
Abstract
Background
The hypoxia-responsive long non-coding RNA, RP11-367G18.1, has recently been reported to induce histone 4 lysine 16 acetylation (H4K16Ac) through its variant 2; however, the underlying molecular mechanism remains poorly understood.
Methods
RNA pull-down assay and liquid chromatography-tandem mass spectrometry were performed to identify RP11-367G18.1 variant 2-binding partner. The molecular events were examined utilizing western blot analysis, real-time PCR, luciferase reporter assay, chromatin immunoprecipitation, and chromatin isolation by RNA purification assays. The migration, invasion, soft agar colony formation, and in vivo xenograft experiments were conducted to evaluate the impact of RP11-367G18.1 variant 2–YY1 complex on tumor progression.
Results
In this study, RNA sequencing data revealed that hypoxia and RP11-367G18.1 variant 2 co-regulated genes were enriched in tumor-related pathways. YY1 was identified as an RP11-367G18.1 variant 2-binding partner that activates the H4K16Ac mark. YY1 was upregulated under hypoxic conditions and served as a target gene for hypoxia-inducible factor-1α. RP11-367G18.1 variant 2 colocalized with YY1 and H4K16Ac in the nucleus under hypoxic conditions. Head and neck cancer tissues had higher levels of RP11-367G18.1 and YY1 which were associated with poor patient outcomes. RP11-367G18.1 variant 2–YY1 complex contributes to hypoxia-induced epithelial–mesenchymal transition, cell migration, invasion, and tumorigenicity. YY1 regulated hypoxia-induced genes dependent on RP11-367G18.1 variant 2.
Conclusions
RP11-367G18.1 variant 2–YY1 complex mediates the tumor-promoting effects of hypoxia, suggesting that this complex can be targeted as a novel therapeutic strategy for cancer treatment.
Similar content being viewed by others

Background
Long non-coding RNAs (lncRNAs), transcripts longer than 200 nucleotides with limited protein-coding potential, play a critical role in gene regulation. LncRNAs participate in multiple processes regulating gene expression, such as chromatin organization via histone modification, transcription factor recruitment, and maintenance of mRNA stability [1]. Increasing evidence has revealed the crucial roles of lncRNAs in various cellular processes, such as cell migration, stemness, and genome maintenance [2]. Dysregulation of lncRNAs can drive tumor progression and serve as a prognostic marker [3, 4].
Hypoxia, deficiency of oxygen, is a common condition in solid tumors that facilitates tumor growth, angiogenesis, and metastasis. Hypoxia-inducible factors (HIFs) are the key transcriptional regulators of gene expression. HIF-1α is prominently upregulated under hypoxia and modulates the epithelial–mesenchymal transition (EMT)-activating transcription factors, histone modifiers, and lncRNAs [5,6,7]. Several hypoxia-responsive lncRNAs have been reported to mediate HIF-1α signaling via diverse mechanisms [8]. These hypoxia-responsive lncRNAs interact with protein complexes, epigenetic regulators, and microRNAs to regulate hypoxic gene expression [9]. Although lncRNAs have been reported to guide chromatin-modifying complexes, lncRNA-associated histone marks are not yet fully understood.
Emerging role of the alternative splicing of lncRNAs has gained attention in cancer research. LncRNAs can undergo alternative splicing to produce different variants having different functional mechanisms and regulate tumorigenesis in a transcript-dependent manner [10]. Recently, we identified a hypoxia-responsive lncRNA, RP11-367G18.1 (ENSG00000230943), that is associated with poor outcomes in patients with head and neck squamous cell carcinoma (HNSC) [11]. Variant 2 of RP11-367G18.1 (ENST00000452675.1) is a key regulator of EMT and histone 4 lysine 16 acetylation (H4K16Ac). The interacting partner of RP11-367G18.1 variant 2, which mediates its function under hypoxia, remains ambiguous. In this study, we found that the RP11-367G18.1 variant 2–YY1 complex contributes to hypoxia-induced H4K16Ac activation and cancer progression.
Methods
Cell culture
Human cell lines with low (MCF7 breast cancer and FADU HNSC cell lines) and high (H1299 non-small lung cancer and MDA-MB-231 breast cancer cell lines; Figure S1A) HIF-1α levels were purchased from the American Type Culture Collection (Manassas, VA, USA). All cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum at 37 °C and 5% CO2. For hypoxic conditions, cells were cultured in 1% O2, 5% CO2, and 94% N2 for 18 h. Cells were then tested for mycoplasma.
Plasmid construction
Expression constructs for RP11-367G18.1 variant 2 were constructed as previously described [11]. Plasmids pHA-HIF-1α, pHA-HIF-1α (ΔODD), and pHA-HIF-1α (LCLL) expressing wild-type and mutant HIF-1α were obtained from Dr. L. E. Huang (University of Utah, Salt Lake City, UT, USA) [12]. Expression construct Myc-DDK-tagged-YY1 containing cDNA encoding YY1 was cloned into a pCMV6-Entry vector. For knockdown experiments, the target sequences of RP11-367G18.1 variant 2 (5’-GGTTCTACTTCCTGGCAAGTA-3’), RP11-367G18.1 variant 1 (5’- GGTCCTCTTCAATGTACAATC-3’), YY1 (5’-GCCTCTCCTTTGTATATTATT-3’), HIF-2α (5’-CAGTACCCAGACGGATTTCAA-3’), and Scrambled control (5’-CCTAAGGTTAAGTCGCCCTCG-3’) were cloned into pLV2-U6-Puro and pLKO.1-puro vectors, respectively. To generate reporter constructs, the YY1 promoter fragments were cloned into a pGL3-basic vector. Hypoxia response element (HRE) mutants of the reporter constructs were cloned using QuikChange Lightning (Agilent, Santa Clara, CA, USA).
Transfection, luciferase reporter assay, and lentivirus-mediated gene knockdown
Cells were seeded overnight and transfected using Lipofectamine 2000 (Thermo Fisher Scientific), following the manufacturer’s instructions. For the luciferase reporter assay, the reporter constructs and HIF-1α-expressing constructs were co-transfected into FADU cells under normoxia or hypoxia. Luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) and further normalized to Renilla luciferase activity. For knockdown experiments, plasmids containing short hairpin RNA were co-transfected with pMD.G and pCMVΔR8.91 plasmids into HEK293T cells for 48 h to generate a lentivirus, as previously described [13]. To generate stable clones, cells were infected with lentivirus for 24 h and selected with puromycin for two weeks.
RNA-sequencing (RNA-seq) and data analysis
Briefly, RNA from treated cells was extracted using RNeasy, and the TapeStation System (Agilent, Santa Clara, CA, USA) was used for RNA quality control. RNA-seq libraries were generated using a KAPA Hyperprep Kit containing RiboMinus. The libraries were then sequenced via 150 nucleotide paired-end running on Illumina HiSeq/Illumina Novaseq/MGI2000 instrument. The reads were mapped to the reference GRCh38 using HISAT2 [14]. To define differentially expressed genes, we set up a cut-off of fold-change ≥ 1.5 and a false-discovery rate < 0.05. Gene Ontology (GO) analysis, including biological process, cellular component and molecular function categories, was conducted for functional annotation. Hallmark pathway enrichment analysis was conducted via Gene Set Enrichment Analysis (GSEA).
RNA pull-down assay
RNA pull-down assay was performed using a previously described protocol, with minor modifications [15]. Briefly, biotin-labeled RP11-367G18.1 variant 2 was transcribed using the Biotin RNA Labeling Mix and T7 RNA polymerase. Biotinylated RP11-367G18.1 variant 2 was treated with RNaseOUT and purified using an RNeasy Mini Kit (QIAGEN, CA, USA). Twenty µg of RNA was mixed with 50 µL of streptavidin beads in an RNA capture buffer (20 mM Tris-HCl [pH 7.5], 1 M NaCl, and 1 mM EDTA) for 30 min at room temperature. Beads were then washed with NT2 buffer, added to the cell extracts, and incubated at 4 °C. After 6 h of incubation, the mixture was washed thrice. Samples were eluted and resolved via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). RP11-367G18.1 variant 2-specific bands were excised and subjected to mass spectrometry (MS) analysis.
Liquid chromatography-tandem MS (LC-MS/MS) analysis and protein identification
Biotinylated sense RP11-367G18.1 variant 2-specific bands were excised and trypsinized for peptide extraction. LC-MS experiments were performed using an LTQ-Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). The peptide mixtures were reconstituted in buffer A (0.1% formic acid) and treated on a C18 column (75 μm × 250 mm). Peptides were separated using 3 μm C18-AQ particles (100 μm × 15 cm) and mobile phase A (water with 0.1% formic acid) and a segmented gradient in 120 min up to 80% mobile phase B (acetonitrile with 0.1% formic acid) at a rate of 500 nL/min. Survey scans with 120,000 resolution and a mass range of m/z 300–1600 were performed in a data-dependent mode, and the top 10 precursors were selected. Peptide sequences were searched for trypsin specificity with 0–2 missed cleavage sites. A precursor ion mass tolerance of 10 ppm and fragment ion mass tolerance of 0.6 kDa were used. Variable modifications included carbamidomethylation and oxidation. The data were processed using MaxQuant software and filtered with a false-discovery rate of 1%.
RNA fluorescent in situ hybridization (FISH) and immunofluorescence staining
Cells were fixed in 4% formaldehyde for 15 min and permeabilized in phosphate-buffered saline (PBS) containing 0.1% Triton X-100. Hybridization was performed using FAM dye-labeled RP11-367G18.1 variant 2 probes (Table S1) at 37 °C overnight. For colocalization analysis, after RNA FISH, cells were fixed with 2% formaldehyde and incubated with primary antibodies against H4K16Ac or YY1 at 4 °C overnight. The next day, cells were washed thrice with PBS and incubated with the Alexa Fluor-594-labeled secondary antibody (1:2000 dilution; Abcam, Cambridge, UK) for 1 h, followed by 4′,6-diamidino-2-phenylindole staining (1:1000 dilution; Invitrogen). Cells were then observed under a Leica TCS SP8X confocal microscope.
The Cancer Genome Atlas (TCGA) data analysis
Clinical data on gene expression in patients with HNSC were downloaded and analyzed [16, 17]. For survival analysis, the overall survival of patients was examined using the Kaplan–Meier method and compared using the log-rank test. All analyses were conducted using Prism version 8.01 (Graph Pad Software Inc., CA, USA).
Western blot analysis and quantitative real-time PCR
Proteins and histones were extracted from the cells, and the protein concentration was measured using the Bradford method, as previously described [11]. Western blot analysis was performed using SDS-PAGE with antibodies against HIF-1α, N-cadherin (BD Biosciences, Bedford, MA, USA), E-cadherin, HA, histone H3, HIF-2α, LDHA (Cell Signaling Technology, Danvers, MA, USA), YY1 (Santa Cruz Biotechnology, TX, USA), vimentin, Flag (Sigma-Aldrich, St Louis, MO, USA), plakoglobin, H4K16Ac, Glut1 (Millipore, Burlington, MA, USA), Streptavidin (HRP), H4K5Ac, H4K8Ac, H4K12Ac, and histone H4 (Abcam), and β-actin (Genetex, Alton Pkwy Irvine, CA, USA). To determine the transcript expression levels, RNA was purified using TRIzol reagent and cDNA was synthesized using the MultiScribe Reverse Transcriptase system. Quantitative real-time PCR was performed using Fast SYBR Green Master Mix (Thermo Fisher Scientific) as previously described [18]. All primers used for quantitative real-time PCR are listed in Table S2 and S5. Relative expression levels were normalized to that of 18 S rRNA.
Transwell migration, invasion, and soft agar colony formation assays
For the migration assay, cells (3 × 104) in a serum-free medium were seeded into the upper transwell chamber with 8-µm pores and complete medium was added to the lower chamber for 12 h incubation. For the invasion assay, cells (5 × 104) in a serum-free medium were seeded onto Matrigel-coated Transwell (Becton Dickinson, Mountain View, CA, USA) and incubated for 20 h. Migrated or invaded cells were fixed with methanol and stained with crystal violet (Sigma-Aldrich). For the colony formation assay, cells were plated at 5,000 cells/well on soft agar, as previously described [19]. After 14 days of incubation, cells were stained with crystal violet and counted under a light microscope.
Chromatin immunoprecipitation (ChIP) and chromatin isolation by RNA purification (ChIRP) assays
ChIP assays were performed using anti-IgG, anti-H4K16Ac, and anti-YY1 antibodies, as previously described [11, 13]. For the ChIRP assay, anti-sense oligonucleotide probes corresponding to RP11-367G18.1 variant 2 and LacZ were designed using a probe designer (https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer; Table S3). Anti-sense probes were synthesized with a biotin tag at the 3′-end, as previously described [20]. Cells were harvested, rinsed with PBS, and cross-linked in 1% glutaraldehyde/PBS for 10 min at room temperature. One-tenth volume of 1.25 M glycine was added to the reaction and incubated at room temperature for 5 min. Cells were washed twice and resuspended in Pierce IP Lysis Buffer containing Protease Inhibitor Cocktail and SUPERase•In RNase Inhibitor (Thermo Fisher Scientific). Lysates were sonicated using a Bioruptor at 4 °C for 30 min and subjected to ChIRP. Probes (100 pmol) were mixed with 1 mL of the cell lysate and incubated for 4 h at 37 °C with shaking. C-1 magnetic beads (Invitrogen) were washed twice with the lysis buffer and added to the cell lysates for 60-min hybridization at 37 °C. Beads were then washed for RNA and DNA isolation. DNA samples were examined via quantitative real-time PCR using specific primers (Table S4).
In vivo xenograft experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee (2020122504) of the Chang Gung Memorial Hospital. FADU cells (2 × 106) were subcutaneously injected into five-week-old BALB/c nu/nu mice (National Science Council Animal Center, Taipei, Taiwan; n = 5 per group). After inoculation for 30–35 days, the xenografted mice were sacrificed for tumor volume evaluation.
Statistical analysis
All experiments were performed at least thrice. Error bars represent the standard deviation (SD) of the data. Statistical comparisons were made using Student’s t-test. P-value < 0.05 was considered to be statistically significant.
Results
Hypoxia- and RP11-367G18.1 variant 2-upregulated genes are involved in cancer progression
To explore the biological role of hypoxia-induced RP11-367G18.1 variant 2, RNA-seq was performed to identify the differentially expressed genes. RNA-seq revealed that 2,055 genes (942 upregulated and 1,113 downregulated) were significantly different between hypoxic and normoxic conditions in MCF7 cells. A total of 2,534 genes (1,866 upregulated and 668 downregulated) were differentially expressed in MCF7 cells overexpressing RP11-367G18.1 variant 2 and control cells (Fig. 1A). Upregulated genes were then classified via GO analysis. Hypoxia-upregulated genes were involved in glycolytic, glucose catabolic, and protein hydroxylation processes. RP11-367G18.1 variant 2-upregulated genes were associated with the MHC protein complex, antigen processing and presentation of endogenous antigen, and lumenal side of endoplasmic reticulum membrane (Fig. 1B). We identified 306 genes that were upregulated by both hypoxia and RP11-367G18.1 variant 2 overexpression (Fig. 1C and D). These upregulated genes were located in the cytoplasm, involved in the immune response, and mainly functioned in receptor binding (Figure S1B). Moreover, GSEA revealed that hypoxia- and RP11-367G18.1 variant 2-upregulated genes were enriched in the hypoxia, EMT, angiogenesis, and inflammation pathways (Fig. 1E).

Hypoxia and RP11-367G18.1 variant 2 co-upregulated genes involve in tumor progression. (A) RNA-seq revealed that genes were differentially expressed following hypoxia (up) and RP11-367G18.1 variant 2 overexpression (bottom) in MCF7 cells. (B) Bar charts show the GO analysis results of upregulated genes under hypoxia (up) and RP11-367G18.1 variant 2 overexpression (bottom). (C) Venn graph showed the number of hypoxia- and RP11-367G18.1 variant 2-upregulated genes in MCF7 cells. (D) Heatmap analysis of hypoxia and RP11-367G18.1 variant 2 co-upregulated genes (n = 306). (E) GSEA revealed that hypoxia and RP11-367G18.1 variant 2 co-upregulated genes were enriched in the hypoxia, EMT, angiogenesis, and inflammatory response pathways. For hypoxic conditions, cells were cultured in 1% O2, 5% CO2, and 94% N2 for 18 h. FC, fold-change; N, normoxia; H, hypoxia; V2, variant 2
HIF-1α-regulated YY1 interacts with RP11-367G18.1 variant 2 to activate H4K16Ac
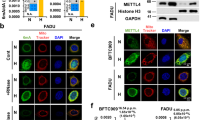
We previously demonstrated that RP11-367G18.1 variant 2 specifically regulated the H4K16Ac mark [11]. To identify the binding partner of RP11-367G18.1 variant 2 that contributed to H4K16Ac activation, we performed an RNA pull-down assay using biotinylated RP11-367G18.1 variant 2. Biotinylated RP11-367G18.1 variant 2 or beads (as the negative control) were incubated with whole-cell extracts of H1299 cells and pulled down using streptavidin beads. RP11-367G18.1 variant 2-interacting proteins were analyzed using gradient gel electrophoresis and Coomassie blue staining (Fig. 2A, left). Specific bands (1–11) in RP11-367G18.1 variant 2 pull-down samples were excised for LC-MS/MS analysis (Fig. 2A, right). YY1 has been reported to interact with histone acetyltransferases to activate gene transcription [21]. Therefore, we focused on YY1 in this study and found that the levels of H4K16Ac were decreased by YY1 knockdown and increased by YY1 overexpression in H1299 cells (Fig. 2B and C). LC-MS/MS analysis results were validated using an RNA pull-down assay. YY1, but not histone H4 or histone H3, was pulled down by biotinylated RP11-367G18.1 variant 2. However, anti-sense RNA, RP11-367G18.1 variant 1, or the bead control did not show any interaction with YY1 (Fig. 2D and S2A). YY1-induced H4K16Ac activation was attenuated by RP11-367G18.1 variant 2 knockdown (Fig. 2E). RP11-367G18.1 variant 1 and 2 were induced by hypoxia (Figure S2B). Interestingly, both mRNA and protein levels of YY1 were upregulated under hypoxic conditions (Fig. 2F and G). However, knockdown of HIF-2α did not affect the expression levels of YY1 under hypoxic conditions, indicating that HIF-1α plays a crucial role in the regulation of YY1 (Figure S2C). HIF can bind to HRE, which contains the sequence 5’-(A/G)CGTG-3’, to activate the transcription of hypoxic target genes. Three putative HREs are located in the proximal promoter of YY1. To study whether YY1 was regulated by hypoxia at the transcriptional level, reporter constructs containing the wild-type and mutant HREs in the YY1 promoter were cloned for luciferase reporter assay (Fig. 2H, left). Reporter constructs containing wild-type HREs responded to hypoxia, HIF-1α overexpression, and constitutively active HIF-1α (ΔODD) overexpression. Reporter construct containing mutant HREs (-493 to -489 bp upstream of the transcription start site of YY1 gene; mut3) did not respond to hypoxia and HIF-1α overexpression (Fig. 2H). HIF-1α, but not HF1-2α, bound to HREs in the YY1 promoter under hypoxia (Fig. 2I and S2D), suggesting that YY1 was directly modulated by HIF-1α. Moreover, cells with high endogenous HIF-1α levels exhibited high expression levels of YY1 and RP11-367G18.1 variant 2 (Figure S1A). We observed that H4K16Ac marks were elicited by hypoxic condition (Fig. 3A). Notably, immunofluorescence staining revealed that RP11-367G18.1 variant 2 was co-localized with YY1 and H4K16Ac in the nucleus (Fig. 3B). Knockdown of RP11-367G18.1 variant 2 or YY1 did not have any impact on the expression levels or subcellular distributions of each other (Figure S3A and B). H4K16Ac was activated following RP11-367G18.1 variant 2 and YY1 overexpression in FADU cells (Fig. 3C). These results indicated that the RP11-367G18.1 variant 2–YY1 complex activated H4K16Ac under hypoxia.

RP11-367G18.1 variant 2 interacts with YY1 to activate H4K16Ac. (A) RNA pull-down assay and Coomassie blue staining revealed 11 RP11-367G18.1 variant 2-specific bands (left). Protein identity of the 11 bands was analyzed via LC-MS/MS and was shown in a table (right). (B) Knockdown of YY1 suppressed H4K16Ac levels in H1299 cells. (C) Overexpression of YY1 increased H4K16Ac levels in H1299 cells. (D) YY1 was pulled down by biotinylated sense RP11-367G18.1 variant 2. Beads or biotinylated anti-sense RP11-367G18.1 variant 2 were used as the negative control. (E) YY1-induced H4K16Ac activation was suppressed following RP11-367G18.1 variant 2 knockdown. (F) mRNA levels of YY1 were upregulated under hypoxia in FADU and MCF7 cells. (G) Protein levels of YY1 were upregulated under hypoxia in FADU and MCF7 cells. (H) Reporter constructs containing wild-type and mutant HREs in YY1 promoter were shown (left). Reporter assay revealed that HRE (-493/-489) was responsive to hypoxia in YY1 promoter (right). (I) ChIP assay revealed that HIF-1α bound to the YY1 proximal promoters containing HRE under hypoxia. For hypoxic conditions, cells were cultured in 1% O2, 5% CO2, and 94% N2 for 18 h. Scr, Scrambled; Cont, control; V2, variant 2. Data are represented as the mean ± SD. *P < 0.05

RP11-367G18.1 variant 2-YY1 complex activates H4K16Ac. (A) Hypoxia enhanced H4K16Ac activation. (B) Immunofluorescence staining revealed that RP11-367G18.1 variant 2 was colocalized with YY1 and H4K16Ac in FADU cells under hypoxia. (C) Overexpression of RP11-367G18.1 variant 2 and YY1 enhanced H4K16Ac activation. For hypoxic conditions, cells were cultured in 1% O2, 5% CO2, and 94% N2 for 18 h
High expression levels of RP11-367G18.1 and YY1 are linked to worse outcomes in patients with HNSC
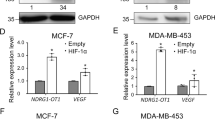
We previously reported that RP11-367G18.1 expression was associated with short survival period in patients with HNSC [11]. To further evaluate the clinical significance of RP11-367G18.1 and YY1, we examined the data on gene expression and survival status using TCGA database. We found that patients with HNSC with high YY1 expression had poor overall survival (Figure S4A). HNSC tumor tissues showed higher expression levels of RP11-367G18.1 and YY1 than the normal tissues (Fig. 4A and B). YY1 expression was positively correlated with RP11-367G18.1 and HIF-1α expression levels (Fig. 4C and S4B). Patients with high expression levels of both RP11-367G18.1 and YY1 had worse overall survival than patients with low expression levels of RP11-367G18.1 and YY1 (Fig. 4D and S4C). Moreover, the combination of RP11-367G18.1, YY1, and HIF-1α also resulted in the worse overall survival of patients with HNSC (Fig. 4E and S4D). These results highlighted the unfavorable prognostic roles of RP11-367G18.1 and YY1 in patients with HNSC.

RP11-367G18.1 and YY1 are associated with worse outcomes in patients with HNSC. (A and B) Expression levels of RP11-367G18.1 and YY1 were higher in tumor tissues than those in normal tissues in patients with HNSC from TCGA dataset. (C) Positive correlation between RP11-367G18.1 and YY1 in HNSC tissues was shown. (D) Patients with HNSC with high expression levels of RP11-367G18.1 and YY1 showed the worse overall survival. (E) Patients with HNSC with high expression levels of RP11-367G18.1, YY1, and HIF-1α showed the worse overall survival
RP11-367G18.1 variant 2–YY1 complex mediates hypoxia-induced EMT
To explore the role of the RP11-367G18.1 variant 2–YY1 complex in EMT, transwell migration and invasion assays were performed. The data revealed that RP11-367G18.1 variant 2-enhanced cell migration and invasion were repressed by YY1 knockdown (Figure S5A). Knockdown of RP11-367G18.1 variant 2 inhibited YY1-induced cell migration and invasion (Figure S5B). Importantly, overexpression of HIF-1α (ΔODD) suppressed the expression of epithelial markers (E-cadherin and plakoglobin), induced the expression of mesenchymal markers (vimentin and N-cadherin), and promoted cell migration and invasion. These effects were reversed by RP11-367G18.1 variant 2 or YY1 knockdown (Fig. 5A and B). Similarly, hypoxia-induced EMT was suppressed by RP11-367G18.1 variant 2 or YY1 knockdown (Fig. 5C and D). These results suggested that the RP11-367G18.1 variant 2–YY1 complex mediates hypoxia-induced EMT.

RP11-367G18.1 variant 2–YY1 complex mediates hypoxia-induced EMT. (A and B) Knockdown of RP11-367G18.1 variant 2 or YY1 suppressed EMT, cell migration, and invasion of FADU and MCF7 cells overexpressing HIF-1α (ΔODD). (C and D) Knockdown of RP11-367G18.1 variant 2 or YY1 suppressed EMT, cell migration, and invasion of FADU and MCF7 cells under hypoxia. Glut1 and LDHA, the hypoxia-inducible genes, served as positive control. For hypoxic conditions, cells were cultured in 1% O2, 5% CO2, and 94% N2 for 18 h. Scr, Scrambled; Cont, control; V2, variant 2; N, normoxia; H, hypoxia. Data are represented as the mean ± SD. *P < 0.05
Hypoxia induces tumorigenicity via the RP11-367G18.1 variant 2–YY1 complex
We explored the role of the RP11-367G18.1 variant 2–YY1 complex in tumorigenicity and found that RP11-367G18.1 variant 2-induced colony formation was inhibited by the knockdown of YY1 in FADU cells (Fig. 6A). Knockdown of RP11-367G18.1 variant 2 inhibited YY1-enhanced colony formation in H1299 cells (Fig. 6B). Furthermore, ectopic expression of HIF-1α (ΔODD) facilitated colony formation, which was suppressed by RP11-367G18.1 variant 2 or YY1 knockdown (Fig. 6C). We further explored the effect of the RP11-367G18.1 variant 2–YY1 complex on hypoxia-induced tumor growth in vivo. Consistently, knockdown of RP11-367G18.1 variant 2 or YY1 suppressed the tumor growth of xenografted FADU cells overexpressing HIF-1α (ΔODD) (Fig. 6D), suggesting that hypoxia-induced tumorigenicity was attributable to the RP11-367G18.1 variant 2–YY1 complex.

RP11-367G18.1 variant 2–YY1 complex is essential for hypoxia-induced tumorigenicity. (A) Knockdown of YY1 decreased the colony formation of FADU cells overexpressing RP11-367G18.1 variant 2. (B) Knockdown of RP11-367G18.1 variant 2 suppressed the colony formation of FADU cells overexpressing YY1. (C) Knockdown of RP11-367G18.1 variant 2 or YY1 suppressed the colony formation of H1299 cells overexpressing HIF-1α (ΔODD). (D) Knockdown of RP11-367G18.1 variant 2 or YY1 suppressed HIF-1α (ΔODD)-enhanced tumor volume of FADU cell-derived xenografts. Scr, Scrambled; Cont, control; V2, variant 2. Data are represented as the mean ± SD. *P < 0.05
RP11-367G18.1 variant 2 is essential for H4K16Ac activation and YY1 binding to the promoters of hypoxia-induced genes
To validate the role of the RP11-367G18.1 variant 2-YY1 complex in regulating target genes that were co-upregulated by hypoxia and RP11-367G18.1 variant 2 (as identified from the enriched pathways in Fig. 1E), we conducted real-time PCR analysis. Our findings revealed that the expression levels of both RP11-367G18.1 variant 2 and YY1 transcripts were significantly elevated after a two-hour exposure to hypoxic conditions. The levels of H4K16Ac marks reached their peak after an 8-hour period of hypoxia. Subsequently, the downstream target genes were upregulated after 8 h of hypoxic exposure (Fig. 7A and B, S6A, and S6B). Hypoxia led to the upregulation of HK2, TGFBI, VEGFC, and LIF, as well as the hypoxia-inducible gene Glut1, which served as a positive control. Interestingly, the expression of HK2, TGFBI, VEGFC, and LIF (except for Glut1) was found to be suppressed upon knockdown of RP11-367G18.1 variant 2 or YY1 (Fig. 7C and S6C). However, knockdown of RP11-367G18.1 variant 1 did not exhibit the same effect on the expression of these genes (Figure S6D). We examined H4K16Ac levels in the promoters of HK2, TGFBI, VEGFC, LIF, and Glut1. ChIP assay revealed decreased levels of H4K16Ac in the promoters of HK2, TGFBI, VEGFC, and LIF under hypoxia following RP11-367G18.1 variant 2 knockdown. Remarkably, knockdown of RP11-367G18.1 variant 2 significantly decreased the binding of YY1 to the promoters of HK2, TGFBI, VEGFC, and LIF under hypoxia. This implies that the binding of YY1 to the promoters of hypoxia and RP11-367G18.1 variant 2 co-upregulated genes is dependent on RP11-367G18.1 variant 2 (Fig. 7D and S6E). The 306 hypoxia and RP11-367G18.1 variant 2 co-upregulated genes comprised a total of 233 (76.1%) protein-coding genes (Fig. 1C and S7A). Under hypoxic conditions, 233 protein-coding genes were induced, with 136 of them (58.4%) being suppressed by YY knockdown (Figure S7B and C). These results indicated that the RP11-367G18.1 variant 2–YY1 complex was essential for the regulation of hypoxia and RP11-367G18.1 variant 2 co-upregulated genes.

RP11-367G18.1 variant 2–YY1 complex regulates gene expression. (A) The transcript expression levels of RP11-367G18.1 variant 2, YY1, HK2, TGFBI, VEGFC, and LIF were measured at the indicated time points (in hours) under hypoxic conditions in FADU cells. (B) The proteins expressions levels of HIF-1α, YY1 and H4K16Ac were measured at the indicated time points (in hours) under hypoxic conditions in FADU cells. (C) Knockdown of RP11-367G18.1 variant 2 or YY1 inhibited the expression of HK2, TGFBI. VEGFC, and LIF under hypoxia in FADU cells. (D) Knockdown of RP11-367G18.1 variant 2 suppressed the level of H4K16Ac and the binding of YY1 to the proximal promoters of HK2, TGFBI, VEGFC, and LIF under hypoxic conditions. Glut1, a hypoxia-inducible gene, served as positive control. For hypoxic conditions, cells were cultured in 1% O2, 5% CO2, and 94% N2 for 18 h. Cont, control; V2, variant 2. Data are represented as the mean ± SD. Student’s t-test, *P < 0.05
Discussion
Specific expression of lncRNAs under hypoxic conditions is usually linked to the clinicopathologic characteristics of solid tumors [22]. Hypoxia-responsive lncRNAs usually play oncogenic roles via diverse mechanisms. LncRNA PDIA3P1 sponges miR-124-3p to activate NF-κB pathway to facilitate mesenchymal transition in glioma [23]. The hypoxic lncRNA, KB-1980E6.3, interacts with IGF2BP1 to stabilize c-Myc mRNA and maintain the stemness of breast cancer cells [24]. In addition, lncRNA variants may perform different biological functions [25]. LncRNA-PXN-AS1 generates two lncRNA variants, PXN-AS1-L and PXN-AS1-S, via alternative splicing. PXN-AS1-L protects against PXN mRNA degradation, thereby promoting hepatocellular carcinoma progression. In contrast, PXN-AS1-S dissociates the translation elongation factors from PXN mRNA, thereby inhibiting PXN mRNA translation and suppressing tumorigenesis [26].
We previously reported that RP11-367G18.1 promoted EMT and H4K16Ac activation via its variant 2, but not variant 1 [11]. In this study, we investigated the molecular mechanisms underlying the role of RP11-367G18.1 variant 2 in epigenetic regulation. Our data suggested that YY1 interacted with the RP11-367G18.1 variant 2. Both RP11-367G18.1 variant 2 and YY1 were regulated by hypoxia/HIF-1α. YY1 bound to the promoter of genes and activated H4K16Ac by associating with RP11-367G18.1 variant 2 under hypoxia. The RP11-367G18.1 variant 2–YY1 complex regulated hypoxia-inducible genes implicated in several biological and cellular processes, such as EMT, angiogenesis, metabolism, and inflammatory responses (Fig. 8).

Schematic representation of the mechanism by which the RP11-367G18.1 variant 2–YY1 complex regulates hypoxia-inducible gene expression. In a hypoxic tumor microenvironment, RP11-367G18.1 variant 2 and YY1 are regulated by HIF-1α. RP11-367G18.1 variant 2 and YY1 form a complex and activate H4K16Ac to increase the expression levels of hypoxia-inducible genes, TGFBI, VEGFC, HK2, and LIF
LncRNAs are localized in the nucleus, cytoplasm, and several cellular compartments and linked to their cellular functions. Nuclear lncRNAs may perform histone modifications or transcriptional regulation depending on their interactions with DNA, RNA, and proteins [27]. LncRNAs can recruit histone modifiers modulating gene transcription [28]. LncRNA kcnq1ot1 interacts with histone methyltransferase G9a and the PRC2 complex to increase the trimethylation of H3K9 and H3K27, thus silencing lineage-specific transcription [29]. LncRNA-JADE increases the expression of Jade1, a scaffold protein of HBO1 histone acetylase, thereby inducing the acetylation of H4 [30]. Although histone H4 acetylation can be observed in the promoters of active genes, H4K16Ac is associated with both transcriptional activation and repression [31]. H4K16Ac is involved in various cellular processes, such as chromatin accessibility, DNA damage response, and autophagy [32,33,34]. Recent studies have suggested an association between H4K16Ac and tumorigenesis [35, 36].
Transcription factor, YY1, is ubiquitously expressed in mammalian cells and acts as a transcriptional activator and repressor [21]. Some DNA-binding transcription factors have been reported to bind to RNA [37]. YY1 binds to single-stranded RNA with low specificity [38]. YY1 can bind to both lncRNA Xist and DNA via different sequences, which is essential for tethering Xist to the inactive X nucleation center [39]. YY1 physically interacts with lncRNA Sox2ot to suppress the differentiation of neural progenitors by inhibiting Sox2 expression [40]. Interestingly, both histone acetyltransferases and histone deacetylases (HDACs) interact with YY1. YY1 interacts with p300 and CREB-binding protein to activate transcription [21]. YY1 recruits HDAC2 to deacetylate histone H3, thereby suppressing chondromodulin-I expression [41]. In addition, YY1 recruits the histone H4-specific methyltransferase, PRMT1 to the YY1-activated promoter [42]. These studies suggest that YY1 regulates gene transcription in a context-dependent manner. Nevertheless, LC-MS/MS analysis revealed that no histone modifier was pulled down by biotinylated RP11-367G18.1 variant 2. Hence, YY1-related histone modifiers mediating RP11-367G18.1 variant 2-activated H4K16Ac need to be investigated further in future studies.
Conclusions
In conclusion, our results revealed that the RP11-367G18.1 variant 2–YY1 complex promotes cancer progression in a hypoxic tumor microenvironment. Moreover, the RP11-367G18.1 variant 2–YY1 complex enhances hypoxia-inducible gene expression via H4K16Ac activation. Therefore, the RP11-367G18.1–YY1 complex can potentially be used as a therapeutic target for HNSC treatment.
Data Availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Fernandes JCR, Acuna SM, Aoki JI, Floeter-Winter LM, Muxel SM. Long non-coding RNAs in the regulation of Gene expression: physiology and disease. Noncoding RNA. 2019;5(1).
Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157.
Liu SJ, Dang HX, Lim DA, Feng FY, Maher CA. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer. 2021;21(7):446–60.
Jiang MC, Ni JJ, Cui WY, Wang BY, Zhuo W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am J Cancer Res. 2019;9(7):1354–66.
Nakamura N, Shi X, Darabi R, Li Y. Hypoxia in cell reprogramming and the epigenetic regulations. Front Cell Dev Biol. 2021;9:609984.
Hapke RY, Haake SM. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020;487:10–20.
Peng X, Gao H, Xu R, Wang H, Mei J, Liu C. The interplay between HIF-1alpha and noncoding RNAs in cancer. J Exp Clin Cancer Res. 2020;39(1):27.
Wang X, Zhao D, Xie H, Hu Y. Interplay of long non-coding RNAs and HIF-1alpha: a new dimension to understanding hypoxia-regulated tumor growth and metastasis. Cancer Lett. 2021;499:49–59.
Peng PH, Hsu KW, Chieh-Yu Lai J, Wu KJ. The role of hypoxia-induced long noncoding RNAs (lncRNAs) in tumorigenesis and metastasis. Biomed J. 2021;44(5):521–33.
Ouyang J, Zhong Y, Zhang Y, Yang L, Wu P, Hou X et al. Long non-coding RNAs are involved in alternative splicing and promote cancer progression. Br J Cancer. 2021.
Peng PH, Lai JC, Chang JS, Hsu KW, Wu KJ. Induction of epithelial-mesenchymal transition (EMT) by hypoxia-induced lncRNA RP11-367G18.1 through regulating the histone 4 lysine 16 acetylation (H4K16Ac) mark. Am J Cancer Res. 2021;11(6):2618–36.
Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95(14):7987–92.
Peng PH, Chieh-Yu Lai J, Hsu KW, Wu KJ. Hypoxia-induced lncRNA RP11-390F4.3 promotes epithelial-mesenchymal transition (EMT) and metastasis through upregulating EMT regulators. Cancer Lett. 2020;483:35–45.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–15.
Xing Z, Lin A, Li C, Liang K, Wang S, Liu Y, et al. LncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell. 2014;159(5):1110–25.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, et al. TANRIC: an interactive Open platform to explore the function of lncRNAs in Cancer. Cancer Res. 2015;75(18):3728–37.
Hsu KW, Huang CY, Tam KW, Lin CY, Huang LC, Lin CL et al. The application of non-invasive apoptosis detection sensor (NIADS) on histone deacetylation inhibitor (HDACi)-Induced breast Cancer Cell Death. Int J Mol Sci. 2018;19(2).
Qi Y, Lai YL, Shen PC, Chen FH, Lin LJ, Wu HH, et al. Identification and validation of a miRNA-based prognostic signature for cervical cancer through an integrated bioinformatics approach. Sci Rep. 2020;10(1):22270.
Hsu KW, Lai JC, Chang JS, Peng PH, Huang CH, Lee DY, et al. METTL4-mediated nuclear N6-deoxyadenosine methylation promotes metastasis through activating multiple metastasis-inducing targets. Genome Biol. 2022;23(1):249.
Verheul TCJ, van Hijfte L, Perenthaler E, Barakat TS. The why of YY1: mechanisms of Transcriptional Regulation by Yin Yang 1. Front Cell Dev Biol. 2020;8:592164.
Kuo TC, Kung HJ, Shih JW. Signaling in and out: long-noncoding RNAs in tumor hypoxia. J Biomed Sci. 2020;27(1):59.
Wang S, Qi Y, Gao X, Qiu W, Liu Q, Guo X, et al. Hypoxia-induced lncRNA PDIA3P1 promotes mesenchymal transition via sponging of mir-124-3p in glioma. Cell Death Dis. 2020;11(3):168.
Zhu P, He F, Hou Y, Tu G, Li Q, Jin T, et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40(9):1609–27.
Chen J, Liu Y, Min J, Wang H, Li F, Xu C, et al. Alternative splicing of lncRNAs in human diseases. Am J Cancer Res. 2021;11(3):624–39.
Yuan JH, Liu XN, Wang TT, Pan W, Tao QF, Zhou WP, et al. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat Cell Biol. 2017;19(7):820–32.
Long Y, Wang X, Youmans DT, Cech TR. How do lncRNAs regulate transcription? Sci Adv. 2017;3(9):eaao2110.
Hanly DJ, Esteller M, Berdasco M. Interplay between long non-coding RNAs and epigenetic machinery: emerging targets in cancer? Philos Trans R Soc Lond B Biol Sci. 2018;373:1748.
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32(2):232–46.
Wan G, Hu X, Liu Y, Han C, Sood AK, Calin GA, et al. A novel non-coding RNA lncRNA-JADE connects DNA damage signalling to histone H4 acetylation. EMBO J. 2013;32(21):2833–47.
Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16(11):1467–81.
Samata M, Alexiadis A, Richard G, Georgiev P, Nuebler J, Kulkarni T, et al. Intergenerationally maintained histone H4 lysine 16 acetylation is instructive for future gene activation. Cell. 2020;182(1):127–44. e23.
Singh M, Bacolla A, Chaudhary S, Hunt CR, Pandita S, Chauhan R et al. Histone acetyltransferase MOF orchestrates outcomes at the crossroad of Oncogenesis, DNA damage response, proliferation, and Stem Cell Development. Mol Cell Biol. 2020;40(18).
Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500(7463):468–71.
Chen J, Liu G, Wu Y, Ma J, Wu H, Xie Z, et al. CircMYO10 promotes osteosarcoma progression by regulating miR-370-3p/RUVBL1 axis to enhance the transcriptional activity of beta-catenin/LEF1 complex via effects on chromatin remodeling. Mol Cancer. 2019;18(1):150.
Wang L, Li X, Zhang W, Yang Y, Meng Q, Wang C, et al. miR24-2 promotes malignant progression of Human Liver Cancer Stem cells by enhancing tyrosine kinase src epigenetically. Mol Ther. 2020;28(2):572–86.
Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, et al. Transcription factor trapping by RNA in gene regulatory elements. Science. 2015;350(6263):978–81.
Wai DC, Shihab M, Low JK, Mackay JP. The zinc fingers of YY1 bind single-stranded RNA with low sequence specificity. Nucleic Acids Res. 2016;44(19):9153–65.
Jeon Y, Lee JT. YY1 tethers xist RNA to the inactive X nucleation center. Cell. 2011;146(1):119–33.
Knauss JL, Miao N, Kim SN, Nie Y, Shi Y, Wu T, et al. Long noncoding RNA Sox2ot and transcription factor YY1 co-regulate the differentiation of cortical neural progenitors by repressing Sox2. Cell Death Dis. 2018;9(8):799.
Aoyama T, Okamoto T, Fukiage K, Otsuka S, Furu M, Ito K, et al. Histone modifiers, YY1 and p300, regulate the expression of cartilage-specific gene, chondromodulin-I, in mesenchymal stem cells. J Biol Chem. 2010;285(39):29842–50.
Rezai-Zadeh N, Zhang X, Namour F, Fejer G, Wen YD, Yao YL, et al. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev. 2003;17(8):1019–29.
Funding
This study was supported by grants from the Ministry of Science and Technology Summit and Frontier grants (MOST 109-2320-B-182A-022, MOST 109-2628-B-039-006, MOST 110-2320-B-182A-019, MOST 110-2628-B-039-007, MOST 111-2628-B-039-007-MY3, MOST 111-2314-B-182A-034, NSTC 112-2314-B-182A-056), Chang Gung Memorial Hospital (NMRPG3K0511, NRRPG3L0151, NRRPG3M0081, NRRPG3N0041), China Medical University (CMU110-MF-10, CMU111-MF-05, CMU112-MF-06), and the “Drug Development Center, China Medical University” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project (Ministry of Education, Taiwan).
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision: WCC and KWH. Formal analysis, and investigation: PHP, JLC, HHW, WHY, LJL, JCYL, JSC, JLS, HTW, FTH, WCC, and KWH. Resources: HHW, WHY, LJL, JCYL, JSC, JLS, HTW, FTH, and WCC. Writing - Original Draft: PHP and JLC. Writing - Review & Editing: FTH, WCC, and KWH. All authors contributed substantially to the conception and design of the study. All authors have read and revised the manuscript critically for intellectual content and have approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethics approval
All animal experiments were approved by the Institutional Animal Care and Use Committee (2020122504) of the Chang Gung Memorial Hospital.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peng, PH., Chen, JL., Wu, HH. et al. Interplay between lncRNA RP11-367G18.1 variant 2 and YY1 plays a vital role in hypoxia-mediated gene expression and tumorigenesis. Cancer Cell Int 23, 266 (2023). https://doi.org/10.1186/s12935-023-03067-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-03067-6