
Abstract
The incidence of melanoma, the most lethal form of skin cancer, has increased due to ultraviolet exposure. The treatment of advanced melanoma, particularly metastatic cases, remains challenging with poor outcomes. Targeted therapies involving BRAF/MEK inhibitors and immunotherapy based on anti-PD1/anti-CTLA4 antibodies have achieved long-term survival rates of approximately 50% for patients with advanced melanoma. However, therapy resistance and inadequate treatment response continue to hinder further breakthroughs in treatments that increase survival rates. This review provides an introduction to the molecular-level pathogenesis of melanoma and offers an overview of current treatment options and their limitations. Cells can die by either accidental or regulated cell death (RCD). RCD is an orderly cell death controlled by a variety of macromolecules to maintain the stability of the internal environment. Since the uncontrolled proliferation of tumor cells requires evasion of RCD programs, inducing the RCD of melanoma cells may be a treatment strategy. This review summarizes studies on various types of nonapoptotic RCDs, such as autophagy-dependent cell death, necroptosis, ferroptosis, pyroptosis, and the recently discovered cuproptosis, in the context of melanoma. The relationships between these RCDs and melanoma are examined, and the interplay between these RCDs and immunotherapy or targeted therapy in patients with melanoma is discussed. Given the findings demonstrating melanoma cell death in response to different stimuli associated with these RCDs, the induction of RCD shows promise as an integral component of treatment strategies for melanoma.
Similar content being viewed by others

Introduction
Melanoma, a life-threatening malignancy primarily affecting the skin, affects different primary sites, with cutaneous, ocular, and mucosal sites accounting for 93.3%, 5.5%, and 1.3% of reported cases, respectively [1]. The 5-year survival rate for all stages of cutaneous melanoma has been reported to be 94% by the Surveillance, Epidemiology, and End Results (SEER) Program. In contrast, survival rates for metastatic cutaneous melanoma were significantly lower, at 39.4% diagnosed between 2016 and 2018 [2]. In a study involving 428 patients with metastatic melanoma treated with checkpoint inhibitors from 2007 to 2018, the 5-year survival rates for cutaneous, acral, uveal (ocular), and mucosal melanoma were 46%, 34%, 21%, and 22%, respectively [3]. The various prognoses of these subtypes reflect distinct pathogenesis and genetic alterations. Understanding the underlying pathogenesis is crucial for overcoming treatment obstacles in unresponsive metastatic melanomas. In the first part of this article, we provide an overview of the pathogenesis and current treatment options and limitations for melanoma.
In the second part of the article, the relationship between regulated cell death (RCD), namely, autophagy-dependent cell death, pyroptosis, necroptosis, ferroptosis and cuproptosis, and melanoma is discussed. Autophagy-dependent cell death strictly requires autophagy induction. Pyroptosis is a proinflammatory type of RCD characterized by apoptotic body-like protrusions on the plasma membrane established during gasdermin pore formation. Necroptosis is regulated by various cytokines and pattern recognition receptors (PRRs). Ferroptosis specifically depends on high intracellular iron concentrations and is characterized by the accumulation of lipid peroxides. Cuproptosis is a copper-dependent form of cell death regulated by mitochondrial ferredoxin 1 (FDX1)-mediated protein lipoylation. All these RCDs differ from apoptosis and have been studied in the melanoma context, either in vitro or in vivo, showing a promising role in inducing the death of melanoma cells. This review provides a comprehensive overview of these RCDs and their interactions with BRAF/MEK inhibitors or immune blocker therapy. Since the current treatment choice for melanoma, including BRAF/MEK inhibitors or check blocker therapy, has drawbacks, understanding these nonapoptotic RCDs may help to identify new therapeutic targets for melanoma treatment.
Pathogenesis and treatment limitations of melanoma
Overview of pathogenesis
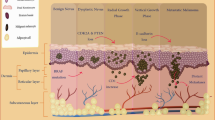
Melanoma is a malignancy originating from melanocytes located in the basal layer of the epidermis. Because melanocytes are also in the digestive tract, urogenital tract and mucous glands, noncutaneous melanoma can affect these tissues, and noncutaneous melanoma represents approximately 5% of all cases of melanoma [4]. Melanomas may develop in or near a previously existing precursor lesion, including a common nevus, dysplastic nevus, congenital nevus, and blue nevus, or in healthy-appearing skin. It has two growth phases: radial and vertical phases. When melanoma tumors are thin, superficial and primarily confined to the epidermis, they are in the radial growth phase and indolent. With the development of the vertical growth phase, which can arise de novo or from lesions in the radial growth phase, melanoma cells invade deep into tissues and show metastatic potential.
The incidence of melanoma is increasing worldwide in white populations, especially where fair-skinned people receive excessive sun exposure; for example, 20–30 cases per 100,000 individuals per year are reported in the United States, and 50–60 cases per 100,000 individuals per year are reported in Australia; in contrast, the melanoma incidence is lower than 5 cases per 100,000 individuals per year in Asia and Africa [5, 6]. Excessive sun exposure, indoor tanning booths, the number of typical nevi, the presence of atypical nevi, a personal history of melanoma, and a family history of melanoma all increase the risk of melanoma. Traditionally, invasive cutaneous melanoma is morphologically classified into 4 subtypes: superficial spreading melanoma, nodular melanoma, lentigo malignant melanoma, and acral lentiginous melanoma [7]. With the discovery of genetic alterations in melanocytic tumors, the new classification incorporates the clinical, pathological, and genomic characteristics of melanoma. Therefore, the 2018 World Health Organization (WHO) classification of melanocytic tumors indicates nine evolutionary pathways into three major categories of melanomas as determined by the intensity of chronic ultraviolet radiation exposure/cumulative solar damage, ranging from low cumulative solar damage (CSD)-related (pathway 1), high CSD-related (pathways 2 and 3), and non-CSD-related melanoma (pathways 4–9) [8, 9] (Table 1). In addition, the American Joint Committee on Cancer (AJCC) staging system, which is based on tumor size, regional involvement of lymph nodes, and distant metastasis, is commonly used in clinical practice to record disease severity, guide treatment options, and predict prognosis [10].
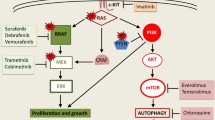
The WHO classification of melanoma not only reflects the pathway concept of melanoma pathogenesis but also reveals distinct molecular signatures associated with different anatomical locations and levels of patient sun exposure. Melanoma lesions associated with low ultraviolet (UV) exposure/CSD (pathway 1) are located mainly on the trunk and extremities, and approximately 45 ~ 50% of these tumors carry a BRAF mutation (Table 1) [6, 8]. Melanoma lesions associated with high UV exposure/CSD (pathways 2 and 3) are located mainly in the head and neck region and show a moderate frequency of NRAS (NRAS is a proto-oncogene and a GTPase) mutations, which are found in approximately 10–20% of cutaneous melanomas [21]. Non-sun-related melanoma lesions (pathways 4 to 9) are located mainly on acral and mucosal sites and do not carry BRAF, NRAS, or neurofibromin 1 (NF1) mutations (triple wild-type) and exhibit a low frequency of c-KIT (KIT proto-oncogene, receptor tyrosine kinase) mutations (15%) [9, 22]. All BRAF, NRAS and NF1 mutations can activate the mitogen-activated protein kinase (MAPK) pathway and are generally introduced in the early stages of tumor evolution as driver mutations [23]. In addition to these driver mutations, a combination of genetic alterations leads to cancer development. In cutaneous melanoma, subsequent mutations in the telomerase reverse transcriptase (TERT) promoter and in regulators of the cell cycle, such as cyclin-dependent kinase inhibitor 2A (CDKN2A), are introduced before the mutation of TP53, which is associated with advanced stages of primary tumor progression [23].
Current treatment options and limitations
Approximately 90% of melanomas are diagnosed as primary tumors without any evidence of metastasis, with a tumor-specific 10-year survival of 75–95% [6]. For primary melanoma without metastasis, excision with a safety margin remains the standard of care [24]. Sentinel lymph node dissection should be performed as a staging procedure in patients with tumor thickness ≥ 1.0 mm or ≥ 0.8 mm with additional histological risk factors [24]. Positive lymph node involvement of melanoma is classified as at least stage III disease. For melanoma with distant metastasis (stage IV) irrespective of local tumor resectability and stage III melanoma, systemic therapy is proposed [24]. However, before 2010, no randomized controlled trial had demonstrated a survival advantage in people with advanced melanoma, and the median overall survival for patients with stage IV melanoma was less than one year [25]. At that time, chemotherapy was the only systemic treatment for metastatic melanoma, with a low response rate of 12.1–17.6% for dacarbazine [26,27,28,29], the only U.S. Food and Drug Administration (FDA)-approved chemotherapy for melanoma. In contrast, chemotherapy is now considered the last-line treatment and is used in patients with resistance to immunotherapy and targeted therapy. To date, the first-line systemic therapy has been immunotherapy, regardless of BRAF mutational status, or alternatively, a combination of BRAF and MEK inhibitors for patients carrying a BRAF mutation [24].
Immunotherapy exploits one’s own personal immune system to kill cancer cells. Since the FDA approval of ipilimumab (anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA4) antibody) to treat metastatic melanoma in 2011 [30], immune checkpoint blocker therapy has represented the primary immunotherapy for melanoma, as it targets CTLA4 and PD-1 on T cells or programmed cell death ligand 1 (PD-L1) on tumor cells, enabling them to escape antitumor responses [24]. Established to evaluate the combined use of anti-PD1 and anti-CTLA4 antibodies for advanced cutaneous melanoma, the phase IIIb/IV CheckMate 511 study reported a 53% response rate and a 61% 3-year overall survival rate [31]. Another trial, CheckMate 067, evaluating the effects of nivolumab (an anti-PD1 antibody) plus ipilimumab and nivolumab alone versus ipilimumab alone for patients with previously untreated unresectable stage III or stage IV melanoma showed 6.5-year overall survival rates of 57%, 43%, and 25% in patients with BRAF-mutant melanoma and 46%, 42%, and 22% in those with BRAF-wild-type melanoma, respectively [32]. From the above experience, it is suggested that the combined use of anti-PD1 and anti-CTLA4 antibodies yields a better response than the solitary use of either antibody, and approximately half of patients at an advanced stage of melanoma can benefit long-term from immunotherapy.
In patients carrying the BRAF V600E mutation, the combination of BRAF and MEK inhibitors such as vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) resulted in a better response than BRAF inhibitors alone and led to a treatment response in a small subset of patients with disease progression receiving BRAF inhibitors alone. Compared to the 53% response rate to vemurafenib alone reported by Sosman et al. [33], an 87% response rate in patients who had never received a BRAF inhibitor and a 15% response rate in patients whose disease had recently progressed while taking vemurafenib alone were recorded in a trial to evaluate the combined use of vemurafenib and cobimetinib [34]. Despite the improved response rate of combined use of BRAF and MEK inhibitors compared with single use, the combination of BRAF and MEK inhibitors for advanced melanoma in patients carrying the BFAF V600E mutation yielded a 50–70% response rate but an approximately 30% 5-year survival rate [24, 34, 35].
Apart from unsatisfactory long-term survival by either immunotherapy or targeted therapy, the treatment options for some types of melanomas, including acral melanoma, mucosal melanoma and uveal melanoma, remain very limited. Due to ill-defined tumor margins, local recurrence often occurs after surgery in patients with acral and mucosal melanoma [36]. Despite effective local therapy, nearly 50% of patients with uveal melanoma develop metastatic disease [37]. In addition, in acral melanoma, the BRAF V600 mutation rate is 10–35%, which limits the use of BRAF/MEK inhibitors (Table 1). C-Kit inhibitors such as imatinib used for metastatic melanomas, mostly from acral and mucosal melanoma with c-KIT alterations, showed an overall response rate of 23% [38]. In addition to uncommon BRAF and c-KIT mutations (Table 1), the unsatisfactory response to currently available targeted therapy with BRAF and c-KIT inhibitors, the paucity of large studies and the rarity of these subtypes also restrict their standard use in patients with acral, mucosal or uveal melanoma [36, 39]. Furthermore, the tumor mutation burden (TMB) of these subtypes of melanoma was usually lower than that of cutaneous melanoma, corresponding to no CSD in the genetic pathology, such that the response to immune checkpoint blocker therapy was unsatisfactory, with a 15.6–43% response rate to the combined use of anti-PD1 and anti-CTLA4 antibodies [40, 41]. In addition, considering that melanoma at AJCC low-risk stages I and IIA accounts for most of the deaths because of the high number of patients with this diagnosis and some so-called high-risk patients with stage III or IIB-C disease who are exposed to systemic treatment but do not need it, finding a better biomarker linked to prognosis is necessary [24].
Therefore, to increase the response rate of immune checkpoint blocker therapy, nivolumab (anti-PD1 antibody) plus an anti-lymphocyte-activation gene 3 (LAG3) antibody (relatlimab) received approval in the United States for the treatment of unresectable or metastatic melanoma in adult patients in March 2022 [42]. Tebentafusp, a bispecific antibody that targets the glycoprotein 100 (gp100) protein on tumor cells and CD3 on T cells, received approval for the treatment of unresectable or metastatic uveal melanoma in January 2022 [43]. The first viral oncolytic vaccine, talimogene-laherparepvec (T-VEC), was even approved by the FDA as early as 2015 for patients with unresectable metastatic melanoma with improved local control but a mild systemic effect [44]. The ideal implantation of combined BRAF/MEK inhibitors and immune checkpoint blocker therapy, the involvement of oncolytic viral vaccines with immunotherapy, new drug development and nanotechnology-based administration systems for melanoma treatment are ongoing [45]. The development of novel treatments for targeting biomarkers responsible for melanoma progression, as alternatives or as complementary treatments to immunotherapy, is an outcome of recent investigations [46]. One strategy for using potential novel agents is the induction of RCD. Different types of RCD can affect cancer progression and response to therapy, and evasion from RCD is one of the important characteristics of cancer cells[47]. Furthermore, RCD is essential for controlling the immunosuppressive tumor microenvironment (TME) [48]. In addition, developing a RCD-related gene expression score for use as a reliable biomarker to predict prognosis and guide treatment choices is a possible strategy to offer personalized therapy [49].
Regulated cell death in the melanoma context
RCD plays a critical role in cell homeostasis, tissue remodeling, and tumorigenesis. Apoptosis is the most well-characterized type of RCD and is the major RCD modality of cancer cells. Clinically, many traditional chemotherapeutic agents have been designed to induce apoptosis in cancer cells. Therefore, evasion from apoptosis not only enables defective cells with dangerous mutations to undergo tumorigenesis but also promotes therapy resistance. With the discovery of more types of RCD, it is now evident that cancer cells can succumb to other RCD pathways in addition to apoptosis. Research on nonapoptotic pathways of RCD as alternative therapeutic targets is an attractive option to overcome the failure of apoptosis induction in cancer cells. The importance of nonapoptotic cell death signaling pathways, such as autophagy-dependent cell death, necroptosis, ferroptosis, pyroptosis, and cuproptosis, in melanoma formation and progression is discussed in the following section. In addition, the connections among these RCDs, melanoma and immunotherapy are discussed.
Autophagy-dependent cell death
Autophagy is a cellular catabolic mechanism wherein proteins, bulk cytoplasm, and/or organelles are sequestered inside double-membrane intracellular vesicles for subsequent recycling via lysosomes (Fig. 1). Therefore, autophagy plays a crucial role in maintaining cellular homeostasis via the elimination of unfolded proteins and damaged organelles. Autophagy has often been considered a system that suppresses tumor development during the initial stage of carcinogenesis. However, the advancement of cancer, including melanoma, has been linked to the tumor-promoting function of autophagy. Autophagy-related 5 (ATG5) downregulation promotes the proliferation of BRAFV600E melanocytes, supporting the suppressive role of autophagy in tumorigenesis [50]. In contrast, enhanced basal autophagy in melanoma cells carrying the BRAFV600E mutation, causing chronic ER stress, is related to chemoresistance, which can be reversed by chemical chaperones (4-phenylbutyric acid), accompanied by decreased basal autophagy and increased susceptibility to cell death induction [51]. One reason for the tumor-promoting role of autophagy is that active autophagy is an adaptive mechanism to microenvironment insults for melanoma cells. Therefore, inhibition of autophagy makes melanoma cells vulnerable to fluctuating oxygen pressure [52] and low pH [53], both of which are associated with tumor progression and tumor metastasis. Furthermore, autophagy may be a mechanism through which melanoma cells mitigate the effects of drug activity and drug-induced alterations in the TME. Therefore, the autophagy machinery exhibits a significant association with clinical outcomes and plays a substantial role in the development of drug resistance. In a phase II trial of temozolomide (an oral alkylating agent) and sorafenib (an oral multikinase inhibitor), patients with melanoma displaying high autophagy activity had a worse clinical outcome [54]. Conversely, autophagy inhibition with either hydroxychloroquine or inducible shRNA against ATG5 resulted in significantly augmented temozolomide-induced cell death in aggressive melanoma spheroids [55]. Another example is that the efficacy of vemurafenib (a BRAF inhibitor) is enhanced by the ectopic expression of miR-216b to attenuate autophagy both in vitro and in vivo [56]. In addition, natural products are attractive sources of molecules that effectively kill melanoma cells. For example, 7-hydroxydehydronuciferine, a dehydroaporphine that is isolated from the leaves of Nelumbo nucifera Gaertn cv. Rosa-plena, induces cytotoxicity through apoptosis- and autophagy-dependent cell death in the context of melanoma both in vitro and in vivo [57].

Schematic diagram of autophagy-dependent cell death and related research in melanoma. Autophagy is a process initiated by ULK1 complex formation, followed by membrane isolation and phagospore formation. After fusion with lysosome, autophagosome ends up and engulfed substrates are digested in autophagosome. Related researches are expressed in white squares with reference number in parenthesis. The exogenous small molecules are marked in blue squares especially. For example, vemurafenib (a BRAF inhibitor) resistance is associated with elevated expression of Beclin-1, ATG5 and UVRAG, and its efficacy can be increased by ectopic expression of miR-216b to inhibit ATG5. The manipulation of autophagy can also influence the immune response in melanoma, such as increased local infiltration of NK cells in melanoma after inhibition of Beclin-1, ATG5 or p62 correlated with increased expression of CCL5 and improved prognosis. Loss of BNIP3 decreased macrophage phagocytosis of dying melanoma cells. Besides, autophagy activity is associated with MDSC- mediated suppression of anti-melanoma immunity, melanoma adaption to fluctuating O2 or pH, and outcome of targeted therapy with temozolomide and sorafenib
Because of the relatively high responsiveness of melanoma to immunotherapy, studies have focused on the relationship among autophagy, melanoma and the immune response, revealing that the role of autophagy is dynamic and multifaceted and induces different effects in different scenarios. The loss of BCL2-interacting protein 3 (BNIP3, an inducer of autophagy) in melanoma cells did not alter apoptosis induction but attenuated the phagocytosis-mediated clearance of dying melanoma cells [58]. Forced expression of microtubule-associated protein 1 light chain 3 beta (MAP1LC3B, encoding an autophagy initiator) restored the susceptibility of phosphatase and tensin homolog (PTEN)-deficient melanoma cells to T-cell-mediated cell killing [59]. The mouse model showed that the T-cell-mediated response was the same for melanoma cells with functional autophagy machinery, melanoma cells without genes related to autophagy, and melanoma in which autophagy is blocked by chloroquine [60]. In contrast to the neutral or positive role on T-cell and phagocytic immunity, increased local infiltration of natural killer (NK) cells after inhibition of Beclin-1, ATG5 or p62/Sequestosome 1 (SQSTM1) in melanoma cells correlated with increased expression of the chemokine C–C motif chemokine ligand 5 (CCL5) has been reported. High expression of CCL5 has also been correlated with prolonged patient survival [61, 62]. Finally, inhibiting autophagy in myeloid-derived suppressor cells (MDSCs), a group of immune cells that accumulate in tumors to dampen the immune reaction, shows the potential to slow melanoma expansion and trigger strong antimelanoma immune reactions in mice [63].
Pyroptosis
Pyroptosis is a lytic programmed cell death characterized by pore formation in the plasma membrane via the oligomerization of cleaved gasdermin. It was first discovered in 1992 in macrophages infected with the gram-negative bacterial pathogen Shigella flexneri [64], and the term pyroptosis was coined in 2001 [65]. Initially, pyroptosis was thought to involve only the death of monocytes caused by caspase-1 activation [66]. However, research into pyroptosis has increased, revealing a wide range of triggering conditions, such as cancer. This expansion in pyroptosis-related environments coincided with the identification of the gasdermin family. The gasdermin superfamily includes gasdermin A/B/C/D (GSDMA/B/C/D), gasdermin E (GSDME, also known as DFNA5), and DFNB59 (Pejvakin, PJVK) in the context of human biology [67]. Except for Pejvakin, these proteins share a common structural arrangement featuring two conserved segments: the pore-forming domain in the N-terminus and the repressor domain in the C-terminus. Theoretically, gasdermins form pores after the N-terminal domain is dissociated from the C-terminal domain via caspase or granzyme cleavage. Among these gasdermins, GSDMD and GSDME are the most intensively studied in the context of pyroptosis. Generally, pyroptosis can be initiated through one of the following four routes: (1) the canonical pathway: inflammasome assembly triggers caspase-1 activation, leading to GSDMD cleavage and subsequent release of IL-1β and IL-18; (2) the noncanonical pathway: caspase-4/5/11 can be activated through direct binding of the N-terminal caspase-activation and recruitment domain (CARD) to intracellular lipopolysaccharide (LPS) [68] with subsequently activated caspase-4/5/11 cleaving GSDMD to release N-GSDMD, which forms pores on the cell plasma membrane; (3) the caspase 3/8-mediated pathway: previously, caspases 8 and 3 were thought to be initiator and effector caspases, respectively, in the apoptosis pathway; however, chemotherapeutic drugs have been shown to induce caspase-3-mediated pyroptosis [69], and GSDMD can be cleaved by caspase 8 to execute pyroptosis, while caspase 8 autoprocessing is a feasible possibility [70]; and (4) the granzyme-mediated pathway: granzymes A and B, which are released by NK cells and cytotoxic T lymphocytes, respectively, cleave GSDMB and GSDME to initiate pyroptosis (Fig. 2). In the caspase 3-mediated noncanonical pyroptosis pathway activated by chemotherapeutic agents, DNA-binding and DNA-modifying drugs such as doxorubicin, cisplatin, and actinomycin-D, along with topoisomerase inhibitors such as topotecan, CPT-11, etoposide, and mitoxantrone, induce pyroptosis in GSDME-positive cancer cells such as SH-SY5Y neuroblastoma and MeWo skin melanoma cells [71]. GSDME expression can lead to a switch from tumor necrosis factor (TNF)-induced apoptosis to pyroptosis mediated through caspase 3 activity [71]. Antibiotic chemotherapy, including daunorubicin, doxorubicin, epirubicin, and actinomycin-D, was also reported to increase the nuclear translocation of PD-L1, promote GSDMC expression and activate caspase-8. Activated caspase 8 further cleaved GSDMC and induced pyroptosis in breast cancer cells [69]. In addition to inhibiting antitumor immunogenicity to enable PD-L1 to bind PD-1 on T lymphocytes, PD-L1 on melanoma cells trans-interacts with PD-1 on melanoma cells, leading to cell proliferation and in vivo tumor growth [72]. Pyroptosis has been reported to be triggered by nuclear PD-L1 under hypoxic conditions and has been observed in various cancer cell types, including breast, liver, lung, and ovarian cancers and melanoma [69]. Moreover, the GSDME protein levels in cancer cells are not as invariable as those in normal cells [73], and BRAF/MEK inhibitors can promote the cleavage of GSDME and the release of high-mobility group box 1 (HMGB1), which are markers of pyroptotic cell death in melanoma cells [74]. After the development of resistance to BRAF/MEK inhibitors, these resistant melanoma cell lines are susceptible to pyroptosis, which can be induced by etoposide or doxorubicin [74], and the combination of temozolomide and chloroquine [75]. The combined use of a phosphoinositide-dependent kinase 1 (PDPK1) inhibitor (GSK2334470) and a MEK inhibitor (trametinib) suppressed NRAS-mutant xenograft growth and induced GSDME-associated pyroptosis in NRAS-mutant melanoma model mice [76]. In addition, reactive oxygen species (ROS) induce pyroptosis via BAX (BCL2 associated X) recruitment to release cytochrome c and subsequent caspase-3 activation. In contrast to the lack of an effect mediated by iron supplementation on tumor growth reduction in GSDME-knockdown mice, iron-induced ROS production via iron dextran decreased melanoma growth in GSDMEwt mice, demonstrating that ROS-induced pyroptosis is GSDME dependent [77].

Pyroptosis and its role in melanoma. Pyroptosis can be activated by four pathways: (1) the canonical pathway with inflammasome assembly and caspase-1 activation (2) the noncanonical pathway with caspase-4/5/11 activation by direct binding of its N-terminal CARD domain to intracellular LPS. (3) Caspase 3/8-mediated pathway with GSDMC or GSDME cleavage (4) Granzyme-mediated pathway with granzyme A and B, secreted from cytotoxic T lymphocytes and NK cells respectively, can cleave GSDMB and GSDME, respectively. Research of pyroptosis on melanoma was expressed in white squares with reference number in parenthesis. The exogenous molecules are marked in blue squares especially. GSDME cleavage by activated caspase 3 has been observed by chemotherapeutic drugs, BRAF and MEK inhibitors/ PDPK1 and MEK inhibitors, and ROS production with iron dextran or raptinal, a caspase 3 activator. Caspase-1 DNA was ever included in anticancer DNA vaccine to induce pyroptosis of melanoma cells. Pyroptosis can also be induced by temozolomide and chloroquine via autophagy inhibition and inflammasome activation
Since pyroptosis is a form of inflammatory programmed necrosis, the role of pyroptosis in the TME has been studied. After treatment with BRAF/MEK inhibitors, melanoma cells lacking GSDME exhibited impaired infiltration of tumor-associated T cells, the number of activated dendritic cells was diminished, and a higher incidence of tumor regrowth after cessation of drug treatment was recorded [74]. Pyroptosis-associated genes were utilized in modeling aimed at forecasting melanoma prognosis, predicting immunotherapy responses, and discerning immune microenvironment attributes [78], with applicability extending to metastatic melanoma as well [79]. The increased expression of pyroptosis-related genes correlated with increased infiltration of tumor-associated B cells, plasma cells, CD8+ T cells, activated memory CD4+ T cells, regulatory T cells (Tregs), and M1 macrophages, while the levels of resting NK cells, M2 macrophages, M0 macrophages, and resting mast cells were reduced. This pattern of gene expression and cell contents corresponded to a more favorable prognosis [78]. Nevertheless, pyroptosis might exhibit tumor-promoting characteristics stemming from inflammasome activation in the context of chronic inflammation. This possibility was supported by the finding of a significantly reduced incidence of lung cancer and decreased lung cancer mortality in a trial after the activity of the major product of inflammasome activation, IL‐1β, was inhibited by a specific antibody, canakinumab [80]. A recently proposed hypothesis suggests that the results hinge on whether inflammasome activation occurs predominantly within the tumor or within immune cells. Persistent inflammasome activation within the TME leads to tumor promotion and immune suppression. In contrast, when inflammasomes are activated within the immune system, specifically within dendritic cells, they contribute to attenuated melanoma progression [81]. In addition, active caspase 1, a genetic adjuvant in DNA vaccination against cancers, promoted pyroptosis to kill melanoma cells in mice [82]. In addition, the caspase-3 direct activator raptinal, a bifluorene–dicarbaldehyde compound, induced pyroptosis in both human and mouse melanoma cell line models and delayed tumor growth in vivo; this study suggests that the release of damage-associated molecular patterns (DAMPs) and inflammatory cytokines is dependent on caspase activity and GSDME expression [83]. Therefore, pyroptosis induction may be a strategy to treat melanoma, but to determine how to manipulate pyroptosis to eliminate its tumor suppression effect, more study is needed.
Necroptosis
Necroptosis is a caspase-independent cell death pathway recognized as programmed necrosis. Furthermore, necroptosis can be triggered by the substantial production of ROS, hyperactivation of poly (ADP-ribose) polymerase 1 (PARP1) and depletion of ATP [84, 85]. Through activating death receptors, Toll-like receptors or cytosolic nucleic acid sensors that induce type I interferon (IFN-I) and TNFα production in an autocrine feedback loop, necroptosis can be triggered. Initially, receptor-interacting protein kinase 1 (RIPK1) is deubiquitylated by CYLD lysine 63 deubiquitinase (CYLD) and can recruit receptor-interacting protein kinase 3 (RIPK3). Then, the RIPK1/RIPK3 complex recruits and phosphorylates mixed lineage kinase domain-like pseudokinase (MLKL). Finally, phosphorylated MLKL oligomerizes and forms a large pore on the plasma membrane, leading to necroptotic cell death [86] (Fig. 3). Crosstalk between the extrinsic pathway of apoptosis and necroptosis is mediated through the activation of death receptors, and necroptosis can be activated when the intracellular apoptosis signaling pathway is inhibited [87]. A shift from the autophagic flux response to necroptotic cell death has been observed when nitrogen-doped titanium dioxide (N-TiO2) nanoparticles were photoactivated in melanoma cells [88]. Several investigations have reported that the potential to trigger necroptosis in melanoma might be inhibited due to low expression levels of both CYLD [89] and RIPK3 in melanoma cell lines [90, 91].

Schematic diagram of necroptosis and its effects on melanoma. Necroptosis can be triggered by activation of death receptors or Toll-like receptors. Through interaction of activated RIPK1 with RIPK3, and subsequently, RIPK3 phosphorylates MLKL. Pore formation on cell membrane is the final step of necroptosis. Research of necroptosis on melanoma was expressed in white squares with reference number in parenthesis. The exogenous molecules are marked in blue squares especially. While unsure effect of RIPK1 inhibition on melanoma metastasis, direct intratumor delivery of MLKL mRNA can inhibit melanoma tumor growth and metastasis and augment the efficiency of immune blockade therapy. Besides, NTiO2, BAY 87–2243 and CBL0137 are the agents with the ability to induce necroptosis in melanoma cells
With the manipulation of upstream RIPK1 and the effector protein of MLKL in the necroptosis pathway, the relevance of necroptosis to melanoma progression and metastasis is not clear. For example, a novel RIPK1 inhibitor, PK68, significantly suppressed the lung metastasis of melanoma cells in mice [92] The inhibition of RIPK1 in mice by knock-in inactivated RIPK1 D138N or the murine-potent inhibitor GNE684 showed no effect in reducing lung metastases of B16 melanoma cells, although it mitigated collagen antibody-induced arthritis, skin inflammation caused by mutation of Shank-associated RH domain interactor (SHARPIN, which is associated with NF-kappa-B activation and regulation of inflammation), or colitis caused by deletion of Nemo (also known as IKK-γ. a subunit of activated NF-κB) [93]. However, direct intramelanoma delivery of either the RIPK3 gene via adenovirus or mRNA encoding MLKL, a necroptosis executioner, elicited both necroptosis and potent antitumor immunity in melanoma model mice [94, 95]. The combination of MLKL mRNA and anti-PD1 treatment revealed better antitumor activity compared with anti-PD-1 alone, and the effect depended on CD4 and CD8 T cells under the control of type I interferon signaling and basic leucine zipper transcription factor ATF-like 3 (Batf3)-dependent dendritic cells [95]. Anti-PD-1 unresponsiveness in mouse models of melanoma was reversed by a small molecule, curaxin CBL0137, which potently activated Z-form nucleic acid binding protein 1 (ZBP1)-dependent necroptosis by triggering Z-DNA formation in tumor-infiltrating fibroblasts, irrespective of the potential for low necroptotic gene expression in melanoma cells [96]. In addition, necroptosis and ferroptosis may be the mechanisms underlying melanoma cell death simultaneously; for example, BAY 87-2243-induced melanoma cell death due to ROS accumulation through mitochondrial complex I inhibition was attenuated by necrostatin (a necroptosis inhibitor), knockdown of RIPK1 or MLKL, ferrostatin (a ferroptosis inhibitor) or knockdown of GPX4 but not treatment with the pancaspase inhibitor z-VAD-FMK [97].
Ferroptosis
Ferroptosis is an orchestrated caspase-independent mechanism underlying cell death distinguished by the excessive generation of ROS and accumulation of iron-associated lipid peroxides [98,99,100]. Free radical attack on polyunsaturated fatty acids in the membrane results in the formation of lipid hydroperoxides (L-OOH), which can be converted into highly reactive lipid alkoxy radicals (L-O•) by ferrous ions and thus induce the initiation of ferroptosis. L-OOH can be reduced to lipid alcohol (L-OH) by selenoprotein glutathione peroxidase 4 (GPX4) in the presence of glutathione (GSH), an undependable hydrophilic cellular antioxidant that prevents lipid peroxidation. Therefore, ferroptosis can be triggered by inhibition of GSH biosynthesis or inhibition of GPX4 (Fig. 4). System Xc−, a transporter responsible for exchanging cysteine (Cys) and glutamic acid (Glu), comprises the catalytic subunit xCT (also known as solute carrier family 7 member 11, SLC7A11), which is the light chain, and the regulatory subunit 4F2hc (also known as solute carrier family 3 member 2, SLC3A2), which is the heavy chain. These subunits are interconnected by disulfide bonds, forming a pivotal upstream hub within the System Xc − /GSH/GPX4 pathway. This pathway relies on the System Xc− − mediated import of cysteine, which is utilized for the biosynthesis of glutathione (GSH). Therefore, erastin, which interferes with system Xc−, is a ferroptosis inducer.

The relationship between ferroptosis and melanoma. Ferroptosis is characterized by the overwhelming production of ROS and accumulation of iron-dependent lipid peroxides. Research of ferroptosis on melnoma was expressed in white squares with reference number in parenthesis. The exogenous molecules are marked in blue squares especially. Ferroptosis can be induced in melanoma cells by GPX4 inhibition in the situation of BRAF inhibitor resistance or with the application of plant-derived phyto-sesquiterpene lactone. Nobiletin can also induce ferroptosis in melanoma cells through another pathway of GSK3β-mediated Keap1/Nrf2/HO-1 signaling. In addition, IFNγ release after immunotherapy can drive ferroptosis by downregulating the expression of system Xc−. which can be further enhanced by the delivery of miR-21-3p-loaded gold nanoparticles
Cancer cells are reported to be highly vulnerable to disruptions to thiol metabolism and an overabundance of iron [101]. In melanoma, BRAF inhibitors can sensitize melanoma cells to agents that cause ferroptosis, which reduces the abundance of SLC7A11 transcripts [102]. The sensitivity of melanoma cells to ferroptosis relies on their enhanced dependence on oxidative phosphorylation (OXPHOS), which is upregulated by BRAF inhibitors in BRAF-mutant melanoma cells, resulting in the accumulation of ROS [103, 104]. With the development of resistance to BRAF inhibitors, ferroptosis plays a role in melanoma cell viability. Using a mouse model and melanoma cell lines generated from mouse tumors treated with vemurafenib (BRAF inhibitor) or not, it has been shown that the acquisition of drug resistance is associated with an increase in mitochondrial OXPHOS, growth dependency on the glutamine supply and a compensated increase in glutathione levels, which is associated with strong activation of the nuclear factor erythroid 2-related factor-2 (NRF2) pathway and increased xCT expression [105]. The increase in the GSH level and increased xCT expression indicated that these melanoma cells may be more resistant to ferroptosis induction. In contrast, by clustering gene expression in 53 human melanoma cell lines, melanoma dedifferentiation was linked to resistance to MAPK inhibitors and immunotherapy, lower basal levels of GSH and increased sensitivity to ferroptosis [106]. The basal GSH levels were lower in vemurafenib-resistant cell lines than in matched drug-naïve cells [106]. Moreover, loss of GPX4 induced the death of BRAF-mutant therapy-resistant cells via ferroptosis in vitro and prevented tumor relapse after treatment in vivo [107]. Although the basal GSH level in MAPK inhibitor-resistant melanoma cells was not conclusive by Khamari et al. [105] or Tsoi et al. [106], all their studies revealed the need for GSH level increases for melanoma cells to overcome ferroptosis, and the induction of ferroptosis may be a therapeutic target, even in MAPK inhibitor-resistant melanoma cells. In addition, nobiletin, isolated from citrus peel, induced ferroptosis in human skin melanoma cells through the GSK3β-mediated Keap1/Nrf2/HO-1 signaling pathway [108]. The phyto-sesquiterpene lactone DET and its derivative DETD-35 cause lipid ROS to accumulate, which leads to ferroptotic cell death in both BRAF-sensitive and BRAF-resistant V600E melanoma cells [109]. These studies show that natural products or extracts from plants are possible treatment alternatives to MAPK inhibitors.
In contrast to alternative forms of cellular demise, such as apoptosis, pyroptosis, and necroptosis, the factors of which are clearly recognized by the immune system [110], whether inducing or instigating ferroptosis through external or internal mechanisms can lead to a similar "physiological function" as these other forms of RCD is unclear. Notably, GPX4 can abrogate lipoxygenase and cyclooxygenase function by lowering lipid peroxide levels [111]. It seems likely that GPX4 activity can exert a large effect on the release of both proinflammatory and anti-inflammatory lipids [110] when GPX4 activity is hampered. A previous investigation demonstrated that CD8+ T cells activated by immunotherapy released IFNγ, causing a reduction in the expression of SLC3A2 and SLC7A11, subsequently leading to high lipid peroxidation specific to ferroptosis. As a result, ferroptosis is induced within tumor cells [112]. Systemic delivery of miR-21-3p-loaded gold nanoparticles increased the efficacy of anti-PD-1 antibodies by promoting ferroptosis in preclinical melanoma model mice [113]. Administration of a biomineralized nanovaccine containing Fe3+ and the photosensitizer IR820 through intratumoral injection triggered ferroptosis and localized immunogenic cell death. This approach enhanced the effectiveness of CTLA-4 blockade therapy in model mice [114]. In addition, a model established with ferroptosis-related genes or long noncoding RNAs (lncRNAs) was generated to predict the prognosis of melanoma, showing the correlation of increased local immune cell infiltration with an increased response to immunotherapy [115, 116]. All these studies suggest that the combination of ferroptosis induction-based therapy and immunotherapy is a potential option for treating melanoma.
Cuproptosis
Cuproptosis, a recently discovered RCD modality, was first described in a 2022 publication by Tsvetkov et al. [117]. This distinct mode of cellular demise is activated by the accumulation of intracellular copper, which results in the clustering of mitochondrial lipoylated proteins and the disruption of Fe-S cluster proteins. In contrast to other types of controlled cell death, such as apoptosis, necroptosis, and ferroptosis, inhibitors such as ferrostatin-1 and necrostatin-1 and antioxidants such as N-acetyl cysteine exert no effect on cuproptosis. In addition, cuproptosis functions through the mitochondrial respiration chain complex, not through ATP production, as demonstrated by the fact that treatment with copper ionophores did not significantly reduce basal or ATP-linked respiration. In the study by Tsvetkov et al., intracellular copper shuttled by elesclomol, a copper ionophore, directly bound to lipoylated mitochondrial proteins, leading to their aggregation and subsequent loss of Fe-S cluster proteins. This proteotoxic stress condition ultimately resulted in cuproptosis (Fig. 5). The lipoylated mitochondrial proteins identified, including DBT, GCSH, DLST, and DLAT, were involved in regulating carbon entry points to the tricarboxylic acid (TCA) cycle [118, 119].

Schematic diagram of cuproptosis and correlation between cuproptosis and melanoma. Copper ions can cross the cell membrane into the intracellular space with a copper ionophore, and their concentration can be regulated by copper importers/exporters (SLC31A1/ATP7A and B). FDX1 not only reduces divalent copper to more toxic monovalent copper but also regulates protein lipoylation. Intracellular copper can bind lipoylated proteins directly, leading to their aggregation and further occurrence of cuproptosis. Copper ions also contribute to Fe-S cluster loss, which is another mechanism of cuproptosis. Related researches are expressed in white squares with reference number in parenthesis. Elesclomol, a copper ionophore, can induce cuproptosis, and was shown to eliminate slow-cycling melanoma cells. A higher expression level of LIPT1, responsible for transferring lipoic acid to the E2 subunits of AKGDH and pyruvate dehydrogenase PDH, is related to longer survival after immunotherapy
Ferredoxin 1 (FDX1), a reductase that converts Cu2+ to its toxic form, Cu1+, and a direct binding target of elesclomol [120], was established as the upstream positive regulator of DLAT lipoylation, and it was essential for copper binding. Furthermore, mass spectrometry analysis indicated that copper ionophore treatment led to the loss of Fe–S cluster proteins in an FDX1-dependent manner.
The discovery of cuproptosis has sparked numerous studies aimed at understanding its role in tumor development and prognosis, particularly in melanoma. Through July 2023, 10 articles discussing cuproptosis and melanoma had been published on PubMed [121,122,123,124,125,126,127,128,129,130]. Eight of these studies described the correlation between cuproptosis-related genes (CRGs) and melanoma prognosis [121,122,123, 126,127,128,129,130], and the other two studies presented a discussion of the impact of cuproptosis-related lncRNAs [124, 125]. CRGs were assessed in cutaneous melanoma samples in five studies [121, 126,127,128, 130] and uveal melanoma in two studies [123, 129]. In addition to prognosis prediction, the expression of CRGs was related to the regulation of the TME with differential immune cell infiltration [121,122,123, 126,127,128,129,130], response to immunotherapy [126, 130], and different chemotherapeutic and targeted drug sensitivities [122, 128, 130].
Based on the work of Tsvetkov et al., the CRGs include FDX1, LIPT, LIPT1, DLD, DLAT, PDHA1, PDHB, MTF1, GLS, CDKN2A, SLC31A1, ATP7A, and ATP7B [117]. These CRGs not only serve as prognostic indicators but are also used to identify intersecting genes, such as AIM2, LAG, SLC39A6, TMEM117, PTPRC, and KIF14, for building prognostic models [122]. Among these genes, LIPT1 has emerged as a advantageous prognostic marker for individuals with melanoma [121]. Additionally, the expression of the LIPT1 gene has been linked to PD-L1 expression, regulatory T-cell infiltration, and longer survival in melanoma patients who were treated with immunotherapy [121]. Moreover, CRGs have been used to identify pyroptosis-related lncRNAs, leading to the establishment of prognostic models [124, 125]. The immune microenvironment landscape correlated with these lncRNA prognostic models, revealing significant differences in regulatory T-cell infiltration rates and better immunotherapy responses in lower-risk groups [124, 125].
Although the role of cuproptosis in melanoma progression is attractive, the exact mechanism is not known by CRG analysis, and the upstream regulators and downstream effectors remain unclear. In-depth studies are needed to decipher the mechanisms underlying cuproptosis-related proteins, their interactions with other RCD pathways, and the potential therapeutic applications of manipulating cuproptosis as a target. For example, FDX1 has been identified as a positive upstream regulator of cuproptosis, and its expression is decreased in high-risk melanoma, as classified by CRGs; however, its knockdown inhibited melanoma cell proliferation in vitro [122]. Similarly, before the discovery of cuproptosis, copper was noted to cause cell death in the 1980s [131]. Copper ionophores were utilized to treat cancer, and the cytotoxicity was inferred to be ROS mediated at that time [132]. Elesclomol was shown to transport copper to the mitochondria to induce oxidative stress in melanoma cells in a 2012 study [133]. Elesclomol can selectively kill slow-cycling melanoma cells, which are considered multidrug resistant [134]. However, the clinical use of elesclomol combined with paclitaxel yielded mixed results in clinical trials [135, 136]. Clinical trials investigating disulfiram (a copper ionophore) to treat melanoma resulted in unsatisfactory data and were unpublished [137]. The mitochondrial morphological change remains to be determined during cuproptosis [138]. In addition, another copper ionophore, thiomaltol induced rapid lysosomal accumulation of copper, concurrent with the onset of apoptosis. This finding implied a mechanism other than cuproptosis and ROS accumulation to explain copper cytotoxicity [139]. In fact, there was one study stating that copper cytotoxicity was associated with the disturbance of proteostasis due to protein misfolding and aggregation by the interaction of copper with proteins [140]. GSH can protect cells from preventing copper ion interactions with proteins [140]. Regardless of cuproptosis, copper-induced oxidative stress or protein misfolding and aggregation, copper can form a cogroup of many enzymes and proteins that plays a crucial role in maintaining mitochondrial homeostasis [138]. All these findings necessitate further investigation of the mechanism regulating cuproptosis, especially the impact on mitochondrial dysfunction, and its possible role in tumor metabolic pathway switching.
Common mechanisms among these nonapoptotic RCDs
Although pyroptosis, ferroptosis, necroptosis, and cuproptosis present distinct biological processes with unique morphological features and triggers (Table 2), there are certain commonalities among them. For example, the caspase family not only functions in apoptosis but also regulates other types of RCDs, depending on the cellular context and stimuli. Pyroptosis can be triggered by caspase 1 in the canonical pathway, caspase 4/5/11 in the noncanonical pathway, or caspase 3/8 in GSDMC- or GSDME-expressing tumor cells, including melanoma cells. Although necroptosis is a caspase-independent RCD, its activation depends on caspase 8 inactivation. Similarly, the execution of autophagy-dependent cell death is caspase independent, but caspases can modulate it, such as the induction of autophagy-dependent cell death in fibroblasts and monocytoid cells by caspase 8 inhibition, which requires ATG7 and beclin-1 [141]. Ferroptosis and cuproptosis are both caspase-independent RCDs. In addition to caspase, ROS are another important modulator that induces these RCDs. ROS caused by iron supply can lead to pyroptosis of melanoma cells by GSDME cleavage [77]. The accumulation of ROS induces necroptosis and can form positive feedback [142]. The occurrence of ferroptosis depends on lipid peroxidation generated by ROS attack. Although the cell death mechanism is not related to ROS in cuproptosis, copper can induce cell death through an increase in ROS[143]. Since mitochondria are the main source of ROS, the generation of these RCDs is often associated with mitochondrial dysfunction.
BRAF-mutant melanoma cells can become resistant to BRAF/MEK inhibitors after long-term exposure. However, these resistant cells can be manipulated to be sensitive to the induction of pyroptosis and ferroptosis [74, 107, 109]. Inhibition of autophagy activity can increase the efficiency of BRAF inhibitors [56]. The performance of necroptosis or cuproptosis in BRAF inhibitor-resistant melanoma cells is not well evidenced. Apart from targeted therapy, the efficiency of immunotherapy in melanoma has been shown to be enhanced by the induction of necroptosis or ferroptosis [95, 113]. Inhibition of autophagy processing, expression of pyroptosis- or cuproptosis-related genes also showed an impact on the TME and local immunity [59, 62, 78, 121,122,123, 126,127,128,129,130]. These RCDs can be triggered at the same time, such as ferroptosis and necroptosis induction by BAY-2743 [97]. Ferroptosis inducers sorafenib and erastin can also enhance cuproptosis in primary liver cancer cells by increasing copper-dependent lipoylated protein aggregation [144]. All these findings support the possible interchangeability of these RCDs on the dying pathway of melanoma cells, and what we know is not enough.
Conclusions and perspectives
Effective treatment for metastatic melanoma has been historically challenging, with a 5-year survival rate of only 15–20% recorded between 1992 and 2011 [145]. However, with the introduction of targeted therapy and immunotherapy in the 2010s, the 3-year survival rate increased to 39.4% [2]. Despite these advancements, the continuously increasing incidence of melanoma highlights the ongoing need for more effective therapeutic approaches.
In the first part of this review, we provide an overview of the molecular-level pathogenesis of melanoma and describe current treatment options, focusing on systemic therapy involving targeted therapy and immune blocker therapy for advanced melanoma. Although significant progress has been made, certain limitations persist, such as poor response rates to immune checkpoint blockers and targeted therapy in patients with subtypes such as acral, mucosal, and uveal melanoma. Additionally, secondary treatment resistance and the need for reliable biomarkers to guide treatment selection remain important challenges.
The application of immunotherapy and targeted therapy has ushered in a new era of melanoma treatment. However, these approaches alone are not sufficient due to treatment resistance and loss of response. Synergistic therapy, combining different treatment modalities, holds great potential and may become the future mainstay of cancer therapy. As evasion from RCD is a key feature of tumorigenesis, targeting various nonapoptotic RCDs with pharmacological small-molecule compounds is a promising therapeutic avenue (Table 3).
The second part of this review focuses on summarizing five nonapoptotic RCDs in melanoma: autophagy-dependent cell death, pyroptosis, necroptosis, ferroptosis, and cuproptosis. Although melanoma cells show vulnerability to the induction of RCD, it is challenging to induce cells to succumb to a specific type of RCD due to the complexity and crosstalk among RCD pathways, particularly in cancer cells such as melanoma. Simultaneous regulation of multiple RCD subroutines may help overcome resistance to specific types of RCD [47]. Thus, future research should be directed at exploring the interplay between different RCD pathways, as it holds promise for melanoma treatment as a part of combination therapy with immunotherapy and in targeted therapy. In addition, prognostic models using pyroptosis, ferroptosis, and cuproptosis-related genes or lncRNAs respectively have been reported [77,78,79, 115, 116, 123, 125], and further collaboration may offer reliable biomarkers to treat melanoma.
Overall, a comprehensive understanding of the molecular mechanisms underlying melanoma development, combined with the exploration of novel therapeutic strategies targeting RCD pathways, shows the potential to further enhance treatment outcomes and address existing challenges in melanoma management.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article.
Abbreviations
- AJCC:
-
American Joint Committee on Cancer
- ATG5:
-
Autophagy related 5
- ATP7A/B:
-
ATPase copper transporting alpha/beta
- Batf3:
-
Basic leucine zipper transcriptional factor ATF-like 3
- BAX:
-
BCL2 associated X
- BNIP3:
-
BCL2-interacting protein 3
- BRAF:
-
B-Raf proto-oncogene, serine/threonine kinase
- CARD:
-
Caspase-activation and recruitment domain
- CCL5:
-
C-C motif chemokine ligand 5
- CDKN2A:
-
Cyclin dependent kinase inhibitor 2A
- c-KIT:
-
KIT proto-oncogene, receptor tyrosine kinase
- CRGs:
-
Cuproptosis-related genes
- CSD:
-
Cumulative solar damage
- CTLA4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CYLD:
-
CYLD lysine 63 deubiquitinase
- DAMP:
-
Damage-associated molecular pattern
- DBT:
-
Dihydrolipoamide branched chain transacylase E2
- DLAT:
-
Dihydrolipoamide S-acetyltransferase
- DLD:
-
Dihydrolipoamide dehydrogenase
- DLST:
-
Dihydrolipoamide S-succinyltransferase
- EDNRB:
-
Endothelin B receptor
- FDX1:
-
Ferredoxin 1
- GSDMA/B/C/D/E:
-
Gasdermin A/B/C/D/E
- GCSH:
-
Glycine cleavage system protein H
- GLS:
-
Glutaminase
- GPX4:
-
Glutathione peroxidase 4
- GSH:
-
Glutathione
- GSK3β:
-
Glycogen synthase kinase-3 beta
- HMGB1:
-
High mobility group box 1
- HO-1:
-
Heme oxygenase-1
- IFN-I:
-
Type I interferon
- Keap1:
-
Kelch-like ECH-associated protein 1
- KIF14:
-
Kinesin family member 14
- LAG3:
-
Lymphocyte-activation gene 3
- LC3:
-
Microtubule-associated protein 1A/1B-light chain 3
- LIAS:
-
Lipoic acid synthetase
- LIPT1:
-
Lipoyltransferase 1
- L-O:
-
Lipid alkoxy radicals
- L-OH:
-
Lipid alcohols
- L-OOH:
-
Lipid hydroperoxides
- MAP1LC3B:
-
Microtubule associated protein 1 light chain 3 beta
- MAPK:
-
Mitogen-activated protein kinase
- MDSC:
-
Myeloid-derived suppressor cells
- MEK:
-
Mitogen-activated protein kinase kinase
- MLKL:
-
Mixed lineage kinase domain-like
- MTF1:
-
Metal response element-binding transcription factor 1
- N-TiO2 :
-
Nitrogen-doped titanium dioxide
- NF1:
-
Neurofibromin 1
- NRAS:
-
NRAS proto-oncogene, GTPase
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- OXPHOS:
-
Oxidative phosphorylation
- PD-1:
-
Programmed cell death protein 1
- PDHA1:
-
Pyruvate dehydrogenase E1 component subunit alpha 1
- PDHB:
-
Pyruvate dehydrogenase E1 component subunit beta
- PI3K:
-
Phosphoinositide-3-kinase
- PRRs:
-
Pattern recognition receptors
- PTEN:
-
Phosphatase and tensin homolog
- PTPRC:
-
Protein tyrosine phosphatase receptor-type C
- PARP1:
-
Poly (ADP-ribose) polymerase 1
- RIPK1/3:
-
Receptor-interacting protein kinase 1/3
- SEER:
-
Surveillance, Epidemiology, and End Results
- SHARPIN:
-
Shank associated RH domain interactor
- SLC31A1:
-
Solute carrier family 31 member 1
- SLC39A6:
-
Solute carrier family 39 member 6
- SLC3A2:
-
Solute carrier family 3 member 2
- SLC7A11:
-
Solute carrier family 7 member 11
- SQSTM1:
-
Sequestosome 1
- TERT:
-
Telomerase reverse transcriptase
- TMB:
-
Tumor mutation burden
- TMEM117:
-
Transmembrane protein 117
- ULK1:
-
Unc-51-like kinase 1
- UVRAG:
-
UV radiation resistance-associated gene
- ZBP-1:
-
Z-form nucleic acid binding protein 1
References
Chang AE, Karnell LH, Menck HR. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. Cancer. 1998;83(8):1664–78.
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
Klemen ND, Wang M, Rubinstein JC, Olino K, Clune J, Ariyan S, Cha C, Weiss SA, Kluger HM, Sznol M. Survival after checkpoint inhibitors for metastatic acral, mucosal and uveal melanoma. J Immunother Cancer. 2020;8(1): e000341.
McLaughlin CC, Wu XC, Jemal A, Martin HJ, Roche LM, Chen VW. Incidence of noncutaneous melanomas in the US. Cancer. 2005;103(5):1000–7.
Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob JJ, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A, et al. Melanoma. Nat Rev Dis Primers. 2015;1:15003.
Garbe C, Amaral T, Peris K, Hauschild A, Arenberger P, Bastholt L, Bataille V, Del Marmol V, Dreno B, Fargnoli MC, et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: diagnostics—update 2019. Eur J Cancer. 2020;126:141–58.
Situm M, Buljan M, Kolic M, Vucic M. Melanoma–clinical, dermatoscopical, and histopathological morphological characteristics. Acta Dermatovenerol Croat. 2014;22(1):1–12.
Rabbie R, Ferguson P, Molina-Aguilar C, Adams DJ, Robles-Espinoza CD. Melanoma subtypes: genomic profiles, prognostic molecular markers and therapeutic possibilities. J Pathol. 2019;247(5):539–51.
Teixido C, Castillo P, Martinez-Vila C, Arance A, Alos L. Molecular Markers and Targets in Melanoma. Cells. 2021; 10(9):2320.
Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, Lazar AJ, Faries MB, Kirkwood JM, McArthur GA, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. 2017;67(6):472–92.
Lee JH, Choi JW, Kim YS. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br J Dermatol. 2011;164(4):776–84.
Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol. 2014;9:239–71.
Xiong M, Charifa A, Chen CSJ. Lentigo maligna melanoma. 2018.
Dal Pozzo CA, Cappellesso R. The morpho-molecular landscape of spitz neoplasms. Int J Med Sci. 2022;23(8):4211.
Molho-Pessach V, Hartshtark S, Merims S, Lotem M, Caplan N, Alfassi H, Maly A, Goldstein G, Muskatel RS. Giant congenital melanocytic naevus with a novel CUX1-BRAF fusion mutation treated with trametinib. Br J Dermatol. 2022;187(6):1052–4.
Salgado CM, Basu D, Nikiforova M, Bauer BS, Johnson D, Rundell V, Grunwaldt LJ, Reyes-Múgica M. BRAF mutations are also associated with neurocutaneous melanocytosis and large/giant congenital melanocytic nevi. Pediatr Dev Pathol. 2015;18(1):1–9.
Charbel C, Fontaine RH, Malouf GG, Picard A, Kadlub N, El-Murr N, How-Kit A, Su X, Coulomb-L’Hermine A, Tost J. NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Invest Dermatol. 2014;134(4):1067–74.
Ricci C, Ambrosi F, Grillini M, Serra M, Melotti B, Gruppioni E, Altimari A, Fiorentino M, Dika E, Lambertini M. Next-generation sequencing revealing TP53 mutation as potential genetic driver in dermal deep-seated melanoma arising in giant congenital nevus in adult patients: a unique case report and review of the literature. J Cutan Pathol. 2020;47(12):1164–9.
Ferrara G, Argenziano G. The WHO 2018 classification of cutaneous melanocytic neoplasms: suggestions from routine practice. Front Oncol. 2021;11: 675296.
Griewank KG, Müller H, Jackett LA, Emberger M, Möller I, van de Nes JA, Zimmer L, Livingstone E, Wiesner T, Scholz SL, Cosgarea I, Sucker A, Schimming T, Hillen U, Schilling B, Paschen A, Reis H, Mentzel T, Kutzner H, Rütten A, Murali R, Scolyer RA, Schadendorf D. SF3B1 and BAP1 mutations in blue nevus-like melanoma. Mod Pathol. 2017 ;30(7):928–39.
Sanna A, Harbst K, Johansson I, Christensen G, Lauss M, Mitra S, Rosengren F, Häkkinen J, Vallon-Christersson J, Olsson H, et al. Tumor genetic heterogeneity analysis of chronic sun-damaged melanoma. Pigment Cell Melanoma Res. 2020;33(3):480–9.
Torres-Cabala CA, Wang WL, Trent J, Yang D, Chen S, Galbincea J, Kim KB, Woodman S, Davies M, Plaza JA, et al. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol. 2009;22(11):1446–56.
Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, Ruben B, North J, Pincus L, Yeh I, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell. 2018;34(1):45-55 e4.
Garbe C, Amaral T, Peris K, Hauschild A, Arenberger P, Basset-Seguin N, Bastholt L, Bataille V, Del Marmol V, Dreno B, et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: treatment—update 2022. Eur J Cancer. 2022;170:256–84.
Kennedy LB, Salama AKS. A marathon not a sprint: improving outcomes for patients with metastatic melanoma in 2022 and beyond. JCO Oncol Pract. 2022;18(5):353–4.
Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, Gore M, Aamdal S, Cebon J, Coates A, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18(1):158–66.
Ringborg U, Rudenstam CM, Hansson J, Hafström L, Stenstam B, Strander H. Dacarbazine versus dacarbazine-vindesine in disseminated malignant melanoma: a randomized phase II study. Med Oncol Tumor Pharmacother. 1989;6(4):285–9.
Chiarion Sileni V, Nortilli R, Aversa SM, Paccagnella A, Medici M, Corti L, Favaretto AG, Cetto GL, Monfardini S. Phase II randomized study of dacarbazine, carmustine, cisplatin and tamoxifen versus dacarbazine alone in advanced melanoma patients. Melanoma Res. 2001;11(2):189–96.
Young AM, Marsden J, Goodman A, Burton A, Dunn JA. Prospective randomized comparison of dacarbazine (DTIC) versus DTIC plus interferon-alpha (IFN-alpha) in metastatic melanoma. Clin Oncol (R Coll Radiol). 2001;13(6):458–65.
McDermott D, Haanen J, Chen TT, Lorigan P, O’Day S, Investigators MDX. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010–20). Ann Oncol. 2013;24(10):2694–8.
Lebbe C, Meyer N, Mortier L, Marquez-Rodas I, Robert C, Rutkowski P, Butler MO, Eigentler T, Menzies AM, Smylie M, et al. Two dosing regimens of nivolumab (NIVO) plus ipilimumab (IPI) for advanced (adv) melanoma: three-year results of CheckMate 511. J Clin Oncol. 2021;39(15_suppl):9516.
Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J Clin Oncol. 2022;40(2):127–37.
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14.
Ribas A, Gonzalez R, Pavlick A, Hamid O, Gajewski TF, Daud A, Flaherty L, Logan T, Chmielowski B, Lewis K, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol. 2014;15(9):954–65.
Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, Chiarion Sileni V, Schachter J, Garbe C, Bondarenko I, et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N Engl J Med. 2019;381(7):626–36.
Jung S, Johnson DB. Management of acral and mucosal melanoma: medical oncology perspective. Oncologist. 2022;27(8):703–10.
Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. 2017;101(1):38–44.
Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, Corless CL, Li L, Li H, Sheng X, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904–9.
Wang JZ, Lin V, Toumi E, Wang K, Zhu H, Conway RM, Madigan MC, Murray M, Cherepanoff S, Zhou F, et al. Development of new therapeutic options for the treatment of uveal melanoma. FEBS J. 2021;288(21):6226–49.
Zheng Q, Li J, Zhang H, Wang Y, Zhang S. Immune checkpoint inhibitors in advanced acral melanoma: a systematic review. Front Oncol. 2020;10: 602705.
Heppt MV, Amaral T, Kahler KC, Heinzerling L, Hassel JC, Meissner M, Kreuzberg N, Loquai C, Reinhardt L, Utikal J, et al. Combined immune checkpoint blockade for metastatic uveal melanoma: a retrospective, multi-center study. J Immunother Cancer. 2019;7(1):299.
Paik J. Nivolumab plus relatlimab: first approval. Drugs. 2022;82(8):925–31.
Dhillon S. Tebentafusp: first approval. Drugs. 2022;82(6):703–10.
Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene Laherparepvec (T-VEC): an intralesional cancer immunotherapy for advanced melanoma. Cancers (Basel). 2021;13(6):1383.
Lopes J, Rodrigues CMP, Gaspar MM, Reis CP. Melanoma management: from epidemiology to treatment and latest advances. Cancers (Basel). 2022;14(19):4652.
Kozyra P, Krasowska D, Pitucha M. New potential agents for malignant melanoma treatment-most recent studies 2020–2022. Int J Mol Sci. 2022;23(11):6084.
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, Chen Y, Han B. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7(1):286.
Liu J, Hong M, Li Y, Chen D, Wu Y, Hu Y. Programmed cell death tunes tumor immunity. Front Immunol. 2022;13: 847345.
Qi X, Li Q, Che X, Wang Q, Wu G. Application of regulatory cell death in cancer: based on targeted therapy and immunotherapy. Front Immunol. 2022;13: 837293.
Liu H, He Z, Simon HU. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy. 2014;10(2):372–3.
Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015;22(6):946–58.
Wang Y, Wang Y, Wu J, Wang W, Zhang Y. Oxygen partial pressure plays a crucial role in B16 melanoma cell survival by regulating autophagy and mitochondrial functions. Biochem Biophys Res Commun. 2019;510(4):643–8.
Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hägg M, Linder S, Fais S, Codogno P. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem. 2012;287(36):30664–76.
Amaravadi RK, Schuchter LM, McDermott DF, Kramer A, Giles L, Gramlich K, Carberry M, Troxel AB, Letrero R, Nathanson KL. Phase II trial of temozolomide and sorafenib in advanced melanoma patients with or without brain metastases. Clin Cancer Res. 2009;15(24):7711–8.
Ma X-H, Piao S, Wang D, Mcafee QW, Nathanson KL, Lum JJ, Li LZ, Amaravadi RK. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res. 2011;17(10):3478–89.
Luo M, Wu L, Zhang K, Wang H, Wu S, O’Connell D, Gao T, Zhong H, Yang Y. miR-216b enhances the efficacy of vemurafenib by targeting Beclin-1, UVRAG and ATG5 in melanoma. Cell Signal. 2018;42:30–43.
Wu PF, Chiu CC, Chen CY, Wang HM. 7-Hydroxydehydronuciferine induces human melanoma death via triggering autophagy and apoptosis. Exp Dermatol. 2015;24(12):930–5.
Romano E, Rufo N, Korf H, Mathieu C, Garg AD, Agostinis P. BNIP3 modulates the interface between B16–F10 melanoma cells and immune cells. Oncotarget. 2018;9(25):17631–44.
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–16.
Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, Rostker F, Krummel M, Debnath J. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest. 2016;126(12):4417–29.
Noman MZ, Paggetti J, Moussay E, Berchem G, Janji B. Driving natural killer cells toward the melanoma tumor battlefield: autophagy as a valuable therapeutic target. Oncoimmunology. 2018;7(8): e1452583.
Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A. 2017;114(44):E9271–9.
Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, Polyzos A, Xilouri M, Wielockx B, Gogas H, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest. 2018;128(9):3840–52.
Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358(6382):167–9.
D’Souza CA, Heitman J. Dismantling the Cryptococcus coat. Trends Microbiol. 2001;9(3):112–3.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128.
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–92.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, Nie L, Chen Y, Wang YC, Liu C, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22(10):1264–75.
Demarco B, Grayczyk JP, Bjanes E, Le Roy D, Tonnus W, Assenmacher CA, Radaelli E, Fettrelet T, Mack V, Linkermann A, et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv. 2020; 6(47):eabc3465.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103.
Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162(6):1242–56.
De Schutter E, Croes L, Ibrahim J, Pauwels P, Op de Beeck K, Vandenabeele P, Van Camp G. GSDME and its role in cancer: From behind the scenes to the front of the stage. Int J Cancer. 2021;148(12):2872–83.
Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, Berger AC, Hartsough EJ, Rodeck U, Alnemri ES, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10(2):254–69.
Ahmed F, Tseng H-Y, Ahn A, Gunatilake D, Alavi S, Eccles M, Rizos H, Gallagher SJ, Tiffen JC, Hersey P. Repurposing melanoma chemotherapy to activate inflammasomes in the treatment of BRAF/MAPK inhibitor resistant melanoma. J Invest Dermatol. 2022;142(5):1444-55.e10.
Cai W, Nguyen MQ, Wilski NA, Purwin TJ, Vernon M, Tiago M, Aplin AE. A genome-wide screen identifies PDPK1 as a target to enhance the efficacy of MEK1/2 inhibitors in NRAS mutant melanoma. Cancer Res. 2022;82(14):2625–39.
Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, Sun RY, Zhou D, Han J, Wu Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28(12):1171–85.
Wang YY, Shi LY, Zhu ZT, Wang QJ. A new pyroptosis model can predict the immunotherapy response and immune microenvironment characteristics and prognosis of patients with cutaneous melanoma based on TCGA and GEO databases. Ann Transl Med. 2022;10(6):353.
Wu G, Chen B, Jiang J, Chen Y, Chen Y, Wang H. Identification of a pyroptosis-based model for predicting clinical outcomes from immunotherapy in patients with metastatic melanoma. Cancer Med. 2023;12(4):4921–37.
Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–42.
Emran AA, Tseng HY, Coleman MC, Tiffen J, Cook S, McGuire HM, Gallagher S, Feng C, Hersey P. Do innate killing mechanisms activated by inflammasomes have a role in treating melanoma? Pigment Cell Melanoma Res. 2020;33(5):660–70.
Arakelian T, Oosterhuis K, Tondini E, Los M, Vree J, van Geldorp M, Camps M, Teunisse B, Zoutendijk I, Arens R, et al. Pyroptosis-inducing active caspase-1 as a genetic adjuvant in anti-cancer DNA vaccination. Vaccine. 2022;40(13):2087–98.
Vernon M, Wilski NA, Kotas D, Cai W, Pomante D, Tiago M, Alnemri ES, Aplin AE. Raptinal induces gasdermin E-dependent pyroptosis in naïve and therapy-resistant melanoma. Mol Cancer Res. 2022;20(12):1811–21.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–14.
Florean C, Song S, Dicato M, Diederich M. Redox biology of regulated cell death in cancer: a focus on necroptosis and ferroptosis. Free Radic Biol Med. 2019;134:177–89.
Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–21.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9.
Mohammadalipour Z, Rahmati M, Khataee A, Moosavi MA. Differential effects of N-TiO(2) nanoparticle and its photo-activated form on autophagy and necroptosis in human melanoma A375 cells. J Cell Physiol. 2020;235(11):8246–59.
Ke H, Augustine CK, Gandham VD, Jin JY, Tyler DS, Akiyama SK, Hall RP, Zhang JY. CYLD inhibits melanoma growth and progression through suppression of the JNK/AP-1 and β1-integrin signaling pathways. J Invest Dermatol. 2013;133(1):221–9.
Geserick P, Wang J, Schilling R, Horn S, Harris P, Bertin J, Gough P, Feoktistova M, Leverkus M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015;6(9): e1884-e.
Rossi A, Pakhomova ON, Pakhomov AG, Weygandt S, Bulysheva AA, Murray LE, Mollica PA, Muratori C. Mechanisms and immunogenicity of nsPEF-induced cell death in B16F10 melanoma tumors. Sci Rep. 2019;9(1):431.
Hou J, Ju J, Zhang Z, Zhao C, Li Z, Zheng J, Sheng T, Zhang H, Hu L, Yu X. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 2019;10(7):493.
Patel S, Webster JD, Varfolomeev E, Kwon YC, Cheng JH, Zhang J, Dugger DL, Wickliffe KE, Maltzman A, Sujatha-Bhaskar S. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2020;27(1):161–75.
Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, Chiang K, Daniels BP, Baker D, Oberst A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol. 2019;4(36):eaaw2004.
Van Hoecke L, Van Lint S, Roose K, Van Parys A, Vandenabeele P, Grooten J, Tavernier J, De Koker S, Saelens X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat Commun. 2018;9(1):3417.
Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N, Wang S, Gautam A, Williams RM, Crawford JC, et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature. 2022;606(7914):594–602.
Basit F, van Oppen LM, Schöckel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S, Kopitz C, Heroult M, Hgm Willems P, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8(3): e2716.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79.
Fearnhead HO, Vandenabeele P, Vanden BT. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017;24(12):1991–8.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Toyokuni S. Iron and thiols as two major players in carcinogenesis: friends or foes? Front Pharmacol. 2014;5:200.
Gagliardi M, Cotella D, Santoro C, Corà D, Barlev NA, Piacentini M, Corazzari M. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019;10(12):902.
Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23(3):302–15.
Schöckel L, Glasauer A, Basit F, Bitschar K, Truong H, Erdmann G, Algire C, Hägebarth A, Willems PH, Kopitz C, et al. Targeting mitochondrial complex I using BAY 87–2243 reduces melanoma tumor growth. Cancer Metab. 2015;3:11.
Khamari R, Trinh A, Gabert PE, Corazao-Rozas P, Riveros-Cruz S, Balayssac S, Malet-Martino M, Dekiouk S, Joncquel Chevalier Curt M, Maboudou P, et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018;9(3):325.
Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33(5):890-904.e5.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247–50.
Feng S, Zhou Y, Huang H, Lin Y, Zeng Y, Han S, Huang K, Liu Q, Zhu W, Yuan Z, et al. Nobiletin induces ferroptosis in human skin melanoma cells through the GSK3β-mediated Keap1/Nrf2/HO-1 signalling pathway. Front Genet. 2022;13: 865073.
Chang MT, Tsai LC, Nakagawa-Goto K, Lee KH, Shyur LF. Phyto-sesquiterpene lactones DET and DETD-35 induce ferroptosis in vemurafenib sensitive and resistant melanoma via GPX4 inhibition and metabolic reprogramming. Pharmacol Res. 2022;178: 106148.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–82.
Proneth B, Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019;26(1):14–24.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4.
Guo W, Wu Z, Chen J, Guo S, You W, Wang S, Ma J, Wang H, Wang X, Wang H, et al. Nanoparticle delivery of miR-21–3p sensitizes melanoma to anti-PD-1 immunotherapy by promoting ferroptosis. J Immunother Cancer. 2022;10(6):e004381
Ma S, Liang X, Yang N, Yang J, Zhang J, Pan X, Wei Y, Liu Z, Shen Q. Boosting cancer immunotherapy by biomineralized nanovaccine with ferroptosis-inducing and photothermal properties. Biomater Sci. 2023;11(2):518–32.
Xu C, Chen H. A ferroptosis-related gene model predicts prognosis and immune microenvironment for cutaneous melanoma. Front genet. 2021;12: 697043.
Rao Y, Zhu J, Zheng H, Dong W, Lin Q. A novel melanoma prognostic model based on the ferroptosis-related long non-coding RNA. Front Oncol. 2022;12: 929960.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R, Spangler RD, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61.
Rowland EA, Snowden CK, Cristea IM. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr Opin Chem Biol. 2018;42:76–85.
Solmonson A, DeBerardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem. 2018;293(20):7522–30.
Tsvetkov P, Detappe A, Cai K, Keys HR, Brune Z, Ying W, Thiru P, Reidy M, Kugener G, Rossen J, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol. 2019;15(7):681–9.
Lv H, Liu X, Zeng X, Liu Y, Zhang C, Zhang Q, Xu J. Comprehensive analysis of cuproptosis-related genes in immune infiltration and prognosis in melanoma. Front Pharmacol. 2022;13: 930041.
Liu JY, Liu LP, Li Z, Luo YW, Liang F. The role of cuproptosis-related gene in the classification and prognosis of melanoma. Front Immunol. 2022;13: 986214.
Chen Y, Chen X, Wang X. Identification of a prognostic model using cuproptosis-related genes in uveal melanoma. Front Cell Dev Biol. 2022;10: 973073.
Yang X, Wang X, Sun X, Xiao M, Fan L, Su Y, Xue L, Luo S, Hou S, Wang H. Construction of five cuproptosis-related lncRNA signature for predicting prognosis and immune activity in skin cutaneous melanoma. Front genet. 2022;13: 972899.
Zhou Y, Shu Q, Fu Z, Wang C, Gu J, Li J, Chen Y, Xie M. A novel risk model based on cuproptosis-related lncRNAs predicted prognosis and indicated immune microenvironment landscape of patients with cutaneous melanoma. Front genet. 2022;13: 959456.
Liu D, Yang F, Zhang T, Mao R. Leveraging a cuproptosis-based signature to predict the prognosis and drug sensitivity of cutaneous melanoma. J Transl Med. 2023;21(1):57.
Qin W, Gan F, Jike Y, Jiang M, Li A. Consensus clustering and survival-related genes of Cuproptosis in cutaneous melanoma. Mediators Inflamm. 2023;2023:3615688.
Sun Y, Lei S, Luo X, Jiang C, Li Z. The value of cuproptosis-related differential genes in guiding prognosis and immune status in patients with skin cutaneous melanoma. Front Pharmacol. 2023;14:1129544.
Huang W, Yang F, Zhang Y, Fang Q, Lai Y, Lan Y. A newly established cuproptosis-related gene signature for predicting prognosis and immune infiltration in uveal melanoma. Int J Mol Sci. 2023;24(14):11358.
Hu B, Hounye AH, Wang Z, Qi M, Zhang J. A novel Cuprotosis-related signature predicts the prognosis and selects personal treatments for melanoma based on bioinformatics analysis. Front Oncol. 2023;13:1108128.
Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219(1):1–14.
Oliveri V. Selective targeting of cancer cells by copper ionophores: an overview. Front Mol Biosci. 2022;9: 841814.
Nagai M, Vo NH, Shin Ogawa L, Chimmanamada D, Inoue T, Chu J, Beaudette-Zlatanova BC, Lu R, Blackman RK, Barsoum J, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52(10):2142–50.
Cierlitza M, Chauvistré H, Bogeski I, Zhang X, Hauschild A, Herlyn M, Schadendorf D, Vogt T, Roesch A. Mitochondrial oxidative stress as a novel therapeutic target to overcome intrinsic drug resistance in melanoma cell subpopulations. Exp Dermatol. 2015;24(2):155–7.
O’Day S, Gonzalez R, Lawson D, Weber R, Hutchins L, Anderson C, Haddad J, Kong S, Williams A, Jacobson E. Phase II, randomized, controlled, double-blinded trial of weekly elesclomol plus paclitaxel versus paclitaxel alone for stage IV metastatic melanoma. J Clin Oncol. 2009;27(32):5452–8.
O’Day SJ, Eggermont AM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, Robert C, Schachter J, Testori A, Mackiewicz J, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol. 2013;31(9):1211–8.
Xie J, Yang Y, Gao Y, He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. 2023;22(1):46.
Tian Z, Jiang S, Zhou J, Zhang W. Copper homeostasis and cuproptosis in mitochondria. Life Sci. 2023;334: 122223.
Scrivner O, Dao L, Newell-Rogers MK, Shahandeh B, Meyskens FL, Kozawa SK, Liu-Smith F, Plascencia-Villa G, Jose-Yacaman M, Jia S, et al. The ionophore thiomaltol induces rapid lysosomal accumulation of copper and apoptosis in melanoma. Metallomics. 2022;14(1):mfab074.
Saporito-Magrina CM, Musacco-Sebio RN, Andrieux G, Kook L, Orrego MT, Tuttolomondo MV, Desimone MF, Boerries M, Borner C, Repetto MG. Copper-induced cell death and the protective role of glutathione: the implication of impaired protein folding rather than oxidative stress. Metallomics. 2018;10(12):1743–54.
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304(5676):1500–2.
Hsu SK, Chang WT, Lin IL, Chen YF, Padalwar NB, Cheng KC, Teng YN, Wang CH, Chiu CC. The role of necroptosis in ROS-mediated cancer therapies and its promising applications. Cancers (Basel). 2020;12(8):2185.
Karnik-Henry MS, Wang L, Barch DM, Harms MP, Campanella C, Csernansky JG. Medial temporal lobe structure and cognition in individuals with schizophrenia and in their non-psychotic siblings. Schizophr Res. 2012;138(2–3):128–35.
Wang W, Lu K, Jiang X, Wei Q, Zhu L, Wang X, Jin H, Feng L. Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J Exp Clin Cancer Res. 2023;42(1):142.
Enninga EAL, Moser JC, Weaver AL, Markovic SN, Brewer JD, Leontovich AA, Hieken TJ, Shuster L, Kottschade LA, Olariu A, et al. Survival of cutaneous melanoma based on sex, age, and stage in the United States, 1992–2011. Cancer Med. 2017;6(10):2203–12.
Motaparthi K, Kim J, Andea AA, Missall TA, Novoa RA, Vidal CI, Fung MA, Emanuel PO. TERT and TERT promoter in melanocytic neoplasms: Current concepts in pathogenesis, diagnosis, and prognosis. J Cutan Pathol. 2020;47(8):710–9
Shreberk-Hassidim R, Ostrowski SM, Fisher DE. The complex interplay between nevi and melanoma: risk factors and precursors. Int J Mol Sci. 2023;24(4):3541
Acknowledgements
We are thankful to the organizations that provided financial support.
Funding
We thank the following institutions for providing financial support: The National Science and Technology Council, Taiwan (Grant numbers MOST 109-2314-B-037-069-MY3 and NSTC 112-2314-B-037-029); NSYSU-KMU (joint grants; Grant number NSYSUKMU113-P06); Kaohsiung Medical University, Taiwan (KMU-DK(A)113006); the Kaohsiung Medical University Research Center, Taiwan (Grant number KMU-TC112A04).
Author information
Authors and Affiliations
Contributions
Conceptualization: MYH and CCC; software: TYL; investigation: MYH and SKH; data curation: MYH; writing—original draft preparation: MYH; writing—review and editing: CCC; visualization: CYW and MYH; supervision: CYW and CCC; and funding acquisition: CYW and CCC. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hsieh, My., Hsu, SK., Liu, TY. et al. Melanoma biology and treatment: a review of novel regulated cell death-based approaches. Cancer Cell Int 24, 63 (2024). https://doi.org/10.1186/s12935-024-03220-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03220-9