
Abstract
Background
Non-human primates, such as the rhesus macaques, are the preferred model for down-selecting human malaria vaccine formulations, but the rhesus model is expensive and does not allow for direct efficacy testing of human malaria vaccines. Transgenic rodent parasites expressing genes of human Plasmodium are now routinely used for efficacy studies of human malaria vaccines. Mice have however rarely predicted success in human malaria trials and there is scepticism whether mouse studies alone are sufficient to move a vaccine candidate into the clinic.
Methods
A comparison of immunogenicity, fine-specificity and functional activity of two Alum-adjuvanted Plasmodium falciparum circumsporozoite protein (CSP)-based vaccines was conducted in mouse and rhesus models. One vaccine was a soluble recombinant protein (CSP) and the other was the same CSP covalently conjugated to the Qβ phage particle (Qβ-CSP).
Results
Mice showed different kinetics of antibody responses and different sensitivity to the NANP-repeat and N-terminal epitopes as compared to rhesus. While mice failed to discern differences between the protective efficacy of CSP versus Qβ-CSP vaccine following direct challenge with transgenic Plasmodium berghei parasites, rhesus serum from the Qβ-CSP-vaccinated animals induced higher in vivo sporozoite neutralization activity.
Conclusions
Despite some immunologic parallels between models, these data demonstrate that differences between the immune responses induced in the two models risk conflicting decisions regarding potential vaccine utility in humans. In combination with historical observations, the data presented here suggest that although murine models may be useful for some purposes, non-human primate models may be more likely to predict the human response to investigational vaccines.
Similar content being viewed by others


Background
Malaria remains a major health concern in the tropics despite control efforts using available drugs and insecticides. There is a broad consensus that a highly effective vaccine is needed. Two vaccines developed by Sanaria® (PfSPZ) and GlaxoSmithKline Vaccines (RTS,S), have demonstrated the proof-of-concept that a vaccine can protect against controlled human malaria infection (CHMI) [1]. Both PfSPZ and RTS,S vaccines induce potent immune responses against Plasmodium sporozoites, in particular against the circumsporozoite protein (CSP). Efforts are underway to improve upon the success of PfSPZ and RTS,S, but there is considerable debate on what animal model should be used as the critical path for advancing novel vaccine candidates to human trials. The rodent model for malaria is based on Plasmodium berghei, Plasmodium chabaudi or Plasmodium yoelii parasites that naturally infect African thicket rats [2]. These parasites have been adapted to grow in mouse strains for routine laboratory experiments and provide easy access to blood and liver stages. Rodent models have led to successful translation of many malaria drugs, however the down-selection of vaccines using mice has had mixed benefits. While the irradiated sporozoite vaccine can protect both rodents and humans [3], many sub-unit vaccines have failed to translate protection from mice to humans. A CSP DNA vaccine can induce very potent sterilizing protection against P. berghei and P. yoelii in mice [4, 5], but PfCSP-based DNA vaccines did not protect humans [6]. Likewise, the P. yoelii merozoite surface protein-1 (MSP1) [7] and P. chabaudi apical membrane antigen-1 (AMA1) [8] candidates have been known to protect mice, but human-use formulations of PfMSP1 [9] and PfAMA1 [10] vaccines confer limited protection in humans.
A lack of translation of observations from mice to humans is not surprising given that the common ancestor of humans and mice lived ~75 million years ago and significant differences in the invasion biology of rodent and human malaria parasites exist [11]. Rhesus macaques (Macaca mulatta) and humans share a more recent common ancestor that lived approximately 25 million years ago [12], and several malaria parasites that naturally infect rhesus, such as Plasmodium knowlesi and Plasmodium cynomolgi, are human transmissible [13]. Rhesus challenge models can be used to evaluate vaccines that inhibit parasite growth in both the liver and blood, whereas human trials with such vaccines mandate drug treatment immediately at the onset of blood-stage patency [14]. P. cynomolgi parasite challenge in rhesus allows cross-species protection studies to be conducted on Plasmodium vivax vaccines [15], circumventing the need for a much more complex controlled P. vivax human challenge. Rhesus models have also been useful in testing novel concepts such as multivalent and whole parasite vaccines [16, 17]. There are several instances where the rhesus model has predicted the success of human malaria vaccine trials. The rationale for selection of the AS01 over AS02 adjuvant for the RTS,S vaccine was based on a rhesus trial [18] that was subsequently corroborated by CHMI and in the field [19, 20]. In another study, the adenovirus serotype-35 CSP prime and RTS,S boost improved the IFN-γ+ T cell responses in rhesus, but also showed lower antibody titres than RTS,S alone [21]. These findings were closely replicated in humans with improved T cell responses and reduced antibodies in the prime-boost arm [22]. In addition, the irradiated P. knowlesi sporozoite vaccine protected rhesus against virulent P. knowlesi challenge and CD8+ T cell responses correlated with protection [23]; this has been replicated in CHMI model where irradiated Plasmodium falciparum sporozoite vaccine elicited protection was also characterized by induction of CD8+ T cell responses [3, 24, 25]. The rhesus model has not only predicted the success but also the failure of many experimental malaria vaccines. Formulations of PfMSP1 and PfAMA1 that induced low level in vitro merozoite growth inhibition activity in the rhesus model [26] failed to protect humans [9, 10]. Soluble PvCSP that induced relatively low repeat specific antibodies in rhesus [27] also failed to confer sterile protection against PvCHMI [28]. These studies provide direct evidence that rhesus can accurately predict the immunogenicity and protection outcomes of experimental human malaria vaccines.
Despite excellent predictive capability, the scarcity and cost associated with non-human primates remain problematic and although rhesus malaria parasites are human transmissible, both P. falciparum and P. vivax cannot infect rhesus and hence direct challenge experiments are not feasible. A way to overcome the species barrier is to develop transgenic parasites. While transgenic rhesus parasites have been difficult to produce and select, transgenic rodent parasites carrying P. falciparum and P. vivax antigen genes are now routinely used to answer biological questions [29, 30]. Mouse-human chimeric parasites expressing PfCSP, Pfs25, PfMSP1, and PvCSP have been used to compare the efficacy of human P. falciparum and P. vivax vaccines in mice [31–41]. In particular, transgenic parasites have been very useful to study the role of PfCSP during hepatocyte invasion [42, 43] and challenge models have been optimized to down-select PfCSP vaccines using P. berghei parasites that carry functional PfCSP genes [44, 45]. Considering previous examples where mouse data have not translated to protection in humans, there remains an uncertainty in the validity of using a mouse model for down-selecting P. falciparum and P. vivax malaria vaccines prior to CHMI. To address this issue, immunological responses to two vaccines were compared here in mice and rhesus.
The vaccines tested were derived from a nearly full-length PfCSP construct expressed in Escherichia coli that contains the N-terminal region, 19 NANP, and three NVDP repeats followed by the C-terminal region. Also contained within the protein sequence were two inter-species conserved motifs of PfCSP: ‘region I’ (located just before the central repeats) and the cysteine-rich ‘region II’ (located after the repeats). Both of these conserved regions have been implicated in hepatocyte invasion [42, 46]. In order to produce a comparator particulate vaccine, the soluble CS protein was chemically coupled to recombinant Qβ capsid protein. Qβ protein self-assembles into 25 nm virus-like particles within E. coli cytosol and has been used as vaccine carrier in many human trials [47–50]. During assembly in the cytosol, E. coli RNA is encapsulated inside the Qβ virus-like particles and this bacterial RNA serves as a potent adjuvant since it is a strong TLR7/8 agonist [51, 52]. It was recently reported that soluble CSP chemically conjugated to Qβ particles (Qβ-CSP) induced superior immunogenicity and efficacy compared to soluble CSP in several adjuvants in mice [53]. In the current study, the Qβ-CSP and soluble CSP vaccines were adjuvanted in Alum and used to immunize mice and rhesus. Although human data on these vaccine formulations are not available, rhesus responses were treated as a close surrogate for human responses based on their proven predictive value in previous malaria vaccine trials. Qβ-CSP generally induced higher overall antibody responses than CSP in both mice and rhesus, but differences in epitope specificity, avidity and functionality of antibodies observed between the two species argue that rhesus ought to remain the choice model for the final down-selection step before advancing to human trials.
Methods
Vaccine antigens
CSP expression and purification has been reported previously [45]. The conjugation of CSP to Qβ phage particle was carried out using a cysteine-based cross-linker and has been described earlier [53]. The CSP content of the Qβ-CSP vaccine was determined as the sum of conjugated and unconjugated CSP to ensure the CSP content was equal in all comparator vaccine groups. Method of quantifying CSP in Qβ-CSP has been previously described [53].
Animals
Six- to eight-weeks old female C57Bl/6 mice (Mus musculus) supplied by Jackson Laboratory (Bar Harbor, ME, USA) were housed at the Walter Reed Army Institute of Research (WRAIR) animal facility for the duration of the study (IACUC-approved protocol number 11-MVD-15). The room was maintained at 21–23 °C, with a relative humidity of 3–70%, 1–15 air changes hourly and a 12:12-h light:dark cycle. At the time of this study, all mice were negative for Ectromelia virus, Mouse Cytomegalovirus (MCMV), Mouse Parvovirus (MPV), Murine Norovirus (MNV), Mouse Adenovirus (MAdV-1/MAdV-2), Polyomavirus, Lymphocytic Choriomeningitis Virus (LCMV), Sendai virus, Pneumonia Virus of Mice (PVM), Reovirus 1,2, and 3, Rotavirus, Mouse Hepatitis Virus (MHV), Theiler’s Murine Encephalomyelitis Virus, Mycoplasma pulmonis, CAR bacillus, Clostridium piliforme (Tyzzer’s Disease), and Helicobacter. Mice were singly housed in suspended, polycarbonate shoebox-type cages with filter tops on Beta Chip bedding Q3 (Northeastern Products Corp, Warrensburg, NY, USA), and each cage had shreddable Q4 nesting material (Nestlets, Ancare, Bellmore, NY, USA) and an Igloo or similar tube for enrichment. Mice were fed a Q4 pelleted rodent food (RMH3000, Lab Diet, St Louis, MO, USA) and provided drinking water ad libitum. Sentinel mice used to monitor the health status of the experimental animals tested negative for all monitored pathogens.
Adult male rhesus macaques of Indian origin (M. mulatta), 9–15 years old and 6–15 kg weight were housed at the WRAIR animal facility and used under an IACUC-approved protocol, number 13-MVD-12L. Monkeys used were malaria naïve as assessed by ELISA against CSP antigen, but were used in other non-malaria studies in the past. The environment was maintained at 20–22 °C, with a relative humidity of 30–70%, 10–15 air changes hourly and a 12:12-h light:dark cycle. All animals were free from overt clinical signs of illness, deemed to be in good health and tested negative for Macacine herpesvirus 1, measles, Simian Retrovirus, Simian Immunodeficiency Virus and Simian T-cell Leukemia Virus, and tuberculin skin test. Animals were singly housed, fed a commercial diet (Lab Diet 5038, Purina Mills International, Brentwood, MO, USA) and provided water ad libitum. Environmental enrichment was provided in accordance with WRAIR Veterinary Service Programs standing operating procedures.
Mouse vaccination, bleed and challenge
To determine optimal group size, ELISA titres following three doses of a different CSP-based vaccine were log transformed and the standard deviation fed into the Russ Lenth online power calculator using the 2-sample t test algorithm [54]. The above calculation showed that n = 7 per group would have 80% power to discern ~two-fold differences in mean titre. Mice (n = 7 per group) received three doses of 2.5 µg CSP at 3-week intervals as soluble protein or Qβ-CSP formulated in Alum (300 µg/ml aluminum content; Alhydrogel; Brenntag Biosector, Frederikssund, Denmark). Alum was mixed 1:1 v/v with the antigen by medium vortexing for 3 s every 5 min for 30 min and 100 µl of this vaccine was administered via 50 µl intra-muscular injection into each of the thigh muscles. The antigen dose and vaccine schedule were the same as has been previously used to differentiate between formulations as it gives sub-saturating levels of protection against this specific transgenic parasite strain [44, 45, 53]. Mice were bled at 3 weeks after each immunization, except for the final bleed that was collected at 2 weeks after the last dose. To measure protective efficacy, all mice were challenged with 3000 transgenic P. berghei sporozoites expressing a functional copy of the PfCSP gene at 2 weeks after the last dose, as described previously [42, 44]. Briefly, sporozoites were collected using the Ozaki method [55] and injected into the lateral tail vein of mice. Mice were monitored daily and parasitaemia was detected using thin blood smears beginning at day 4 post-challenge. Blood smears were fixed and stained with Giemsa. Mice found to be parasitaemic for 2 consecutive days were sacrificed and recorded as ‘not protected’ while mice that did not develop blood stage parasitaemia up to 14 days post-challenge were reported as ‘protected’.
Rhesus vaccination and bleeds
Optimal group size was determined as above and n = 5 was shown to have 98% power to discern a two-fold difference in mean titre. Rhesus monkeys (n = 5 per group) were anaesthetized and the thigh area was shaved. A total 0.5 ml of the vaccine was administered intra-muscularly in the outer thigh muscle. Each vaccination contained 25 µg CSP or Qβ-CSP in Alum (300 µg/ml aluminum content). The schedule for vaccination was at 0, 1 and 3 months. Rhesus were bled 2 weeks after each vaccination for serology. The antigen dose for rhesus was chosen based on the proposed human dose of this vaccine, which is ten-fold higher than that used in the mouse model. The vaccine schedule was based on a previous publication on a rhesus monkey trial with RTS,S and an adenovirus-based CSP vaccine [21].
ELISA
ELISA plates (Immulon 2HB) were coated overnight at 4 °C with either the full-length CSP antigen (FL, 50 ng/well), the repeat peptide ((NANP)6C peptide at 20 ng/well) or a recombinant protein encoding the C-terminal region of CSP (50 ng/well) essentially as described previously [44]. The secondary antibody used for the mouse and rhesus ELISA were HRP-conjugated anti-mouse or anti-rhesus IgG, respectively (Southern Biotech, Birmingham, AL, USA). Horse Radish Peroxidase labelled secondary antibodies against Rhesus IgG1 (clone 7H11), IgG2 (clone 3C10) and IgG3 (2G11) were obtained from the NIH Non-human Primate Reagent Resource. The antibody titre was calculated as the serum dilution that produced an absorbance of 1.0 optical density (OD) units using Gen5™ software (Biotek). To measure avidity, the above ELISA protocol was modified with an additional 6 M urea incubation step for 10 min following the primary antibody incubation. Equal volume of phosphate buffered saline (PBS) was added for a control. Avidity index was defined as the titre (OD = 1.0) in the presence of urea divided by the titre in the PBS control. Epitope specific ELISA was done using the following peptides—NT: DNAGTNLYNELEMNYYGKQENWYS; RI + repeat: KLKQPADGNPDPNANPNVDPNANPNVDPNANPNVDPNANP and RII: WSPCSVTCGNGIQVRIKPGSANKPKDELDYANDIEKKICKMEKCSS. All peptides were coated at 200 ng/well and 1:100 serum dilution was added to the well for 1 h. The reaction was developed as above and OD at 415 nm was recorded.
Competitive ELISA
Monoclonal antibodies 2A10 and 5D5 were obtained from the Malaria Research and Reference Reagent Resource Center MR4 (BEI Resources, Manassas, VA, USA). To determine the ability of polyclonal serum to block the binding of CSP-specific monoclonal antibodies (mAb), a competitive ELISA was developed. In brief, ELISA plates were coated overnight at 4 °C with FL antigen (15 ng/well) and then washed with PBS + Tween. Non-specific binding was blocked with PBS + 1% albumin for 1 h at room temperature (RT) and then washed. Seventy-five microliter/well of serially diluted (two-fold) mouse or rhesus serum was added to the plate and incubated for 1 h at RT. The mAbs were labelled with HRP using Lightning-Link HRP conjugation kit (Novus Biologicals, Littleton, CO, USA). Seventy-five microliter (~15 ng) of HRP-conjugated N-terminal mAb 5D5 [56], C-terminal mAb 2F12 [53] or NANP mAb 2A10 [57] was then added directly to the wells and incubated for 1 h at RT. Plates were then washed and developed by the addition of 50 µl/well of ABTS peroxidase substrate system (KPL) for 1 h at RT and stopped by adding 50 µl of 5% SDS. Absorbance was read at 415 nm on a microplate reader (Synergy 4, Biotek) using Gen5™ software (Biotek).
Sporozoite neutralization assay
To assess in vivo protective efficacy of rhesus antibodies in mice, a sporozoite neutralization assay (SNA) was developed. In brief, 50 µl of untreated serum from individual rhesus monkeys was incubated for 10 min with 50 µl of RPMI containing 3000 transgenic P. berghei sporozoites. Mice were then challenged intravenously with 100 µl of the serum/sporozoites mixture. Pre-immune rhesus serum from each of the test monkeys was used as a control. After challenge, mice were monitored for parasitaemia as described above.
Statistical analysis
Graphs show individual data points and/or the mean ± SEM (geometric mean for graphs representing titres) of each group. In all cases, a P value of <0.05 was considered significant, as determined by an unpaired two-tailed t test comparing the soluble and particulate platforms to each other within the species. Statistically significant difference in group means was indicated as * (P < 0.05), ** (P < 0.01), *** (P < 0.001) or **** (P < 0.0001). Graphs were plotted and statistics were assessed by using GraphPad Prism software (La Jolla, CA, USA).
Results
Qβ-CSP analysis and vaccination
Qβ-CSP vaccine was prepared as previously described [53] and analysed by SDS-PAGE. Figure 1 shows that CSP and the Qβ proteins were highly pure and both migrated as a single band on reducing SDS-PAGE. Following conjugation of CSP and Qβ, multiple bands were detectable (Qβ-CSP lane). A ~45 kDa band corresponded to free CSP (black arrow), ~15 kDa band corresponded to the Qβ monomer (red arrow) and ~55 kDa band (blue arrow) corresponded to the conjugated Qβ-CSP molecule. Bands above 55 kDa in the Qβ-CSP lane corresponded to a CSP molecule linked to Qβ multimers that were generated during the cysteine-coupling reaction. Densitometric quantification of conjugation revealed that 19% of Qβ monomers had a CSP molecule conjugated to them. Hence, 34 CSP molecules were conjugated to an assembled Qβ particle that is composed of 180 Qβ monomers [58]. To be able to compare the two vaccines, identical amounts of CSP were administered. For the Qβ-CSP vaccine conjugated and free CSP in the preparation were quantified as previously described [53]. The animal studies were based on previously published reports of CSP vaccine selection in mice [45] and in rhesus [21]. Mice received three vaccinations of 2.5 µg CSP at week 0, 3 and 6 and rhesus received three vaccinations of 25 µg CSP (expected human dose) at month 0, 1 and 3.

Purity of CSP, Qβ protein and conjugate Qβ-CSP. A range of CSP (black arrow, ~45 kDa) and Qβ protein (red arrow, ~15 kDa) amounts were run on reducing SDS-PAGE; loaded amounts are noted above each well. Conjugated Qβ-CSP (15 µl) was loaded in the last lane. Gel stained with Coomassie blue. Bands migrating at ~55 kDa and above (blue arrow) correspond to Qβ-CSP conjugate(s)
Kinetics of antibody response
Comparing the trend of antibody titres over three immunizations, mice (Fig. 2, coloured lines) showed a clearer separation between the Qβ-CSP (green) and CSP (red) vaccine induced FL, NANP and C-term titres as compared to rhesus (black lines). In both models Qβ-CSP showed higher responses than CSP after the first dose, however, mice and rhesus differed substantially in the response to booster vaccines. In mice, successive doses of Qβ-CSP and CSP boosted titres but little or no boosting was observed beyond the second dose in rhesus. Indeed, no boosting of NANP titres were observed in rhesus after the first dose of Qβ-CSP, while in mice boosting post-second and post-third was observed.

Immunogenicity of CSP and Qβ-CSP in mice and rhesus. ELISA results against (a) FL antigen, (b) NANP repeat or (c) C-terminus for serum collected 2–3 weeks after each immunization with CSP (solid symbols) or Qβ-CSP (open symbols). Plotted are group geometric mean antibody titre for mice (coloured lines, red or green, n = 7) and rhesus (black lines, n = 5). Fold increase of Qβ-CSP relative to CSP at each time-point is shown in Table 1
Both models agreed that FL, NANP and C-terminus titres were higher for the Qβ-CSP vaccine than CSP following the first vaccination (Table 1), but the magnitude of the difference between vaccines was maximal for the NANP epitope in mice. The NANP titre for the Qβ-CSP was >900-fold higher than CSP in mice, but only 12-fold higher in rhesus (Table 1). In mice, the Qβ-CSP maintained a clear immunological edge over CSP after the second dose with statistically significant differences in FL and NANP titres. In contrast, the differences between the two vaccines after the second dose were less clear in rhesus (Table 1). The immunogenicity difference between the two vaccines was further reduced in both models after the third dose, but mice continued to show significantly higher FL and NANP response in the Qβ-CSP group. In rhesus, the NANP titre of the Qβ-CSP vaccine group was ~2.4-fold higher than CSP, however this difference was not statistically significant.
Avidity and subtypes
Avidity and IgG subtypes were measured against FL antigen post third immunization. In mice, Qβ-CSP generated significantly higher avidity antibodies relative to CSP, while no avidity differences between the two vaccines were detectable in rhesus after the third dose (Fig. 3). As was previously reported [53], higher levels of IgG2c were induced by the Qβ-CSP vaccine compared to the CSP vaccine (data not shown); however, attempts to measure sub-classes in rhesus using either rhesus-specific or human-specific secondary antibodies were unsuccessful as only the IgG1 antibody (rhesus or human) reacted with rhesus serum.

Serum antibody avidity against FL antigen. Avidity index for mice (solid points, n = 7) and rhesus (open points, n = 5) was calculated as the titre (OD = 1.0) in the presence of urea as a percentage of the titre in the PBS control. Bars represent mean avidity index ± SEM against FL antigen at 2 weeks post-third vaccination. P values are shown for statistically significant differences between CSP and Qβ-CSP determined by two-tailed t test
Region-specific bias
Difference in antibody bias towards specific regions of the vaccine molecule was determined by normalizing individual NANP and C-term titres by the respective FL titre. The NANP bias of Qβ-CSP antibodies was greater than CSP antibodies (NANP:FL titre ratio >2) following the first and second vaccination in mice (Fig. 4a). Even after the third dose, a notable NANP bias was observed for Qβ-CSP in mice, although this was not statistically significant compared to CSP. The higher level of NANP bias induced by Qβ-CSP was replicated in rhesus (NANP:FL titre ratio ~2) (Fig. 4c), albeit only after the first dose. Mice and rhesus displayed no difference in C-terminus bias between the two vaccines (Fig. 4b, d).

Relative immunogenicity of the NANP repeat and C-terminus regions of CSP. Data are expressed as the mean titre ratio (bars) ± SEM (whiskers) for mice (a, b; n = 7) and rhesus (c, d; n = 5) of NANP repeat titres (a, c) or C-terminus titres (b, d) to their respective FL antigen ELISA titre at post-first (P1), post-second (P2) and post-third (P3) vaccination with either CSP (solid bars) or Qβ-CSP (open bars). P values are shown for statistically significant differences between CSP and Qβ-CSP determined by two-tailed t tests
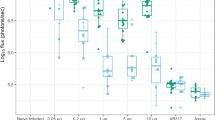
Epitope-specificity
To assess the relative abundance of antibodies against CSP epitopes, post-third vaccination sera were analysed by a direct peptide ELISA and a monoclonal antibody competition ELISA. The ‘NT’ peptide corresponded to a sequence in the N-terminus, the ‘RI + repeat’ peptide was based on the conserved region-I and NVDPNANP repeat sequence, and the ‘RII’ peptide corresponded to the cysteine-rich region-II within the C-terminus. Mouse sera raised against Qβ-CSP vaccine contained higher levels of NT peptide-reacting antibodies than CSP vaccine sera; in contrast, the corresponding rhesus sera showed little NT peptide reactivity (Fig. 5a, b). Mice also showed a higher R1 + repeat and RII peptide response in favour of Qβ-CSP vaccine after the third dose (Fig. 5a, b), while only the RII peptide ELISA showed a significant difference between the two vaccines in rhesus. The ability of post-third mouse and rhesus serum to inhibit binding of mAbs to the FL antigen was also tested in a competition ELISA (Fig. 5c, d). Mouse antibodies showed that the Qβ-CSP vaccine induced higher N-terminus (mAb 5D5) and NANP repeat (mAb 2A10)-specific binding inhibition relative to the CSP vaccine (Fig. 5c). Rhesus antibodies, however, failed to discern any difference between the two vaccines using this competition ELISA (Fig. 5d). Increased mAb 5D5 inhibition (Fig. 5c) by Qβ-CSP mouse sera correlated with the higher NT peptide ELISA responses (Fig. 5a). Overall, more epitope-specific differences between Qβ-CSP versus soluble CSP were seen in mice than in rhesus.

Region- and epitope-specific responses. a, b Points represent optical density from a peptide ELISA at 1:100 serum dilution. Peptides comprised of the N-terminus (NT), conserved region I plus NVDPNANP repeats region (RI + repeat), and the cysteine rich region II of C-terminus (RII). c, d Points represent per cent inhibition of HRP-labelled 5D5 (N-terminus), 2A10 (NANP repeat) or 2F12 (C-terminus) mAb in the presence of individual sera. Inhibition is expressed as the percent decrease in optical density obtained by ELISA with mAb alone. Bars represent group means and whiskers represent standard error of the mean (SEM) for mouse (n = 5) and rhesus (n = 7) serum from 2 weeks post-third vaccination. P values are shown for statistically significant differences between CSP and Qβ-CSP determined by two-tailed t tests
Protection
To evaluate the protective efficacy of the CSP and Qβ-CSP vaccines in mice, a transgenic P. berghei sporozoite line that expressed the full-length CSP gene of P. falciparum was used to infect the mice 2 weeks after the third vaccine dose. All control mice became infected by day 7 and there was no significant difference in the level of protection induced by the CSP and Qβ-CSP vaccines (4/7 versus 5/7; Fig. 6a), even though the FL and NANP titres of the two groups were significantly different. Since no transgenic parasites for rhesus challenge were available, individual rhesus sera were tested using an in vivo SNA in the mouse transgenic parasite challenge model. All mice representing individual rhesus pre-immune sera were infected by day 5, but rhesus sera from 4/5 Qβ-CSP group monkeys showed protection while only 1/5 rhesus sera in the CSP group conferred protection (Fig. 6b). The higher protection in the Qβ-CSP group SNA was loosely associated with a higher titre (although not significant) and smaller spread of NANP repeat antibodies (Fig. 6b).

Protection efficacy of CSP versus Qβ-CSP vaccination. Individual titres for mice (n = 7) and rhesus (n = 5) 2 weeks post-third vaccination. Red symbols represent (a) mice immunized and protected in direct challenge with 3000 transgenic P. berghei sporozoites (Tr-Pb) or (b) mice protected by challenge with 3000 Tr-Pb in SNA with rhesus serum. Blue numbers represent the number of protected mice out of the total. Bars represent group geometric mean. P values are shown for statistically significant differences between CSP and Qβ-CSP determined by two-tailed t tests
Discussion
A comparison of two malaria vaccines was conducted to determine if mice and rhesus models provided congruent antibody read-outs. While there were differences in vaccine schedule and vaccine dose between species, the two models agreed on several immunologic outcomes: (1) Qβ-CSP vaccine had overall higher immunogenicity; (2) within the CSP molecule, the relative immunogenicity of the repeat NANP epitope was specifically improved by particulate presentation on Qβ; and, (3) Qβ-CSP was better at priming antibody responses, but soluble CSP was able to boost antibody levels after subsequent doses, particularly against the NANP repeat. Apart from the similarities, the mouse and rhesus models differed from each other in the magnitude, kinetics, epitope breath and functionality of antibodies, summarized in Table 2. In general, more immunological differences between Qβ-CSP and CSP vaccines were detected in mice. The rhesus model revealed a key difference in functional activity of antibodies induced by the two vaccines using a SNA while mice failed to discern this difference upon direct challenge.
Mouse models are being used at an increasing rate to down-select second-generation malaria vaccines, but it would be costly to ignore preceding investigations where mouse data did not necessarily translate to humans. To the authors’ knowledge, this is the first report where identical malaria vaccine formulations have been compared in C57BL/6 mice and rhesus, both of which are commonly used models in malaria vaccine development. The main difference between the CSP and Qβ-CSP vaccines was the enhanced immunogenicity of Qβ-CSP, which could have resulted from its particulate nature and/or the TLR7/8 agonist activity of the entrapped RNA [59, 60]. Others have also reported that PfCSP antibodies can be readily boosted by a third immunization in mice while no additional boosting beyond the second vaccination was observed in rhesus [18, 26, 34, 61]. Not surprisingly, the boosting pattern of RTS,S in humans is similar to the rhesus after the second dose [62]. Overall, compared to rhesus, mice greatly amplified the immunogenicity differences between soluble CSP and Qβ-CSP, and this may reflect differences in the innate immune response pathways of the two species as was observed using a CpG oligodeoxynucleotide adjuvant for a Leishmania vaccine [63]. Furthermore, monkeys harbour much more genetic and epigenetic variability stemming from differences in individual age, microbiome and physiology, while the impact of these factors on immunogenicity is largely suppressed in inbred mouse strains.
Despite the relatively low immunogenicity of soluble CSP in Alum, mice were protected in a direct challenge experiment. While reasons remain unclear, high-level protection with Alum-adjuvanted soluble CSP has been reported previously in mice [53], but it is also known that Alum-adjuvanted CSP vaccines do not induce significant protection in humans [64–67]. A similar observation has been made in influenza where several Alum-adjuvanted vaccines that induce protective levels of haemagglutinin response in mice do not protect humans [49, 68]. Using a functional SNA, rhesus revealed that Qβ-CSP + Alum was more protective than CSP + Alum. This functional activity was loosely associated with the magnitude and spread of NANP titres that also happen to be the best correlate of protection for CSP-based malaria vaccines in CHMI [69]. A recent report has suggested that antibodies against the N-terminus of CSP may offer additional protection, as they can block a proteolytic processing step during sporozoite invasion [56]. In mice, the Qβ-CSP induced higher N-terminal responses than CSP, but there was no difference in rhesus. These data highlight a fundamental problem with down-selecting human vaccines in mice and cautions against over-interpreting mouse vaccine data that show enhanced epitope-specific responses, enhanced avidity or even enhanced protection, as these results may not translate to higher mammalian species. Rhesus on the other hand are not ideal for mechanistic studies in immunology as attempts to sub-class rhesus macaque IgG using available rhesus (NIH NHP Reagent Resource) or human-specific reagents were unsuccessful. Others have reported similar limitations of using human sub-class reagents to analyse rhesus IgG [70]. Rhesus produces three distinct IgG sub-classes, but there is a much greater diversity among rhesus Ig genes compared to humans [71] and the functional equivalency of rhesus and known human IgG sub-classes has not been established.
While it would be ideal to use rhesus as they best emulate the human host, mice continue to be the pre-eminent in vivo model for malaria vaccine work as evidenced by more than 1900 Pubmed hits compared to 148 on rhesus malaria vaccine research. It was found that the mouse immune system can amplify small immunological differences between formulations. Mice are therefore useful for early decision-making where one formulation must be picked among many. It is important however that the selection criteria in mice be kept fairly stringent, such as using a sterile protection outcome and conservative statistical tests to minimize Type-1 error. In rhesus, a less obvious difference between formulations was seen. Careful balance between sample size and study cost is therefore necessary to reduce the risk of a Type-2 error while down-selecting vaccines in rhesus. These data and past limitations of mouse models suggest that non-human primate immunogenicity in combination with functional assays needs to be a preferred model for final down-selection prior to clinical trials.
Conclusions
This report has direct implications for numerous ongoing efforts to down-select CSP-based malaria vaccine candidates and cautions against the use of only murine models for final down-selection of potential vaccine candidates for human studies. The immunogenicity of two malaria vaccine candidates in mice and rhesus were compared and it was found that, while the two models shared some of the immunologic outcomes, there were key differences between them. Mice showed a clear immunologic superiority of a particulate Qβ-CSP formulation over soluble CSP, while the difference in responses to the soluble and particulate antigens was muted in rhesus. With the caveat that these vaccines have not yet been tested in humans, this study suggests that there are limitations of reliance on data derived solely from the mouse model in predicting the human response to investigational vaccines and underlines the likely superiority of the rhesus model in this context.
References
Teneza-Mora N, Lumsden J, Villasante E. A malaria vaccine for travelers and military personnel: requirements and top candidates. Vaccine. 2015;33:7551–8.
Perkins SL, Sarkar IN, Carter R. The phylogeny of rodent malaria parasites: simultaneous analysis across three genomes. Infect Genet Evol. 2007;7:74–83.
Epstein JE, Tewari K, Lyke KE, Sim BK, Billingsley PF, Laurens MB, et al. Live attenuated malaria vaccine designed to protect through hepatic CD8(+) T cell immunity. Science. 2011;334:475–80.
Sedegah M, Hedstrom R, Hobart P, Hoffman SL. Protection against malaria by immunization with plasmid DNA encoding circumsporozoite protein. Proc Natl Acad Sci USA. 1994;91:9866–70.
Hoffman SL, Sedegah M, Hedstrom RC. Protection against malaria by immunization with a Plasmodium yoelii circumsporozoite protein nucleic acid vaccine. Vaccine. 1994;12:1529–33.
Richie TL, Charoenvit Y, Wang R, Epstein JE, Hedstrom RC, Kumar S, et al. Clinical trial in healthy malaria-naive adults to evaluate the safety, tolerability, immunogenicity and efficacy of MuStDO5, a five-gene, sporozoite/hepatic stage Plasmodium falciparum DNA vaccine combined with escalating dose human GM-CSF DNA. Hum Vaccines Immunother. 2012;8:1564–84.
Ling IT, Ogun SA, Holder AA. Immunization against malaria with a recombinant protein. Parasite Immunol. 1994;16:63–7.
Anders RF, Crewther PE, Edwards S, Margetts M, Matthew ML, Pollock B, Pye D. Immunisation with recombinant AMA-1 protects mice against infection with Plasmodium chabaudi. Vaccine. 1998;16:240–7.
Ogutu BR, Apollo OJ, McKinney D, Okoth W, Siangla J, Dubovsky F, et al. Blood stage malaria vaccine eliciting high antigen-specific antibody concentrations confers no protection to young children in Western Kenya. PLoS ONE. 2009;4:e4708.
Thera MA, Doumbo OK, Coulibaly D, Laurens MB, Ouattara A, Kone AK, et al. A field trial to assess a blood-stage malaria vaccine. N Engl J Med. 2011;365:1004–13.
Benton MJ, Donoghue PC. Paleontological evidence to date the tree of life. Mol Biol Evol. 2007;24:26–53.
Rhesus Macaque Genome S, Analysis C, Gibbs RA, Rogers J, Katze MG, Bumgarner R, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–34.
Ta TH, Hisam S, Lanza M, Jiram AI, Ismail N, Rubio JM. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar J. 2014;13:68.
Hamid MM, Remarque EJ, El Hassan IM, Hussain AA, Narum DL, Thomas AW, et al. Malaria infection by sporozoite challenge induces high functional antibody titres against blood stage antigens after a DNA prime, poxvirus boost vaccination strategy in rhesus macaques. Malar J. 2011;10:29.
Dutta S, Kaushal DC, Ware LA, Puri SK, Kaushal NA, Narula A, et al. Merozoite surface protein 1 of Plasmodium vivax induces a protective response against Plasmodium cynomolgi challenge in rhesus monkeys. Infect Immun. 2005;73:5936–44.
Kusi KA, Remarque EJ, Riasat V, Walraven V, Thomas AW, Faber BW, et al. Safety and immunogenicity of multi-antigen AMA1-based vaccines formulated with CoVaccine HT and Montanide ISA 51 in rhesus macaques. Malar J. 2011;10:182.
Pichyangkul S, Spring MD, Yongvanitchit K, Kum-Arb U, Limsalakpetch A, Im-Erbsin R, et al. Chemoprophylaxis with sporozoite immunization in P. knowlesi rhesus monkeys confers protection and elicits sporozoite-specific memory T cells in the liver. PLoS ONE. 2017;12:e0171826.
Stewart VA, McGrath SM, Walsh DS, Davis S, Hess AS, Ware LA, et al. Pre-clinical evaluation of new adjuvant formulations to improve the immunogenicity of the malaria vaccine RTS, S/AS02A. Vaccine. 2006;24:6483–92.
Kester KE, Cummings JF, Ofori-Anyinam O, Ockenhouse CF, Krzych U, Moris P, et al. Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS, S/AS01B and RTS, S/AS02A in malaria-naive adults: safety, efficacy, and immunologic associates of protection. J Infect Dis. 2009;200:337–46.
Polhemus ME, Remich SA, Ogutu BR, Waitumbi JN, Otieno L, Apollo S, et al. Evaluation of RTS, S/AS02A and RTS, S/AS01B in adults in a high malaria transmission area. PLoS ONE. 2009;4:e6465.
Stewart VA, McGrath SM, Dubois PM, Pau MG, Mettens P, Shott J, et al. Priming with an adenovirus 35-circumsporozoite protein (CS) vaccine followed by RTS, S/AS01B boosting significantly improves immunogenicity to Plasmodium falciparum CS compared to that with either malaria vaccine alone. Infect Immun. 2007;75:2283–90.
Ockenhouse CF, Regules J, Tosh D, Cowden J, Kathcart A, Cummings J, et al. Ad35.CS.01-RTS, S/AS01 heterologous prime boost vaccine efficacy against sporozoite challenge in healthy malaria-naive adults. PLoS ONE. 2015;10:e0131571.
Weiss WR, Jiang CG. Protective CD8+ T lymphocytes in primates immunized with malaria sporozoites. PLoS ONE. 2012;7:e31247.
Seder RA, Chang LJ, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science. 2013;341:1359–65.
Ishizuka AS, Lyke KE, DeZure A, Berry AA, Richie TL, Mendoza FH, et al. Protection against malaria at 1 year and immune correlates following PfSPZ vaccination. Nat Med. 2016;22:614–23.
Pichyangkul S, Tongtawe P, Kum-Arb U, Yongvanitchit K, Gettayacamin M, Hollingdale MR, et al. Evaluation of the safety and immunogenicity of Plasmodium falciparum apical membrane antigen 1, merozoite surface protein 1 or RTS, S vaccines with adjuvant system AS02A administered alone or concurrently in rhesus monkeys. Vaccine. 2009;28:452–62.
Vanloubbeeck Y, Pichyangkul S, Bayat B, Yongvanitchit K, Bennett JW, Sattabongkot J, et al. Comparison of the immune responses induced by soluble and particulate Plasmodium vivax circumsporozoite vaccine candidates formulated in AS01 in rhesus macaques. Vaccine. 2013;31:6216–24.
Bennett JW, Yadava A, Tosh D, Sattabongkot J, Komisar J, Ware LA, et al. Phase 1/2a trial of Plasmodium vivax malaria vaccine candidate VMP001/AS01B in malaria-naive adults: safety, immunogenicity, and efficacy. PLoS Negl Trop Dis. 2016;10:e0004423.
Mlambo G, Kumar N. Transgenic rodent Plasmodium berghei parasites as tools for assessment of functional immunogenicity and optimization of human malaria vaccines. Eukaryot Cell. 2008;7:1875–9.
Salman AM, Mogollon CM, Lin JW, van Pul FJ, Janse CJ, Khan SM. Generation of transgenic rodent malaria parasites expressing human malaria parasite proteins. Methods Mol Biol. 2015;1325:257–86.
Othoro C, Johnston D, Lee R, Soverow J, Bystryn JC, Nardin E. Enhanced immunogenicity of Plasmodium falciparum peptide vaccines using a topical adjuvant containing a potent synthetic Toll-like receptor 7 agonist, imiquimod. Infect Immun. 2009;77:739–48.
Carapau D, Mitchell R, Nacer A, Shaw A, Othoro C, Frevert U, Nardin E. Protective humoral immunity elicited by a needle-free malaria vaccine comprised of a chimeric Plasmodium falciparum circumsporozoite protein and a Toll-like receptor 5 agonist, flagellin. Infect Immun. 2013;81:4350–62.
McCoy ME, Golden HE, Doll TA, Yang Y, Kaba SA, Zou X, et al. Mechanisms of protective immune responses induced by the Plasmodium falciparum circumsporozoite protein-based, self-assembling protein nanoparticle vaccine. Malar J. 2013;12:136.
Kastenmuller K, Espinosa DA, Trager L, Stoyanov C, Salazar AM, Pokalwar S, et al. Full-length P. falciparum circumsporozoite protein administered with poly-ICLC or GLA/SE elicits potent antibody and CD4+ T cell immunity and protection in mice. Infect Immun. 2013;81:789–800.
Noe AR, Espinosa D, Li X, Coelho-Dos-Reis JG, Funakoshi R, Giardina S, et al. A full-length Plasmodium falciparum recombinant circumsporozoite protein expressed by Pseudomonas fluorescens platform as a malaria vaccine candidate. PLoS ONE. 2014;9:e107764.
Mlambo G, Kumar N, Yoshida S. Functional immunogenicity of baculovirus expressing Pfs25, a human malaria transmission-blocking vaccine candidate antigen. Vaccine. 2010;28:7025–9.
Persson C, Oliveira GA, Sultan AA, Bhanot P, Nussenzweig V, Nardin E. Cutting edge: a new tool to evaluate human pre-erythrocytic malaria vaccines: rodent parasites bearing a hybrid Plasmodium falciparum circumsporozoite protein. J Immunol. 2002;169:6681–5.
Mizutani M, Iyori M, Blagborough AM, Fukumoto S, Funatsu T, Sinden RE, et al. Baculovirus-vectored multistage Plasmodium vivax vaccine induces both protective and transmission-blocking immunities against transgenic rodent malaria parasites. Infect Immun. 2014;82:4348–57.
Espinosa DA, Yadava A, Angov E, Maurizio PL, Ockenhouse CF, Zavala F. Development of a chimeric Plasmodium berghei strain expressing the repeat region of the P. vivax circumsporozoite protein for in vivo evaluation of vaccine efficacy. Infect Immun. 2013;81:2882–7.
Bauza K, Malinauskas T, Pfander C, Anar B, Jones EY, Billker O, et al. Efficacy of a Plasmodium vivax malaria vaccine using ChAd63 and modified vaccinia Ankara expressing thrombospondin-related anonymous protein as assessed with transgenic Plasmodium berghei parasites. Infect Immun. 2014;82:1277–86.
Ramjanee S, Robertson JS, Franke-Fayard B, Sinha R, Waters AP, Janse CJ, et al. The use of transgenic Plasmodium berghei expressing the Plasmodium vivax antigen P25 to determine the transmission-blocking activity of sera from malaria vaccine trials. Vaccine. 2007;25:886–94.
Tewari R, Spaccapelo R, Bistoni F, Holder AA, Crisanti A. Function of region I and II adhesive motifs of Plasmodium falciparum circumsporozoite protein in sporozoite motility and infectivity. J Biol Chem. 2002;277:47613–8.
Aldrich C, Magini A, Emiliani C, Dottorini T, Bistoni F, Crisanti A, et al. Roles of the amino terminal region and repeat region of the Plasmodium berghei circumsporozoite protein in parasite infectivity. PLoS ONE. 2012;7:e32524.
Porter MD, Nicki J, Pool CD, DeBot M, Illam RM, Brando C, et al. Transgenic parasites stably expressing full-length Plasmodium falciparum circumsporozoite protein as a model for vaccine down-selection in mice using sterile protection as an endpoint. Clin Vaccine Immunol. 2013;20:803–10.
Schwenk R, DeBot M, Porter M, Nikki J, Rein L, Spaccapelo R, et al. IgG2 antibodies against a clinical grade Plasmodium falciparum CSP vaccine antigen associate with protection against transgenic sporozoite challenge in mice. PLoS ONE. 2014;9:e111020.
Coppi A, Natarajan R, Pradel G, Bennett BL, James ER, Roggero MA, et al. The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J Exp Med. 2011;208:341–56.
Ambuhl PM, Tissot AC, Fulurija A, Maurer P, Nussberger J, Sabat R, et al. A vaccine for hypertension based on virus-like particles: preclinical efficacy and phase I safety and immunogenicity. J Hypertens. 2007;25:63–72.
Bachmann MF, Jennings GT. Therapeutic vaccines for chronic diseases: successes and technical challenges. Philos Trans R Soc Lond B Biol Sci. 2011;366:2815–22.
Low JG, Lee LS, Ooi EE, Ethirajulu K, Yeo P, Matter A, et al. Safety and immunogenicity of a virus-like particle pandemic influenza A (H1N1) 2009 vaccine: results from a double-blinded, randomized Phase I clinical trial in healthy Asian volunteers. Vaccine. 2014;32:5041–8.
Cornuz J, Zwahlen S, Jungi WF, Osterwalder J, Klingler K, van Melle G, et al. A vaccine against nicotine for smoking cessation: a randomized controlled trial. PLoS ONE. 2008;3:e2547.
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–9.
Schmitz N, Beerli RR, Bauer M, Jegerlehner A, Dietmeier K, Maudrich M, et al. Universal vaccine against influenza virus: linking TLR signaling to anti-viral protection. Eur J Immunol. 2012;42:863–9.
Khan F, Porter M, Schwenk R, DeBot M, Saudan P, Dutta S. Head-to-head comparison of soluble vs. Qbeta VLP circumsporozoite protein vaccines reveals selective enhancement of NANP repeat responses. PLoS ONE. 2015;10:e0142035.
Lenth RV. Java applets for power and sample size. http://homepage.divms.uiowa.edu/~rlenth/Power/; 2006. Accessed 27 Feb 2017.
Ozaki LS, Gwadz RW, Godson GN. Simple centrifugation method for rapid separation of sporozoites from mosquitoes. J Parasitol. 1984;70:831–3.
Espinosa DA, Gutierrez GM, Rojas-Lopez M, Noe AR, Shi L, Tse SW, et al. Proteolytic cleavage of the Plasmodium falciparum circumsporozoite protein is a target of protective antibodies. J Infect Dis. 2015;212:1111–9.
Kumar S, Zheng H, Sangweme DT, Mahajan B, Kozakai Y, Pham PT, et al. A chemiluminescent-western blot assay for quantitative detection of Plasmodium falciparum circumsporozoite protein. J Immunol Methods. 2013;390:99–105.
Golmohammadi R, Fridborg K, Bundule M, Valegard K, Liljas L. The crystal structure of bacteriophage Q beta at 3.5 A resolution. Structure. 1996;4:543–54.
Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–96.
Akache B, Weeratna RD, Deora A, Thorn JM, Champion B, Merson JR, et al. Anti-IgE Qb-VLP conjugate vaccine self-adjuvants through activation of TLR7. Vaccines. 2016;4:3.
Tewari K, Flynn BJ, Boscardin SB, Kastenmueller K, Salazar AM, Anderson CA, et al. Poly(I:C) is an effective adjuvant for antibody and multi-functional CD4+ T cell responses to Plasmodium falciparum circumsporozoite protein (CSP) and alphaDEC-CSP in non human primates. Vaccine. 2010;28:7256–66.
Regules JA, Cicatelli SB, Bennett JW, Paolino KM, Twomey PS, Moon JE, et al. Fractional third and fourth dose of RTS, S/AS01 malaria candidate vaccine: a phase 2a controlled human malaria parasite infection and immunogenicity study. J Infect Dis. 2016;214:762–71.
Verthelyi D, Kenney RT, Seder RA, Gam AA, Friedag B, Klinman DM. CpG oligodeoxynucleotides as vaccine adjuvants in primates. J Immunol. 2002;168:1659–63.
Ballou WR, Hoffman SL, Sherwood JA, Hollingdale MR, Neva FA, Hockmeyer WT, et al. Safety and efficacy of a recombinant DNA Plasmodium falciparum sporozoite vaccine. Lancet. 1987;1:1277–81.
Fries LF, Gordon DM, Schneider I, Beier JC, Long GW, Gross M, et al. Safety, immunogenicity, and efficacy of a Plasmodium falciparum vaccine comprising a circumsporozoite protein repeat region peptide conjugated to Pseudomonas aeruginosa toxin A. Infect Immun. 1992;60:1834–9.
Herrington DA, Clyde DF, Davis JR, Baqar S, Murphy JR, Cortese JF, et al. Human studies with synthetic peptide sporozoite vaccine (NANP)3-TT and immunization with irradiated sporozoites. Bull World Health Organ. 1990;68(Suppl):33–7.
Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, et al. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS, S Malaria Vaccine Evaluation Group. N Engl J Med. 1997;336:86–91.
Jegerlehner A, Zabel F, Langer A, Dietmeier K, Jennings GT, Saudan P, Bachmann MF. Bacterially produced recombinant influenza vaccines based on virus-like particles. PLoS ONE. 2013;8:e78947.
White MT, Bejon P, Olotu A, Griffin JT, Riley EM, Kester KE, et al. The relationship between RTS, S vaccine-induced antibodies, CD4(+) T cell responses and protection against Plasmodium falciparum infection. PLoS ONE. 2013;8:e61395.
Ruiz W, McClements WL, Jansen KU, Esser MT. Kinetics and isotype profile of antibody responses in rhesus macaques induced following vaccination with HPV 6, 11, 16 and 18 L1-virus-like particles formulated with or without Merck aluminum adjuvant. J Immune Based Ther Vaccines. 2005;3:2.
Scinicariello F, Engleman CN, Jayashankar L, McClure HM, Attanasio R. Rhesus macaque antibody molecules: sequences and heterogeneity of alpha and gamma constant regions. Immunology. 2004;111:66–74.
Authors’ contributions
TWP and CJG developed and conducted assays; ADM and NAH conducted the rhesus study; MDP and MD conducted the mouse study; FAK produced the vaccine antigen; SD, TWP, CJG, PS, and NCW wrote the paper; SD and PS designed the study. All authors read and approved the final manuscript.
Acknowledgements
This work was supported in part by an appointment to the Postgraduate Research Participation Program at the Walter Reed Army Institute of Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and MRMC. The authors thank Lisa Dlugosz for laboratory support and thank Lorraine Soisson and Carter Diggs for comments on the paper and advice.
Competing interests
SD holds a patent on the CSP vaccine antigen.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval
Research was conducted under an approved animal use protocol in an AAALACi accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition.
Funding
The funding for this work was provided by the USAID Malaria Vaccine Development Program and the Department of the Army. The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Disclaimer
Material has been reviewed by the Walter Reed Army Institute of Research and the US Agency for International Development. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the authors, and are not to be construed as official, or as reflecting true views of the Department of the Army, the Department of Defense, or the US Agency for International Development.
Author information
Authors and Affiliations
Corresponding author
Additional information
Timothy W. Phares and Anthony D. May are first authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Phares, T.W., May, A.D., Genito, C.J. et al. Rhesus macaque and mouse models for down-selecting circumsporozoite protein based malaria vaccines differ significantly in immunogenicity and functional outcomes. Malar J 16, 115 (2017). https://doi.org/10.1186/s12936-017-1766-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-017-1766-3