
Abstract
Background
Plasmodium falciparum resistance to chloroquine (CQ) denied healthcare providers access to a cheap and effective anti-malarial drug. Resistance has been proven to be due to point mutations on the parasite’s pfcrt gene, particularly on codon 76, resulting in an amino acid change from lysine to threonine. This study sought to determine the prevalence of the pfcrt K76T mutation 13 years after CQ cessation in Msambweni, Kenya.
Methods
Finger-prick whole blood was collected on 3MM Whatman® filter paper from 99 falciparum malaria patients. Parasite DNA was extracted via the Chelex method from individual blood spots and used as template in nested PCR amplification of pfcrt. Apo1 restriction enzyme was used to digest the amplified DNA to identify the samples as wild type or sensitive at codon 76. Prevalence figures of the mutant pfcrt 76T gene were calculated by dividing the number of samples bearing the mutant gene with the total number of samples multiplied by 100 %. Chi square tests were used to test the significance of the findings against previous prevalence figures.
Results
Out of 99 clinical samples collected in 2013, prevalence of the mutant pfcrt 76T gene stood at 41 %.
Conclusion
The results indicate a significant [χ2 test, P ≤ 0.05 (2006 vs 2013)] reversal to sensitivity by the P. falciparum population in the study site compared to the situation reported in 2006 at the same study site. This could primarily be driven by diminished use of CQ in the study area in line with the official policy. Studies to establish prevalence of the pfcrt 76T gene could be expanded countrywide to establish the CQ sensitivity status and predict a date when CQ may be re-introduced as part of malaria chemotherapy.
Similar content being viewed by others

Background
Malaria in the 21st Century continues to pose a significant challenge to the health and well-being of populations living in malaria-endemic regions worldwide. As per the latest estimates, 198 million cases of malaria occurred globally in 2013, leading to 584,000 preventable deaths [1]. The burden exerted by this disease is heaviest on the African continent, where 90 % of the deaths due to malaria globally occurred [2, 3]. Of these, 78 % were children under the age of 5 years [1, 4]. In Kenya, malaria accounted for 8.82 million cases out of the 41.8 million of outpatient cases reported across Kenyan healthcare facilities in the year 2013 [5]. Pregnant women and young children make up the most vulnerable group to the disease, with 3 % of all deaths in children ≤5 years of age attributed to malaria [5, 6].
Accurate diagnosis followed by prompt treatment with efficacious anti-malarial drugs remains an important cornerstone in the fight against the disease [7]. Microscopy is recommended as the standard tool for parasitological confirmation of malaria as it is highly adaptable to the poor and marginalized setting in most malaria-endemic countries where the majority of cases occur [1, 7]. Rapid diagnostic test (RDT) strips are also employed in malaria diagnosis and are based on detection of specific parasite antigens in a patient’s blood sample. With proper training, RDTs are easy to use and sensitive in detecting low parasitaemia [8, 9]. Efforts to fight back the severe socio-economic impact of the disease have in recent decades been hampered by emergence of strains of Plasmodium falciparum- and Plasmodium vivax-resistant to past and present anti-malarial drugs [10, 11]. Developed in the 1930s as an effective replacement for quinine, chloroquine (CQ) was quickly adopted as the drug of choice against malaria in all malaria-endemic regions globally [12]. Its key selling points were its low cost, low toxicity and high efficacy in clinical cases, which in the decades following its development made the greatest impact in sub-Saharan Africa where morbidity and mortality due to malaria was drastically reduced [13]. Extensive use and availability of CQ as a monotherapy for malaria prophylaxis led to the emergence of CQ-resistant (CQR) P. falciparum and P. vivax strains [12, 14]. First noted in Southeast Asia and South America in the late 1950s, CQR P. falciparum spread out from these foci to cover all malaria-endemic regions globally, emerging in Africa in the 1970s [14]. Plasmodium vivax resistance to CQ emerged much later, pointing to a different genetic mechanism in acquisition of the CQR phenotype [15]. CQ targets the parasite haematin detoxification pathway in the parasite digestive vacuole (DV) in a two-pronged attack where it adsorbs onto growing hemozoin polymers and also binds to toxic haematin molecules generated as the parasite digests host haemoglobin resulting in accumulation of toxic haematin in the DV [16, 17]. Genetic cross studies between CQR and CQ-sensitive (CQS) laboratory strains of P. falciparum identified a 13 exon gene, pfcrt, on chromosome 7 as the candidate gene for the CQR phenotype [18]. The translated product of pfcrt is a multi-domain transmembrane protein localized on the parasite DV. It is thought to play a role in maintenance of osmotic balance by shuttling one or more essential osmolytes across the DV membrane [18–20]. To counter CQ, P. falciparum is believed to primarily employ a mutant pfcrt product to increase efflux of CQ from the DV [20]. This phenotype is the result of complex genetic changes involving point mutations on ten or more codons of the pfcrt gene [18, 21, 22]. Numerous studies have identified the K76T mutation as the central factor in CQR development [18, 21, 23]. This mutation is present in all CQR laboratory lines and field isolates from various malaria-endemic regions and can reliably be used as a CQR marker alone [23] or in concert with other pfcrt mutations. Other point mutations on pfcrt aside from K76T seem only to modulate CQR as determined by the change on codon 76 [23]. The Pgh-1 (P-glycoprotein homologue 1) encoded by the P. falciparum pfmdr-1 gene bearing specific mutations, particularly the N86Y substitution, has been mentioned as a CQR modulator [24, 25]. Statistical associations between N86Y pfmdr-1 and CQR were drawn initially but this gene was later shown to bear no independent effect on CQR and neither does it strengthen K76T based CQR [23]. Its role in CQR may however relate to parasite fitness adaptation in response to physiological changes from pfcrt point mutations [12].
Therapeutic efficacy studies are critical in guiding drug policy and are recommended every 2 years where possible [1]. In light of emerging resistance to current first line anti-malarial drugs, namely artemisinin, the importance of reconsidering the efficacy of CQ cannot be understated, given the time interval since its withdrawal. Malawi was the first country in Africa to cease administration of CQ in malaria chemotherapy in 1993 following widespread treatment failure [24]. Kenya followed suit in 1999 as did many sub-Saharan countries on the continent [26]. Sulfadoxine-pyrimethamine (SP) replaced CQ as the first-line anti-malarial drug in these countries but was discontinued in 2004 following rapid development of parasite resistance to these drugs and replaced with artemisinin-based combination therapy (ACT) [7]. Where CQ use was effectively discontinued through concerted public awareness efforts, studies have shown that in absence of drug pressure CQR strains of P. falciparum with an associated loss in fitness have steadily been replaced by CQ-sensitive (CQS isolates). As has been the case especially in Malawi, where the withdrawal of CQ was supported by an intensive national sensitization programme, the population frequencies of mutations associated with CQR in pfcrt have reduced from 85 % in 1992 to 13 % in 2000 [24]. Patterns in CQR reversal heavily depend on national drug policies and public awareness campaigns as evidenced by persistence of CQR strains of P. falciparum in certain regions where CQ withdrawal was inconclusive in contrast with regions that efficiently removed CQ out of national circulation [24]. Although the trend has not been as drastic as in Malawi, other reports of significant rises in prevalence of CQS P. falciparum isolates have emerged [27]. This reversal to CQS presents an interesting scenario to drug policy makers in these regions where CQ can be re-introduced as malaria chemotherapy, although current knowledge would advocate for CQ administration in concert with another anti-malarial drug that differs in mechanism of action to slow down development of anti-malarial drug resistance [1, 7, 24]. Sixteen years have passed since Kenya withdrew CQ as the first line anti-malarial drug in 1999. A retrospective study done in Kilifi, Kenya [27] reported a pfcrt 76T prevalence of 63 %, down from 94 %, in the 13 years that had passed since CQ withdrawal. The current study thus sought to survey the current sensitivity levels of P. falciparum to CQ in Msambweni area, coastal Kenya, 16 years after official withdrawal of CQ as the first line of treatment for uncomplicated malaria.
Methods
Study site
The study was carried out in the Kenyan coastal constituency of Msambweni, Kwale County. The area is located about 56 km south of Mombasa city and borders Tanzania to the south. The area sits at less than 300 m above sea level and its climate is hot and humid with annual average rainfall between 900 and 1500 mm. Humidity ranges between 70 and 80 % with an annual mean temperature ranging between 22 and 34 °C. According to the 2010 Kenya Malaria Indicator Survey, Kwale County is a malaria-endemic zone with stable P. falciparum transmission. Malaria in this area accounts for 40 % of all outpatient visits and 40 % of all inpatient admissions. Plasmodium falciparum is transmitted by two main vectors in this area: Anopheles gambiae and Anopheles funestus. These vectors exist all through the year but their population peaks with the short rains in the months between April and June and October and November.
Ethical considerations
The study was be approved by the Kenya Medical Research Institute (KEMRI) Nairobi, Ethical Review Committee (ERC). Informed consent was obtained from parents of eligible children aged between 6 months and 10 years before they were enrolled in the study.
Study population
Patients visiting the outpatient clinic at Msambweni District hospital were enrolled in the study based on the following criteria: consent, mono-infection of P. falciparum with parasitaemia between 1000 and 200,000 parasites/µL of blood, axillary temperature ≥37.5 °C, or with a history of fever, and no history of anti-malarial drug intake during the previous week. Patients were excluded from the study if there was administration of any additional anti-malarial drugs, emergence of any non-malarial febrile illness that would interfere with the classification of the treatment outcome, patient relocation from the study site, and withdrawal from the study.
Experimental design
Sample collection
Pre-treatment (day zero) blood samples were collected from patients by finger prick as dried blood spots on 3MM Whatman® filter papers, packaged individually in zip lock bags with a desiccant and transported to KEMRI headquarters in Nairobi, Kenya.
Pfcrt amplification
DNA extraction was done as described by Warhurst et al. [28]. The amplification of the pfcrt gene was done on a GeneAmp® PCR system 9700 machine. PCR amplification was as follows: in a final reaction volume of 25 µL, the outer PCR consisted of 1× PCR buffer (Roche®) 1.5 mM MgCl2, 200 µM dNTP mix, 100 nM each of CRTP1 (GCGCGCGCATGGCTCACGTTTAGGTGGAG) and CRTP2 (GGGCCCGGCGGATGTTACAAAACTATAGTTACC) and 0.1 µL of Taq polymerase (Bioline®, 5 U/µL). The mixture was topped up to a volume of 25 µL with nuclease-free PCR water. The PCR profile was: 95 °C for 5 min, 92 °C for 30 s, 45 °C for 30 s, 65 °C for 45 s, 45 cycles, final extension at 72 °C for 15 min then held at 4 °C. In a final volume of 25 µL the nested PCR consisted of 1 µL outer PCR product, 1× PCR buffer (Roche®) 1.5 mM MgCl2, 200 µM dNTP mix, 100 nM each of CRTD1 (TGTGCTCATGTGTTTAAACTT) and CRTD2 (CAAAACTATAGTTACCAATTTTG and 0.1 µL of Taq polymerase (Bioline®, 5U/µL). The mixture was topped up to a volume of 25 µL with nuclease-free PCR water. The PCR profile was set at 94 °C for 3 min, 94 °C for 30 s, 48 °C for 30 s, 64 °C for 1 min, 30 cycles, final extension at 64 °C for 5 min then halted at 4 °C.
Restriction digests of the pfcrt gene
Allele specific restriction analysis was done using restriction endonuclease Apo1 (New England Bio Labs). Briefly, in a final volume of 20, 1.5 µL of 10× buffer 3, 0.15 µL of bovine serum antigen, 8 µL of amplified DNA, 0.5 µL of Apo1 restriction endonuclease was added to 9.85 µL of nuclease-free PCR water. Incubation was done for between 12 and 14 h at 50 °C with no agitation. Digested samples were analysed on a 2 % agarose gel (Bioline®) in TAE buffer for 35 min at 80 V, with no-digest controls run in adjacent wells on the gel.
Results
A total of 99 samples collected from the Msambweni District Hospital, Kwale County in 2013 were analysed in this study, all of which were from symptomatic, mono-infected microscopy-positive patients. The mean age of the patients, in years, was 4.93 ± SD 2.76 (IQR: 4.64–5.22). Of these, 46.8 % were female and 53.2 % were males. The geometric mean of the asexual microscopic parasite density was 11,813 parasites/µL of blood (95 % CI: 9690–14,402). All the samples run for pfcrt gene amplified successfully and had the characteristic 145-bp fragment. Of the positive controls amplified alongside the field samples, the P. falciparum W2 clone was resistant to digestion using ApoI restriction enzyme while the D6 clone digestion resulted in two bands, approximately 99 and 46 bp long, as expected. These positive controls were used to score the amplified and digested field samples as either mutant or wild type. Using this criterion, 59 % (95 % CI: 0.4824–0.6826) of the samples were sensitive to ApoI digestion, thus CQ sensitive, while 41 % (95 % CI: 0.3221–0.5126) of the samples carried the allele conferring resistance to CQ (Fig. 1). In the case of mixed samples harbouring both resistant and sensitive clones, ApoI digestion was expected to yield three fragments 145, 99 and 46 bp in length. None of the samples yielded this type of banding pattern thus no mixed samples were identified in this study.

Results obtained after Pfcrt amplicon digestion using ApoI restriction enzyme. Of the 99 samples analysed in the study, 59 % (95 % CI: 0.4824–0.6826) were found to be sensitive to ApoI digestion, thus sensitive to chloroquine, while 41 % were resistant to chloroquine (95 % CI: 0.3221–0.5126)
Discussion
The widespread parasite resistance to CQ on the African continent in the 1990s led to its official ban as a first-line treatment drug for uncomplicated P. falciparum malaria in many African countries [24, 26]. The loss of this effective and cheap anti-malarial drug led policy makers to opt for alternative drugs to treat malaria, with SP being the leading candidate for replacement of CQ [24, 27]. Towards the turn of the Millennium, the prevalence of the mutant pfcrt allele in Kenya stood at 100 % [29] indicating total failure of CQ as a first-line anti-malarial drug. A key reason behind this treatment failure was the sustained use of CQ as a monotherapy in treatment of uncomplicated malaria for decades since its development [12]. This led to the official ban on CQ as a first-line anti-malarial drug and the subsequent introduction of SP and amodiaquine (AQ), as first- and second-line anti-malarial treatment, respectively, in 1999. Cases of resistance to SP were already emerging in the late 1990s and in the years following its use as the first-line anti-malarial there were widespread reports of P. falciparum resistance to SP which necessitated the change in malaria treatment regimen in Kenya in 2004 [30]. Artemether-lumefantrine (AL) was selected as the first-line drug in malaria chemotherapy with oral quinine (QN) being the second-line drug for treatment of uncomplicated falciparum malaria [30, 31]. pfcrt mutations conferring CQ resistance have been reported to enhance sensitivity to QN, a feature regarded as a possible exploitable weakness in the CQ resistance mechanism. In 2010 [7] QN was dropped in favour of a safer and less toxic alternative: dihydroartemisinin-piperaquine (DHA-PPQ) as the second-line drug of choice for malaria chemotherapy.
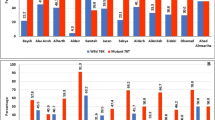
In this study, the authors sought to establish current status of CQ resistance in Msambweni in the 13 years since CQ was officially banned as malaria chemotherapy. Resistance to CQ is linked to a mutation at codon 76 of the P. falciparum pfcrt gene, resulting in an amino acid change from lysine to threonine and this has been proven to be the chief determinant of CQ resistance [18]. Absence of the 76T mutation at this codon has been shown to be a reliable predictor of successful CQ treatment. The current study shows that the prevalence of the mutant pfcrt stands at 41 % within a population sampled in 2013, 14 years after official withdrawal of CQ as the first choice in malaria chemotherapy. This decrease is loosely concordant with the Malawian reduction after CQ withdrawal in 1993 for varied reasons [27]. In a 2009 study carried out within Kilifi, the same geographical area as this survey, the prevalence of the mutant pfcrt allele was 63 % [27] (Fig. 2). Compared to the current study, this represents a significant (P = 0.0029) drop in the level of P. falciparum CQ resistance in the seven-year period between the two studies. Interestingly, the prevalence of the mutant pfcrt allele seems to be very discordant (P < 0.0001) across different areas in Kenya in samples analysed between 2005 and 2008. A study conducted in 2005 reported a pfcrt 76T prevalence of 94 % in Mwea, central Kenya [32]. Around the same time period, other studies published pfcrt 76T prevalence figures of 49 % [33] and 63 % [27] in coastal Kenya and 27.59 % in western Kenya [34]. The stark contrasts noted here could probably be explained by the disparities in enforcement of the ban on CQ as an anti-malarial drug [27]. CQ remained locally available in the years following its official withdrawal as a first-line anti-malarial drug in Kenya [35] although the withdrawal of CQ in the Kilifi area was supported by intensive public awareness programmes as to the diminished efficacy of CQ as an anti-malarial drug [27], and the availability of SP as a more efficacious alternative.

Trends in prevalence of the Pfcrt mutant and wild type alleles across different study sites in Kenya from 1997 through to the current 2013 study. There is a clearly observable reduction in the overall prevalence of the mutant pfcrt allele, which is in line with the results obtained in the current study
The absence of drug pressure is believed to be a key driver in the resurgence of CQS parasites in the field setting [24]. The mutations that confer P. falciparum resistance to CQ come with a significant loss of fitness, causing the CQS parasites to outgrow the mutant strains [24, 32], thus increasing CQ efficacy. Removal of CQ from anti-malarial therapy in Malawi due to widespread treatment failure was followed by a drastic reduction in the prevalence of the mutant pfcrt allele from 85 % in 1993 to 13 % in 2000 [24]. Although the reduction in the Kenyan context has not been as dramatic, the 41 % pfcrt 76T prevalence reported in this study represents an encouraging scenario where the proportion of CQR strains has been surpassed by the proportion of CQS isolates. The slight but significant drop (P = 0.0289) in prevalence of the mutant pfcrt allele from an absolute saturation in 1997 to 94 % (Fig. 2) in a 2005 follow-up study could be attributed to use of AQ as a second-line anti-malarial drug in the official treatment guidelines since AQ selects for pfcrt mutant strains. This is in addition to the alleged incomplete withdrawal of CQ from private dispensaries and pharmacies [32].
Although noteworthy, the 41 % prevalence of the mutant pfcrt allele still needs further investigation to define the CQ resistance patterns across the country with a view to explain any observed discrepancies in CQ sensitivity levels. The reported 41 % prevalence is based on analysis of only the pfcrt gene without comparative prevalence data from the alleged CQR modulator, pfmdr-1 N86Y. The Kilifi report published in 2009 predicted that it would take another 13 years before the CQ resistance levels dipped to a point where policy makers may consider re-introduction of CQ in malaria therapy, albeit in concert with another, different, class of anti-malarial drug. In the 7 years since that prediction was made, there appears to have been a 22 % drop in CQ resistance in the Kilifi area of Kenya. From the results of the current study, it may be predicted that reversion to CQ sensitivity could take longer than initially projected [27] in the Kilifi area. It is possible that sustained presence of the mutant pfcrt allele may also be driven in part by genetic compensatory mechanisms in the CQR parasites allowing them to exist in absence of CQ drug pressure.
Conclusion
The current study has identified an encouraging increase in the prevalence of CQS parasites in the said study area. This could be attributed to a number of factors, chief among which is the reduction or absence of drug pressure to sustain CQR P. falciparum strains. Parasite genetic adaptations in absence of drug pressure could also probably explain the sustained presence of the mutant pfcrt 76T allele in the population. The World Health Organisation (WHO) set a maximum threshold of 10 % drug resistance before an anti-malarial drug can be banned on the basis of failed efficacy. Although lower than previous years, a pfcrt 76T prevalence of 41 % is still much higher than the WHO standard and it would be advisable to investigate if the results of this study can be replicated in other malaria-endemic regions in Kenya. Such investigations could, where resources allow, incorporate other genotyping methods of greater sensitivity [36, 37] in addition to those outlined in this paper to provide a clearer picture of the Kenyan CQR status and test the CQS reversion predictions made previously [27]. This study concludes that there are encouraging indications to suggest the P. falciparum resistance to CQ is steadily declining which could be good news for Kenyan drug policy makers as CQ is cheaper and less toxic than other anti-malarial drugs. If CQ sensitivity can be restored in the Kenyan population, this cheap and effective drug can be in future incorporated into anti-malarial therapy in concert with another anti-malarial drug.
Abbreviations
- KEMRI:
-
Kenya Medical Research Institute
- CQ:
-
chloroquine
- pfcrt :
-
Plasmodium falciparum chloroquine resistance transporter gene
- pfmdr-1 :
-
Plasmodium falciparum multidrug resistance-1 gene
- SP:
-
sulphadoxine-pyrimethamine
- AQ:
-
amodiaquine
- AL:
-
artemether-lumefantrine
- CQR:
-
chloroquine resistant
- CQS:
-
chloroquine sensitive
- DHA-PPQ:
-
dihydroartemisinin-piperaquine
- dNTP:
-
deoxynucleotide triphosphate
- WHO:
-
World Health Organisation
- DNA:
-
deoxyribonucleic acid
- RDT:
-
rapid diagnostic tests
- DV:
-
digestive vacuole
- P-GH1:
-
P-glycoprotein homologue 1
References
WHO. World malaria report 2013. Geneva: World Health Organization; 2013.
WHO. Global Health Observatory data. Number of malaria deaths. 2012. http://www.who.int/gho/malaria/epidemic/deaths/en. Accessed 18 Dec 2014.
WHO. Regional office for Africa. 10 facts on malaria in Africa. 2014. http://www.afro.who.int/en/clusters-a-programmes/dpc/malaria/features/2287-10-facts-on-malaria-in-africa.html. Accessed 20 Dec 2014.
UNICEF. Monitoring the situation of children and women. 2014. http://www.data.unicef.org/child-health/malaria. Accessed 23 Dec 2014.
Ministry of Health 2013. Kenya annual malaria report. Epidemiology of malaria in Kenya, p. 1–2.
The Partnership for Maternal, Newborn and Child Health. Maternal and child health: Kenya. Geneva: World Health Organization; 2010. http://www.who.int/pmnch/media/membernews/2011/20121216_kenyaparliament.pdf.
Ministry of Health and Ministry of Public Health and Sanitation. National guidelines for the diagnosis, treatment and prevention of malaria in Kenya. 2010. p. 2–3.
Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbial Rev. 2002;15:66–78.
Bell D, Wongsrichanalai C, Barnwell J. Ensuring quality and access for malaria diagnosis: how can it be achieved? Nat Rev Microbiol. 2006;4:682–95.
Marsh K. Malaria disaster in Africa. Lancet. 1998;352:924.
Phyo AP, Nkhoma S, Stephniewska K, Ashley EA, Nair S, McGready R, et al. Emergence of artemisinin resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–6.
Wellems TE, Plowe CV. Chloroquine-resistant malaria. J Infect Dis. 2001;184:770–6.
Trape JF, Pison G, Preziosi MP, Enel C, Desgrees du Lou A, Delaunay V, et al. Impact of chloroquine resistance on malaria mortality. C R Acad Sci Paris. 1998;321:689–97.
Olatunde A. Chloroquine resistant Plasmodium falciparum and malaria in Africa. Trans R Soc Ttrop Med Hyg. 1977;71:80–1.
Nomura T, Carlton JM, Baird JK, Del Portillo HA, Fryauff DJ, Rathore D, et al. Evidence for different mechanisms of Chloroquine resistance in 2 Plasmodium species that cause human malaria. J Infect Dis. 2001;183:1653–61.
Dorn A, Vippagunta SR, Matile H, Jaquet C, Vennerstrom JL, Ridley RG. An assessment of drug-haematin binding as a mechanism for inhibition of haematin polymerisation by quinoline antimalarials. Biochem Pharmacol. 1998;55:727–36.
Sullivan DJ, Gluzman IY, Russell DG, Goldberg DE. On the molecular mechanism of chloroquine’s antimalarial action. Proc Natl Acad Sci USA. 1996;93:11865–70.
Fidock DA, Namura T, Tally AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in Plasmodium falciparum digestive vacuole transmembrane protein, Pfcrt and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–71.
Carlton JMR, Fidock DA, Djimdé A, Plowe CV, Wellems TE. Conservation of a novel vacuolar transporter in Plasmodium species and its central role in chloroquine resistance of falciparum but not vivax malaria. Curr Opin Microbiol. 2001;4:415–20.
Roepe DP. Pfcrt mediated drug transport in malarial parasites. Biochemistry. 2011;50:163–71.
Sidhu A, Verdier-Pinard D, Fidock A. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;4:210–3.
Chen N, Kyle DE, Pasay C, Fowler E, Baker J, Peters J, et al. pfcrt allelic types with two novel amino acid mutations in chloroquine-resistant Plasmodium falciparum isolates from the Philippines. Antimicrob Agents Chemother. 2003;47:3500–5.
Djimde A, Ogobara PD, Doumbo K, Cortese JF, Kayentao K, Doumbo S, et al. Molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:257–63.
Kublin JG, Cortese JF, Njunju EM, Mukadam RAG, Wirima JJ, Kazembe PN, et al. Re-emergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. J Infect Dis. 2003;187:1870–5.
Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh 1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;24:906–9.
Shretta R, Omumbo J, Rapuoda B, Snow RW. Using evidence to change antimalarial drug policy in Kenya. Trop Med Int Health. 2000;5:755–64.
Mwai L, Ochong E, Abdirahman A, Kiara SM, Ward S, Kokwaro G, et al. Chloroquine resistance before and after its withdrawal in Kenya. Malar J. 2009;8:106.
Warhurst DC, Awad el Kariem FM, Miles MA. Simplified preparation of malarial blood samples for polymerase chain reaction. Lancet. 1991;337:303–4.
Omar SA. Drug susceptibility and molecular characterization of epidemic and endemic malaria in Kenya. PhD Thesis, University of London; 2002. p. 176–82.
Amin A, Zurovac D, Kangwana B, Greenfield J, Otieno D, Akhwale W, et al. The challenges of changing national malaria drug policy to artemisinin-based combinations in Kenya. Malar J. 2007;6:72.
Ministry of Health. National guidelines for the diagnosis, treatment and prevention of malaria in Kenya. 2006.
Omar SA, Makokha FW, Mohammed FA, Kimani FT, Magoma G. Prevalence of Plasmodium falciparum chloroquine resistant gene markers, pfcrt-76 and pfmdr1-86, eight years after cessation of chloroquine use in Mwea, Kenya. J Infect Dev Ctries. 2007;1:195–201.
Okombo J, Kamau AW, Marsh K, Sutherland CJ, Ochola-Oyiera L. Temporal trends in prevalence of Plasmodium falciparum drug resistance alleles over two decades of changing antimalarial policy in coastal Kenya. Int J Parasitol Drugs Drug Resist. 2014;4:152–63.
Eyase FL, Akala HM, Ingasia L, Cheruiyot A, Omondi A, Okudo C, et al. The role of pfmdr1 and pfcrt in changing chloroquine, amodiaquine, mefloquine and lumefantrine susceptibility in western-Kenya P. falciparum samples during 2008–2011. PLoS One. 2013;8:e64299.
Amin AA, Snow RW. Brands costs and registration status of antimalarial drugs in the Kenyan retail sector. Malar J. 2005;4:36.
Juliano JJ, Kwiek JJ, Cappell K, Mwapasa V, Meshnick SR. Minority variant pfcrt K76T mutations and chloroquine resistance, Malawi. Emerg Infect Dis. 2007;13:872–7.
Juliano JJ, Randrianarivelojosia M, Ramarosandratana B, Ariey F, Mwapasa V, Meshnick RS. Nonradioactive heteroduplex tracking assay for the detection of minority-variant chloroquine-resistant Plasmodium falciparum in Madagascar. Malar J. 2009;8:47.
Authors’ contributions
WCK carried out the sample collection and genotyping work as well as drafting the document. FTK and CH conducted the data analysis and interpretation. EL and LW made significant contributions in the conception and design of the study as well as drafting and critical review of this manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank the director of KEMRI for permission to publish these findings. Our appreciation extends to all the study participants for their involvement in the current study, and to the KEMRI malaria laboratory staff and field workers who assisted with this study. This study was financially supported by KEMRI-Internal Research Grants (IRG #L0211).
Compliance with ethical guidelines
Competing interests The authors declare they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kiarie, W.C., Wangai, L., Agola, E. et al. Chloroquine sensitivity: diminished prevalence of chloroquine-resistant gene marker pfcrt-76 13 years after cessation of chloroquine use in Msambweni, Kenya. Malar J 14, 328 (2015). https://doi.org/10.1186/s12936-015-0850-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-015-0850-9