
Abstract
Background
The prospect of eliminating malaria is challenged by emerging insecticide resistance and vectors with outdoor and/or crepuscular activity. Ivermectin can simultaneously tackle these issues by killing mosquitoes feeding on treated animals and humans. A single oral dose, however, confers only short-lived mosquitocidal plasma levels.
Methods
Three different slow-release formulations of ivermectin were screened for their capacity to sustain mosquito-killing levels of ivermectin for months. Thirty rabbits received a dose of one, two or three silicone implants containing different proportions of ivermectin, deoxycholate and sucrose. Animals were checked for toxicity and ivermectin was quantified periodically in blood. Potential impact of corresponding long-lasting formulation was mathematically modelled.
Results
All combinations of formulation and dose released ivermectin for more than 12 weeks; four combinations sustained plasma levels capable of killing 50% of Anopheles gambiae feeding on a treated subject for up to 24 weeks. No major adverse effects attributable to the drug were found. Modelling predicts a 98% reduction in infectious vector density by using an ivermectin formulation with a 12-week duration.
Conclusions
These results indicate that relatively stable mosquitocidal plasma levels of ivermectin can be safely sustained in rabbits for up to six months using a silicone-based subcutaneous formulation. Modifying the formulation of ivermectin promises to be a suitable strategy for malaria vector control.
Similar content being viewed by others


Background
Increased funding and political commitment have led to outstanding worldwide achievements in malaria control over the last 14 years. The estimated malaria mortality rate in children under five has almost been halved worldwide since 2000 and it is projected to decrease by 61% by 2015 [1]. Yet, malaria is still a formidable public health problem that caused an estimated 198 million cases and 584,000 deaths in 2013 [1]. In many settings, there has been a renewed interest for malaria elimination [2], and the general mood is that of ‘impatient optimism’ [3]. This general optimism has translated into more concrete research [4] and operational [5] agendas to decrease malaria transmission until eradication. The main determinants of malaria transmission are mosquito-related variables [6]; vector control plays a central role in control strategies [7]. It has been the most successful intervention in the past [8] and it is likely to remain so in future elimination endeavours.
The malERA consultative group on vector control identified three main challenges to eradication [9]: 1) the emergence of insecticide resistance affecting all major vector species and all classes of insecticides in two-thirds of endemic countries [7]; 2) the presence of outdoor-biting/resting mosquitoes, not readily targeted by insecticide-treated nets (ITNs) and indoor-residual spraying (IRS) [10,11]; and, 3) the need for new approaches to achieve elimination in areas where vectors exhibit a particularly high vectorial capacity. Additional problems include the selection of vectors with early or crepuscular activity in areas with good ITN coverage [12,13], vector biodiversity and environmental change [14].
One additional source of concern for eradication is the emergence of artemisinin resistance in Southeast Asia. The historic perspective is unsettling as this area has repeatedly been the epicentre from where resistance to anti-malarial drugs has spread to Africa and the rest of the world [15]. An emergency containment plan is in place [16] and even focal elimination of all falciparum malaria has been advocated [17,18]. One further problem is that the main local malaria vectors, Anopheles dirus and Anopheles minimus, show substantial outdoor feeding and biting [19] as well as crepuscular activity and a tendency to bite early at night [20], which limits the effectiveness of ITNs and IRS. Additional vector control methods are urgently needed in the region [19].
Ivermectin (IVM) is a systemic insecticide that reduces the survival of mosquitoes feeding on treated humans, both under laboratory [21,22] and field conditions [23], potentially leading to a disruption in malaria transmission [24]. Mass drug administration (MDA) with IVM has been advocated as a complementary vector control strategy [25,26]. Potential benefits include: i) a novel mechanism of action [27] compared to currently used insecticides, which could circumvent resistance; ii) effectiveness against malaria vectors regardless of place or time of the feeding; and, iii) additional effects inhibiting Plasmodium sporogony [28].
All these characteristics make MDA with IVM a particularly valuable intervention for the vectors in the Mekong region and local elimination endeavours in the light of artemisinin resistance. According to a recent mathematical model [29], a key factor for interrupting transmission would be the time IVM remains in blood above mosquito-killing levels.
The lethal concentration 50 (LC50) is defined as the blood levels needed in order to kill 50% of the mosquitoes feeding on a treated individual, its closest clinical equivalent is the minimum inhibitory concentration used in microbiology labs. The LC50 of IVM for Anopheles gambiae in the first ten days after a blood meal has been estimated by mixing human blood with a known dilution of the drug in a tube and then performing a membrane feeding essay. The LC50 estimated with this method ranges from 14.6 to 26.9 ng/ml [22,28]. Recent evidence from membrane feedings using blood drawn from ivermectin treated volunteers gives a much lower range of 4.69-7.51 ng/ml [30]. This discrepancy could be the result of unknown active metabolites and possibly, blood meal size differences between membrane and skin feeding mosquitoes.
At the approved dose of 200 μg/kg, current oral formulations can only maintain mosquitocidal concentrations for approximately 48 hours [31,32]. Alternatives include a scheme with multiple doses over the course of weeks, which poses logistical challenges, or the administration of a slow-release formulation on one single encounter. Given IVM’s pharmacokinetic properties [33], a slow-release oral tablet could only increase the time with concentrations above LC50 in hours, while injectable, depot formulations could do it for days to weeks. An implantable subcutaneous device [34,35] could sustain key mosquito-killing levels of IVM for months. Subcutaneous implants for contraceptive purposes were licensed more than 30 years ago; they release small amounts of hormones for years and have an excellent security profile [36]. In developing countries suitable for the implementation of slow-release IVM formulations, the acceptance of contraceptive implants is high [37] and seems to be increasing [38].
The main goal of this work was to adapt the design of an IVM slow-release formulation to make it suitable for use in humans and peridomestic animals as an additional malaria vector control intervention. For this, three different IVM-containing silicone implant formulations were screened at different doses in a proof-of-concept animal model. Rabbits were chosen because their weight allows for an easier dose extrapolation to larger mammals.
Methods
The implants
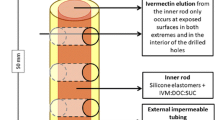
The 40×2-mm implants consist of two concentric cylinders (silicone-covered rod formulation). The external cylinder is a 100% silicone impermeable membrane; the inner cylinder contains silicone and a mixture of IVM, deoxycholate sodium (DOC) and sucrose (SUC). The inner drug-containing matrix contacts the subcutaneous tissue and fluids only at the ends, where the cylinders are cross-sectioned (Figure 1). Water enters at each end, dissolves the IVM-DOC-SUC mixture and creates microchannels in the inner core, allowing for a controlled release of the drug.

Implant’s design and measures. With the current measures, the elution surface of each implant is 6.28 sq mm (2 x π x 12). IVM: ivermectin; DOC: deoxycholate; SUC: sucrose.
IVM is a lipophilic drug. DOC and SUC modify the solubility of IVM and change its release rate from the microchannels in the core. Two previously used proportions of IVM:DOC:SUC known to have an appropriate release profile in mice [35] and dogs [34] were chosen. An third formulation with a high proportion of DOC, which markedly increases the release rate, was also included. This resulted in a different total IVM content for each formulation. The internal cylinder (1.8 × 40 mm) in the implants allows for an elution surface of 5.08 sq mm each (2 π r2). This surface increases arithmetically when using higher doses (two or three implants). The total IVM calculated content of each formulation can be seen in Table 1. Implants were manufactured by Specialty Silicone Fabricators (SSFAb) at their ISO 13485-certified and FDA-registered facility in Tustin, California. The manufacture started my mixing the silicone component with the drug and excipients (DOC and SUC) and molding it in cylindrical shape to form the inner drug matrix core. The outer high consistency rubber layer was extruded separately and applied over the drug core to complete the implants. See references [34,35] for a detailed description of the production methods. The rods were sterilized using an electron beam.
Procedures
See Table 2 for a timeline of all procedures. The subjects were 30 male New Zealand white rabbits of 16 weeks of age. The diet was limited to 250 g/day in an attempt to keep the weight stable. The animals were kept in individual cages. Appropriate environmental enrichment was used. Rabbits were randomly marked in the ear with numbers 1–30 by the provider. Animals were assigned to each formulation (F, M, X) in numerical order. In each formulation group, the rabbits were assigned to a subgroup receiving a dose of one, two or three rods (1F, 2F, 3F, 1M, 2M, 3M, 1X, 2X, 3X). Each subgroup contained three animals (in total 27 intervention plus three controls). Control animals received one, two or three 100% silicone rods. The rods were inserted subcutaneously between the scapulae by means of a Jadelle® trocar under general anaesthesia. When more than one implant was inserted, they were placed forming a ‘V’ shape.
After implantation, the animals were checked daily for clinical signs of IVM toxicity such as sleepiness, ataxia and increase in tone [39]. Blood for IVM quantification was drawn weekly for the first 12 weeks, under sedation, from the marginal ear vein. An interim analysis of plasma levels was performed at 13 weeks and only groups where all subjects maintained at least 10 ng/ml for the whole period were continued until week 25. Blood samples were taken monthly in the remaining groups until euthanasia. IVM plasma levels were determined using a variation of a previously described HPLC-FLD [40,41]. The method was validated in accordance with the European Medicines Agency guidelines [42].
A comprehensive toxicological profile was performed, reviewing the neurological, cardiovascular, respiratory, haematologic, digestive, and urinary systems. For toxicological analysis, additional blood was drawn at baseline and at 12 and 24 weeks. Blood tests included full count, coagulation panel, glucose, electrolytes, muscular and liver enzymes, bilirubin, cholesterol, total proteins, and albumin. A urinary dipstick test was performed before euthanasia. Vital signs were assessed at baseline and in weeks 1, 12 and 24 after implantation together with ECGs and indirect ophthalmoscopic examination. QT intervals in the ECG were corrected using Bazett’s formula. Normal values for vital signs, ECG, haematology, coagulation, and biochemistry were taken from the literature [43] and compared to the mean and 95% CI of the baseline values.
Euthanasia was performed under sedation using T61® (MSD) at 25 weeks. An experienced toxicologist performed full macroscopic autopsies on all rabbits. All procedures were reviewed and approved by the animal experimentation ethics committee of the Universidad de Navarra (Registry number CEEA/135-12).
Pharmacokinetic calculations and statistics
Pharmacokinetic calculations were performed with Mathematica, Version 8.0, Wolfram Research, Inc., Champaign, IL, USA (2010). Areas under the curve (AUC) were calculated using the linear trapezoidal rule.
ANOVA and Chi square test were used to contrast the AUC of the different formulation-dose combinations at 12 weeks and the role of the elution area of the implants on the likelihood of maintaining IVM plasma level above 7 ng/ml (the estimated in vivo LC50) for the same period of time. Furthermore, a univariate linear regression model was performed to assess the capacity of the formula (proportion of DOC x elusion surface) to predict the AUC at 12 weeks.
Comparisons of vital signs and blood test results at baseline and at 1 and 12 weeks after implantation were done using T-test for paired samples. For comparing baseline results with those of 24 weeks, the Wilcoxon paired test was used. Statistical analysis was done with SPSS version 20.0.
Mathematical modelling of potential impact
The mathematical model developed by Slater et al. [29] was used to assess the potential impact of a long-lasting IVM product on mosquito survival and malaria incidence in humans. The model considered a formulation capable of sustaining 8 ng/ml plasma levels for two, four, eight, 12 or 24 weeks administered to 80% of the population over five years of age. This concentration is calculated to increase the daily hazard of mortality to mosquitoes by 4.4 times, resulting in the mean lifespan of a mosquito in the wild decreasing from 7.6 days [44] to 1.7 days.
Results
Plasma levels and pharmacokinetics
All combinations of formulation and dose released IVM for more than 12 weeks. Table 3 shows the median and range of main pharmacokinetics (PK) parameters in the different groups. Figure 2 shows the PK curves of the four groups that maintained IVM levels of at least 10 ng/ml for the first 12 weeks. These were selected to continue until week 25 (3F, 3M, 2X, 3X).

Pharmacokinetic curves of the leading formulations. PK curves (median and range) of the four groups that maintained IVM levels of at least 10 ng/ml for the first 12 weeks and were selected to continue until week 25. The red triangle is the approximate PK curve of one single 150 μg/kg oral dose. The dotted line marks 7 ng/ml, the minimum LC50 determined in vivo for An. gambiae.
Using ANOVA, a statistically significant difference in the AUC at 12 weeks between groups with different elution surface (dose) was found. Additionally, the Chi square test shows a significant difference in the likelihood of maintaining plasma levels above 7 ng/ml between the same groups. Neither of the tests demonstrates statistically significant differences in the same parameters when comparing groups with different formulations (DOC and SUC proportions) (Table 4). The linear regression model shows that the AUC at 12 weeks can be predicted using the product of the DOC proportion and the implant’s elusion surface (DOC% x surface). An increase of this product in 1 sq·mm% correlates with an increase in the AUC at 12 weeks of 26.45 ng · (week)/ml (p < 0.01) (Figure 3).

Linear regression comparing area under the curve at 12 weeks with the product (DOC%•Surface). T (magnitude) 5.34 (p < 0.01). Typified coefficient 0.73. DOC: deoxycholate.
Toxicology
No major adverse effect attributable to the drug was found. See Additional files 1, 2 and 3 for the full toxicology report.
Modelling
Mosquito survival is assumed to be distributed exponentially; therefore, a population of vectors with a mean lifespan of 7.6 days will have a survival curve shown by the dark blue line in Figure 4A. Here, 27% of mosquitoes survive for the ten days required for the parasite to reach its infectious sporozoite stage. After an IVM blood meal containing 8 ng/ml, mean lifespan decreases to 1.7 days – this results in just 0.3% of mosquitoes surviving for ten days. Figure 4B shows how this reduced vector lifespan would impact the number of infectious vectors over time if IVM were given to 80% of the population over five years old. Figure 4C shows the cumulative reduction in clinical incidence in under fives and the number of infectious bites received per individual (the entomological inoculation rate (EIR)) in the six months following treatment. An IVM product with a two-week duration reduces the infectious vector population by over 90% for about four weeks, which results in a 20% reduction in clinical incidence in under fives and EIR. Infectious vector density can be almost totally suppressed (>98% reduction) for three months with an IVM product with a 12-week duration such as these implantables. A formulation with this duration of efficacy is estimated to reduce clinical incidence in under fives and EIR by >60% in the six months following implementation. Figure 4B and C show the expected proportional increase with formulations lasting for longer periods.

Modelling the potential impact of slow-release ivermectin formulations. Panel A: Expected change in vector survival in the presence of ivermectin treatment in the community. Panel B: Expected percentage change in the infectious vector density in time in the presence of different long-lasting ivermectin formulations. Panel C: Reductions in clinical incidence and entomological inoculation rate in the six months following implementation of a long-lasting ivermectin formulation.
Discussion
These results indicate that relatively stable plasma levels of IVM can be safely sustained in mammals for up to 24 weeks using a slow-release formulation. The insecticidal properties of IVM make it attractive as an alternative control tool for malaria [25] and other vector-borne diseases, including leishmaniasis and trypanosomiasis [27]. Its short half-life, however, could limit its widespread application. Modelling shows that the time it remains in plasma at levels lethal to vectors is critical for interrupting transmission [29].
Both the AUC and the time with plasma levels above 7 ng/ml (An. gambiae’s LC50 calculated in vivo) increase with the total elution surface of the formulations. This is a robust parameter likely to play an important role in the development of slow-release formulations due to its dose-dependent effect.
Formulations with a higher content of DOC tended to achieve markedly higher Cmax and AUC both at 12 and 24 weeks (though not statistically significant), possibly at the expense of time with a concentration above 7 ng/ml after 12 weeks, as seen in Figure 2. This is consistent with the increased IVM solubility caused by DOC [35], resulting in faster elution from the rods, i.e., ‘peaked’ PK curves. Formulations with less DOC content have release behaviour closer to zero order.
Using the formula (DOC% x elution surface) allows for a linear prediction of the AUC. Both factors (release area and excipients that increase solubility) seem to have this effect by increasing the Cmax, i.e., the ‘height’ of the curve. Another factor likely to have a linear influence on the AUC and the time above 7 ng/ml is the product (IVM% x elution surface x device volume). It was not possible to probe this theory because implants of equal volume were used. These factors, however, are likely to influence the AUC by increasing the time above target concentration, i.e., the ‘length’ of the curve. These concepts may assist in the design of different slow-release devices.
Given the drug’s lipophilic condition, an extrapolation of these results to humans was not ventured due to the disproportionate increase in adipose tissue when compared to rabbit. This might initially reduce the peak levels reached, but could effectively extend the time above target levels as the drug is release from adipose tissue.
No significant side effect could be attributed to the drug in this study. Although of minor clinical significance, future studies should assess a possible relationship with increased fibrinogen. It was not possible to find suitable data on proportional organ weight for laboratory rabbits of 40 weeks of age or older. The finding of a relative large spleen at 12 weeks post intervention should be interpreted carefully due to this lack of comparative standards and the absence of histological anomalies.
IVM is a safe drug. In ongoing MDA programmes for onchocerciasis and lymphatic filariasis, its benefits exceed the prevalence reduction of the target filariae as it has an impact on several soil-transmitted helminths and ectoparasites. A wider use of the drug as a public health tool has been advocated [45,46]. With appropriate resistance monitoring, assessment of pharmacokinetics and drug interactions issues, a slow-release IVM formulation could have additional value on the prevention and treatment of these [47] and other vector-borne diseases. There is a working delivery infrastructure for the drug used in MDA campaigns. The evaluation of these possibilities warrants joint work of the malaria and NTD communities.
Limitations of this study
This study is intended as a proof of concept. As such it has several limitations. PK results are given as median and range due to the small sample size of each group. Additionally, findings in rabbits should be interpreted with caution due to possible differences in drug distribution and metabolism when compared with other mammals. Also, the rabbits gained an average 12% of their initial weight during the study, which could have affected total body fat and the drug’s PK. The manufacture of prototypes is not a standardized process and some PK variation may be caused by differences in implant weight affecting total drug content. The experiments described in this study were conducted under good laboratory practices (GLP), however, for budgetary reasons; the GLP certification was not sought. The modelling work is based on the hypothetical deployment of a new slow-release IVM product; it aims at elucidating the potential impact of such a long-lasting IVM formulation on malaria transmission. The data used to parameterize the model were based on mosquito mortality data taken from laboratory studies using IVM. The impact of a new formulation of IVM needs to be tested in the field to fully understand the impact on transmission. Potential challenges of implantable devices in the field include the need for a trained worker using a sterile technique for insertion and removal in case of adverse reactions, which might be local or systemic. Additionally, the silicone structure will remain in place after all the IVM and excipients have been released, the silicone itself is medical grade and approved for indefinite implantation, but a strategy should be in place to cover the possible user demand for removal.
Knowledge gaps and future work
Setting a target plasma level was difficult because most LC50 studies employ in vitro membrane feeds of a blood-IVM mixture to mosquitoes. This poses a problem because IVM accumulates in adipose tissue [48], which could lead to a higher concentration in skin capillaries than in a vein. In fact, mosquitoes fed on subjects treated with IVM have a three-fold mortality compared to those feeding through a membrane [29]. A threshold of 7 ng/ml was used as target because it is the lowest determined LC50 for An. gambiae [30]. There is need for a clear determination of the in vivo LC50 for the main vector species.
Importantly, different vectors have different sensitivity to the drug [49,50]. The LC50 varies widely between mosquitoes; Aedes have a much higher LC50 than Anopheles [50] and even different anopheline species in the same area can have different LC50 [49]. This should be taken into account when designing slow-release formulations for any specific area or programme.
Using IVM as a transmission-blocking strategy poses many ethical questions. After all, it would mean exposing individuals to the possible side effects of a drug in the name of a community benefit. Many of these questions have been debated regarding transmission-blocking vaccines. The general consensus is that the indirect personal benefit obtained by reducing transmission at the community level would justify the individual use of the drug [51]. Individuals receiving IVM would also benefit from its wide anti-parasitic effects.
The modelling results reveal the exciting potential of a long-lasting IVM product. Even duration as short as two weeks is estimated to reduce the total number of cases of malaria in under fives by 20% in the following six months. In combination with an ACT, a long-lasting IVM drug could play an important role in interrupting malaria transmission by suppressing the infectious vector population in the months following the intervention and preventing resurgence.
Conclusion
This animal model shows it is technically possible to safely sustain mosquitocidal concentrations of IVM using a slow-release formulation. The total release area and the proportion of the different excipients play an important role in the delivery of the drug. The modelled potential impact on vector population, transmission and clinical incidence is remarkable. These findings warrant further research on slow-release systems for IVM.
Abbreviations
- AUC:
-
Area under the curve
- DOC:
-
Deoxycholate
- EIR:
-
Entomological inoculation rate
- GLP:
-
Good laboratory practices
- IRS:
-
Indoor-residual spraying
- ITNs:
-
Insecticide-treated nets
- IVM:
-
Ivermectin
- LC50 :
-
Lethal concentration 50
- MDA:
-
Mass drug administration
- PK:
-
Pharmacokinetics
- SUC:
-
Sucrose
References
WHO. World Malaria report 2014. Geneva: World Health Organization; 2014.
White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. 2014;383:723–35.
Magill A. We can achieve a world free of malaria. In Impatient Optimists. Bill & Melinda Gates Foundation. 2014. http://www.impatientoptimists.org/Posts/2014/04/We-Can-Achieve-a-World-Free-of-Malaria. Accessed 17 Feb 2015
Alonso PL, Brown G, Arevalo-Herrera M, Binka F, Chitnis C, Collins F, et al. A research agenda to underpin malaria eradication. PLoS Med. 2011;8:e1000406.
Moonen B, Cohen JM, Snow RW, Slutsker L, Drakeley C, Smith DL, et al. Operational strategies to achieve and maintain malaria elimination. Lancet. 2010;376:1592–603.
Macdonald G. The epidemiology and control of malaria. London: Oxford University Press; 1957.
WHO. Global plan for insecticide resistance management in malaria vectors (GPIRM). Geneva: World Health Organization; 2012.
Najera JA, Gonzalez-Silva M, Alonso PL. Some lessons for the future from the Global Malaria Eradication Programme (1955–1969). PLoS Med. 2011;8:e1000412.
The malERA Consultative Group on Vector Control. A research agenda for malaria eradication: vector control. PLoS Med. 2011;8:e1000401.
Govella NJ, Ferguson H. Why use of interventions targeting outdoor biting mosquitoes will be necessary to achieve malaria elimination. Front Physiol. 2012;3:199.
Stevenson J, St Laurent B, Lobo NF, Cooke MK, Kahindi SC, Oriango RM, et al. Novel vectors of malaria parasites in the western highlands of Kenya. Emerg Infect Dis. 2012;18:1547–9.
Moiroux N, Gomez MB, Pennetier C, Elanga E, Djenontin A, Chandre F, et al. Changes in Anopheles funestus biting behavior following universal coverage of long-lasting insecticidal nets in Benin. J Infect Dis. 2012;206:1622–9.
Sougoufara S, Diedhiou SM, Doucoure S, Diagne N, Sembene PM, Harry M, et al. Biting by Anopheles funestus in broad daylight after use of long-lasting insecticidal nets: a new challenge to malaria elimination. Malar J. 2014;13:125.
Ferguson HM, Dornhaus A, Beeche A, Borgemeister C, Gottlieb M, Mulla MS, et al. Ecology: a prerequisite for malaria elimination and eradication. PLoS Med. 2010;7:e1000303.
Time to contain artemisinin resistance [editorial]. Lancet. 2014;383:1438.
WHO. Emergency response to artemisinin resistance in the Greater Mekong subregion: regional framework for action 2013–2015. Geneva: World Health organization; 2013.
Maude RJ, Pontavornpinyo W, Saralamba S, Aguas R, Yeung S, Dondorp AM, et al. The last man standing is the most resistant: eliminating artemisinin-resistant malaria in Cambodia. Malar J. 2009;8:31.
White NJ. Artemisinin resistance–the clock is ticking. Lancet. 2010;376:2051–2.
Kolaczinski J, Macdonald M, Meek S. Vector control to eliminate artemisinin resistant malaria in the Greater Mekong subregion. Lancet Infect Dis. 2014;14:9–11.
Hii J, Rueda LM. Malaria vectors in the Greater Mekong Subregion: overview of malaria vectors and remaining challenges. Southeast Asian J Trop Med Public Health. 2013;44 Suppl 1:73–165. discussion 306–167.
Chaccour C, Lines J, Whitty CJ. Effect of ivermectin on Anopheles gambiae mosquitoes fed on humans: the potential of oral insecticides in malaria control. J Infect Dis. 2010;202:113–6.
Kobylinski KC, Deus KM, Butters MP, Hongyu T, Gray M, da Silva IM, et al. The effect of oral anthelmintics on the survivorship and re-feeding frequency of anthropophilic mosquito disease vectors. Acta Trop. 2010;116:119–26.
Sylla M, Kobylinski KC, Gray M, Chapman PL, Sarr MD, Rasgon JL, et al. Mass drug administration of ivermectin in south-eastern Senegal reduces the survivorship of wild-caught, blood fed malaria vectors. Malar J. 2010;9:365.
Kobylinski KC, Sylla M, Chapman PL, Sarr MD, Foy BD. Ivermectin mass drug administration to humans disrupts malaria parasite transmission in Senegalese villages. Am J Trop Med Hyg. 2011;85:3–5.
Chaccour CJ, Kobylinski KC, Bassat Q, Bousema T, Drakeley C, Alonso P, et al. Ivermectin to reduce malaria transmission: a research agenda for a promising new tool for elimination. Malar J. 2013;12:153.
Foy BD, Kobylinski KC, da Silva IM, Rasgon JL, Sylla M. Endectocides for malaria control. Trends Parasitol. 2011;27:423–8.
Omura S, Crump A. Ivermectin: panacea for resource-poor communities? Trends Parasitol. 2014;30:445–55.
Kobylinski KC, Foy BD, Richardson JH. Ivermectin inhibits the sporogony of Plasmodium falciparum in Anopheles gambiae. Malar J. 2012;11:381.
Slater HC, Walker PG, Bousema T, Okell LC, Ghani AC. The potential impact of adding Ivermectin to a mass treatment intervention to reduce malaria transmission: a modelling study. J Infect Dis. 2014;210:1972–80.
Ouedraogo AL, Bastiaens GJ, Tiono AB, Guelbeogo WM, Kobylinski KC, Ouedraogo A, et al. Efficacy and safety of the mosquitocidal drug ivermectin to prevent malaria transmission after treatment: a double-blind, randomized, clinical trial. Clin Infect Dis. 2015;60:357–65.
Elkassaby MH. Ivermectin uptake and distribution in the plasma and tissue of Sudanese and Mexican patients infected with Onchocerca volvulus. Trop Med Parasitol. 1991;42:79–81.
Gonzalez Canga A, Sahagun Prieto AM, Diez Liebana MJ, Fernandez Martinez N, Sierra Vega M, Garcia Vieitez JJ. The pharmacokinetics and interactions of ivermectin in humans–a mini-review. AAPS J. 2008;10:42–6.
Lo PK, Fink DW, Williams JB, Blodinger J. Pharmacokinetic studies of ivermectin: effects of formulation. Vet Res Commun. 1985;9:251–68.
Cunningham CP, Brown JM, Jacobson GA, Brandon MR, Martinod SR. Evaluation of a covered-rod silicone implant containing ivermectin for long-term prevention of heartworm infection in dogs. Am J Vet Res. 2006;67:1564–9.
Maeda H, Brandon M, Sano A. Design of controlled-release formulation for ivermectin using silicone. Int J Pharm. 2003;261:9–19.
Members of the Caucus on New and Underused Reproductive Health Technologies. Contraceptive implants. Available in the PATH website http://www.path.org/publications/files/RHSC_implants_br.pdf, 2012. Accessed 17 Feb 2015.
Hubacher D, Olawo A, Manduku C, Kiarie J. Factors associated with uptake of subdermal contraceptive implants in a young Kenyan population. Contraception. 2011;84:413–7.
Muchangi J. More Nairobi women prefer implants. In The Star newspaper. Nairobi, Kenya. 2013.
Trailovic SM, Nedeljkovic JT. Central and peripheral neurotoxic effects of ivermectin in rats. J Vet Med Sci. 2011;73:591–9.
Eraslan G, Kanbur M, Liman BC, Cam Y, Karabacak M, Altinordulu S. Comparative pharmacokinetics of some injectable preparations containing ivermectin in dogs. Food Chem Toxicol. 2010;48:2181–5.
Scott EW, McKellar QA. The distribution and some pharmacokinetic parameters of ivermectin in pigs. Vet Res Commun. 1992;16:139–46.
EMA. Guideline on bioanalytical method validation. London: European Medicines Agency; 2011.
Manning PJ. The biology of the laboratory rabbit. 2nd ed. 2014.
Griffin JT, Ferguson NM, Ghani AC. Estimates of the changing age-burden of Plasmodium falciparum malaria disease in sub-Saharan Africa. Nat Commun. 2014;5:3136.
Omura S. Ivermectin: 25 years and still going strong. Int J Antimicrob Agents. 2008;31:91–8.
Speare R, Durrheim D. Mass treatment with ivermectin: an underutilized public health strategy. Bull World Health Organ. 2004;82:562.
Chavasse DC, Post RJ, Lemoh PA, Whitworth JA. The effect of repeated doses of ivermectin on adult female Onchocerca volvulus in Sierra Leone. Trop Med Parasitol. 1992;43:256–62.
Baraka OZ, Mahmoud BM, Marschke CK, Geary TG, Homeida MM, Williams JF. Ivermectin distribution in the plasma and tissues of patients infected with Onchocerca volvulus. Eur J Clin Pharmacol. 1996;50:407–10.
Kobylinski KCP, Alongkot UR, Schuster A, McCardle W, Foy BD, Tarning J, et al. Assessing ivermectin susceptibility of Greater Mekong Subregion malaria vectors. Poster session Annual meeting of the American Society of Tropical Medicine and Hygiene; 2014 Nov 2–6; New Orleans.
Deus KM, Saavedra-Rodriguez K, Butters MP, Black WCT, Foy BD. The effect of ivermectin in seven strains of Aedes aegypti (Diptera: Culicidae) including a genetically diverse laboratory strain and three permethrin resistant strains. J Med Entomol. 2012;49:356–63.
WHO. Malaria transmission blocking vaccines: An ideal public good. Geneva: World Health Organization; 2000.
Acknowledgements
We would like to thank Gloria Abizanda for help handling rabbits, Mark Paulsen for technical advice during implant manufacture and the calculation of IVM content in every device, Juliane Chaccour for editing and all our crowdfunding backers. This work was supported by two crowdfunding campaigns, one done locally in the University of Navarra and a second one called ‘Malaria Mission’, using the online platform Indiegogo, where a list of all donors can be found.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CCh designed the study, participated in experimental work and drafted the manuscript; AIB carried out the HPLC work and contributed to the manuscript; AGR carried out the toxicology work and contributed to the manuscript; DMU performed the statistical analysis and contributed to the manuscript; HS performed the modelling work and contributed to the manuscript; FH performed the PK analysis and contributed to the manuscript; JLDP contributed to the study design, participated in experimental work and contributed to the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Toxicology report. A complete report of the clinical and analytical toxicological assessment of the rabbits.
Additional file 2:
Vital signs and ECG data. Table with all the vital sign and ECG measurements done as part of the toxicology workup.
Additional file 3:
Biochemistry data. Table with all the biochemistry measurements done as part of the toxicology workup.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Chaccour, C., Barrio, Á.I., Royo, A.G.G. et al. Screening for an ivermectin slow-release formulation suitable for malaria vector control. Malar J 14, 102 (2015). https://doi.org/10.1186/s12936-015-0618-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-015-0618-2