberg YJJotRsssB. Controlling the false discovery rate: a practical and powerful approach to multiple testing. 1995, 57:289–300.
Udler MS, McCarthy MI, Florez JC, Mahajan A. Genetic risk scores for diabetes diagnosis and Precision Medicine. Endocr Rev. 2019;40:1500–20.
Kreyszig E, Stroud K, Stephenson GJI. Advanced engineering mathematics. 2008, 9.
Powe CE, Kwak SH. Genetic studies of gestational diabetes and glucose metabolism in pregnancy. Curr Diab Rep. 2020;20:69.
Schäfer SA, Machicao F, Fritsche A, Häring HU, Kantartzis K. New type 2 diabetes risk genes provide new insights in insulin secretion mechanisms. Diabetes Res Clin Pract. 2011;93(Suppl 1):9–24.
Petrie JR, Pearson ER, Sutherland C. Implications of genome wide association studies for the understanding of type 2 diabetes pathophysiology. Biochem Pharmacol. 2011;81:471–7.
Mota-Zamorano S, Robles NR, Lopez-Gomez J, Cancho B, González LM, Garcia-Pino G, Navarro-Pérez ML, Gervasini G. Plasma and urinary concentrations of arachidonic acid-derived eicosanoids are associated with diabetic kidney disease. Excli j. 2021;20:698–708.
Falck JR, Manna S, Moltz J, Chacos N, Capdevila J. Epoxyeicosatrienoic acids stimulate glucagon and insulin release from isolated rat pancreatic islets. Biochem Biophys Res Commun. 1983;114:743–9.
Luther JM, Brown NJ. Epoxyeicosatrienoic acids and glucose homeostasis in mice and men. Prostaglandins Other Lipid Mediat. 2016;125:2–7.
Wang B, Wu L, Chen J, Dong L, Chen C, Wen Z, Hu J, Fleming I, Wang DW. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Signal Transduct Target Ther. 2021;6:94.
Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev. 2011;63:597–609.
Xu X, Tu L, Feng W, Ma B, Li R, Zheng C, Li G, Wang DW. CYP2J3 gene delivery up-regulated adiponectin expression via reduced endoplasmic reticulum stress in adipocytes. Endocrinology. 2013;154:1743–53.
Xu X, Zhao CX, Wang L, Tu L, Fang X, Zheng C, Edin ML, Zeldin DC, Wang DW. Increased CYP2J3 expression reduces insulin resistance in fructose-treated rats and db/db mice. Diabetes. 2010;59:997–1005.
Mota-Zamorano S, Robles NR, González LM, Valdivielso JM, Lopez-Gomez J, Cancho B, García-Pino G, Gervasini G. Genetics Variants in the epoxygenase pathway of Arachidonic Metabolism are Associated with Eicosanoids levels and the risk of Diabetic Nephropathy. J Clin Med 2021, 10.
Ohtoshi K, Kaneto H, Node K, Nakamura Y, Shiraiwa T, Matsuhisa M, Yamasaki Y. Association of soluble epoxide hydrolase gene polymorphism with insulin resistance in type 2 diabetic patients. Biochem Biophys Res Commun. 2005;331:347–50.
Hai-Tao Y, Zhi-Heng G, Yi-Ru C, Yue-Ting L, Hai-Ying Z, Ya-Juan L, Lin X. Gestational diabetes mellitus decreased umbilical cord blood polyunsaturated fatty acids: a meta-analysis of observational studies. Prostaglandins Leukot Essent Fatty Acids. 2021;171:102318.
Ortega-Senovilla H, Schaefer-Graf U, Herrera E. Pregnant women with gestational diabetes and with well controlled glucose levels have decreased concentrations of individual fatty acids in maternal and cord serum. Diabetologia. 2020;63:864–74.
Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–50.
Fisher RA. The correlation between relatives on the supposition of mendelian inheritance. Trans R Soc Edin. 1918;52:399–433.
Moore JH, Williams SM. Epistasis and its implications for personal genetics. Am J Hum Genet. 2009;85(3):309–20.
Bar-Zeev Y, Haile ZT, Chertok IA. Association between prenatal smoking and gestational diabetes Mellitus. Obstet Gynecol. 2020;135:91–9.
Zhang C, Ning Y. Effect of dietary and lifestyle factors on the risk of gestational diabetes: review of epidemiologic evidence. Am J Clin Nutr. 2011;94:1975s–9.
Zhang C, Schulze MB, Solomon CG, Hu FB. A prospective study of dietary patterns, meat intake and the risk of gestational diabetes mellitus. Diabetologia. 2006;49:2604–13.
Acknowledgements
We thank the study participants without whom the study would not have been possible.
Funding
This work was supported by the High-level Hospital Program, Health Commission of Guangdong Province, China (HKUSZH201901025); the project from Shanghai Diabetes Key Laboratory (SHKLD-KF-1701) and the Shanghai Science and Technology Innovation Action Plan (20XD1433300).
Author information
Authors and Affiliations
Contributions
CH supervised the study. SL, DY, JX, MT and XF collected and checked the data. RZ, HZ and XF helped to carry out the experiments. SL performed the data analysis and drafted the manuscript. XY and JG helped with data analysis and prepared Suppl. Figures 3-4. CH, ML and WJ reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The present study was approved by the ethics committee of the University of Hong Kong-Shenzhen Hospital. It was conducted according to the principles of the Declaration of Helsinki as revised in 2013. Written informed consent was obtained from each participant prior to enrolment.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lai, S., Yan, D., Xu, J. et al. Genetic variants in epoxyeicosatrienoic acid processing and degradation pathways are associated with gestational diabetes mellitus. Nutr J 22, 31 (2023). https://doi.org/10.1186/s12937-023-00862-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12937-023-00862-9