
Abstract
Background
Immune checkpoint inhibitors are the most studied forms of immunotherapy for triple-negative breast cancer (TNBC). The Cancer Genome Map (TCGA) and METABRIC project provide large-scale cancer samples that can be used for comprehensive and reliable immunity-related gene research.
Methods
We analyzed data from TCGA and METABRIC and established an immunity-related gene prognosis model for breast cancer. The SDC1 expression in tumor and cancer associated fibroblasts (CAFs) was then observed in 282 TNBC patients by immunohistochemistry. The effects of SDC1 on MDA-MB-231 proliferation, migration and invasion were evaluated. Qualitative real-time PCR and western blotting were performed to identify mRNA and protein expression, respectively.
Results
SDC1, as a key immunity-related gene, was significantly correlated with survival in the TCGA and METABRIC databases, while SDC1 was found to be highly expressed in TNBC in the METABRIC database. In the TNBC cohort, patients with high SDC1 expression in tumor cells and low expression in CAFs had significantly lower disease-free survival (DFS) and fewer tumor-infiltrating lymphocytes (TILs). The downregulation of SDC1 decreased the proliferation of MDA-MB-231, while promoting the migration of MDA-MB-231 cells by reducing the gene expression of E-cadherin and TGFb1 and activating p-Smad2 and p-Smad3 expression.
Conclusion
SDC1 is a key immunity-related gene that is highly expressed TNBC patients. Patients with high SDC1 expression in tumors and low expression in CAFs had poor prognoses and low TILs. Our findings also suggest that SDC1 regulates the migration of MDA-MB-231 breast cancer cells through a TGFb1-Smad and E-cadherin-dependent mechanism.
Similar content being viewed by others


Introduction
Breast cancer is the most common cancer and the leading cause of cancer-related deaths in women [1]. Approximately 3–10% of new breast cancer cases have distant metastasis at the time of diagnosis. In the early stage, 30–40% of patients develop advanced breast cancer, and the five-year survival rate is only 20% [2].
Based on genotype analyses, breast cancer can be divided into the following molecular types: luminal A, luminal B, HER2, basal-like, and normal-like [3]. Triple-negative breast cancer (TNBC) accounts for 13–20% of all breast cancers and is an invasive subtype [4, 5]. The prognosis of patients with TNBC is poor, partly due to the lack of effective targeted drugs; thus, the mainstay of treatment has been traditional chemotherapy [5,6,7,8]. Gene expression profiles have shown that TNBC subtypes were widely distributed from basal to luminal, and most, but not all, TNBC subtypes expressed basal-like properties [4, 9]. Many gene changes have been observed in TNBC, including TP53, PIK3CA, BRCA1, and BRCA2 mutations [10]. BRCA1/2 mutations have been found in 11% of TNBC patients [11]. In addition, TNBC has one of the highest mutation rates of all breast cancer subtypes [12]. TNBC has abundant gene ontologies in immune cell processes including immune cell signaling pathways [13]. It is also considered to be the most immunogenic subtype with higher tumor infiltrating lymphocyte (TIL) density than other subtypes, which is associated with improved prognosis [14,15,16,17] and TIL density can predict the effect of neoadjuvant therapy [18, 19]. Stromal TILs were significantly associated with the OS in patients with cancer [20,21,22,23]. Different immune cell types have been investigated in the prognosis and immunotherapy of a variety of tumors [21, 24,25,26,27]. Higher TILs are associated with improved prognosis of HER2-positive breast cancer and TNBC patients. For every 10% increase in TILs, recurrence and mortality decreased by 15–25% [14,15,16,17]. These data suggest that TNBC is a highly heterogeneous and immunogenic tumor type; therefore, finding new immunity-related targets is critical for prognosis and effective treatment.
We systematically analyzed the expression of immunity-related genes and clinical outcomes of breast cancer patients in the TCGA database, aiming to screen for immunity-related genes related to prognosis and develop a personalized immune prognosis gene panel for breast cancer patients. We further explored the key immune genes, observed their relationship with the prognosis of TNBC and searched possible mechanisms to find new immunotherapeutic targets for TNBC.
Materials and methods
Clinical samples and data collection
Transcriptome RNA sequencing data of 111 normal and 1053 breast cancer samples were downloaded from the TCGA data portal (https://portal.gdc.cancer.gov/repository), and the list of immunity-related genes were obtained through ImmPort [28] (https://www.immport.org/).
Differential gene analysis
R (3.6.3) [29] was used to compare all differentially expressed genes (DEGs) between breast cancer and normal tissues, and the false discovery rate of < 0.05 and the log2 |fold change|> 1 were set as the critical values. Then, the immunity-related DEGs for breast cancer and normal tissues were extracted and compared using R 3.6.3 and the limma, pheatmap, BiocManager, surviva, and survivalROC packages. To explore the potential molecular mechanism of immunity-related DEGs, functional enrichment analysis was carried out using Gene Ontology (GO) pathway analysis (http://www.gsea-msigdb.org/gsea/login.jsp).
Construction of an immunity-related gene prognosis model
The clinical information including the age, tumor-node-metastasis (TNM) stage, tumor size, lymph node metastasis, distant metastasis, and long-term follow-up results of the patients were also extracted from the TCGA database. The association between survival outcome and immunity-related gene expression in breast cancer patients was analyzed to find out the immunity-related genes that were significantly associated with prognosis. By combining the key immunity-related genes obtained with the outcome information of breast cancer patients from the TCGA clinical database, we constructed an immunity-related gene prognosis model by multivariate analysis. The Cox regression coefficient was used to analyze the relationship between gene expression and survival outcome.
Analysis of clinical characteristics and immune cells associated with immunity-related gene prognosis model
The patients in the TCGA clinical database were divided into high- and low-risk groups, according to the median risk value calculated by the model to evaluate its prognostic value. Gene expression data were analyzed again using the TIMER [30] database (https://cistrome.shinyapps.io/timer/), which included 10,897 samples of 32 cancer types of TCGA. The breast cancer sample data downloaded from the TCGA database intersected with the samples from the TIMER database, and the contents of six immune cell types in the intersections were obtained, including B, CD4T+, CD8T+, macrophage, neutrophil, and dendritic cells. We observed the relationship between immune cell infiltration and the risk value in each sample calculated by the immunity-related gene prognosis model.
Analysis of clinical and molecular characteristics associated with key immunity-related genes
We verified the key immunity-related genes in the model using 1093 breast cancer patients with clinical data and survival outcomes in the METABRIC database. The associations were analyzed between key immunity-related genes and survival in the TCGA and METABRIC databases. The key immunity-related genes and clinical features were also observed. SDC1 was selected for subsequent research because it was significantly correlated with survival, TNBC, age, and lymph node metastasis.
Clinical samples and immunohistochemistry in TNBC cohort
The tissue microarray (TMA) was made using formalin-fixed paraffin-embedded tumor samples from 282 consecutive triple-negative breast cancer patients in Peking Union Medical College Hospital (PUMCH). Each tumor specimen contained the tumor and tumor stroma. All the patients had been operated in PUMCH from 2011 to 2014 and had tumors in stages I–III. All the pathological characteristics including tumor size, lymph nodes, tumor grade, hormone receptor (HR) status, and HER2 status were observed. The median follow-up time was 69 months (1–104 months). The TILs in each sample were counted according to the publication A Practical Review for Pathologists and Proposal [31]. All TMA were stained for SDC1 (prediluted, Jinqiaoyatu, Beijing, China), CD3 (PA0553, prediluted; Leica Microsystems, Shanghai, China), CD4 (PA0427, prediluted; Leica Microsystems), CD8 (PA0183, prediluted; Leica Microsystems), CD19 (ZA-0569, Prediluted; Zhongshan Golden Bridge Biotechnology, Beijing, China).
The expression of SDC1 in tumor cells, paracancerous normal mammary duct cells (PNMDCs) and cancer-associated fibroblast (CAFs), as well as frequencies of CD3, CD4, CD8, CD19, and TILs were independently measured by two pathologists. The expression of SDC1 in tumor cells and PNMDCs were observed according to histochemical scoring (H-score) [32]. H-scores of 0–49, 50–99, 100–199, and 200–300 were classified as 0, 1 + , 2 + , and 3 + , respectively. The patients with classifications of 0, 1 + , 2 + were defined as belonging to the negative group, while patients with a classification of 3 + were defined as being in the positive group for tumor cells and PNMDCs. The patients were divided into negative group and positive group for CAFs according to the presence or absence of staining of SDC1. The patients were divided into high and low groups according to a TIL median of 5%, CD3+ TIL median of 20%, CD4+ TIL median of 10%, CD8+ TIL median of 10%, and CD19+ TIL median of 1%.
Cell culture
Human breast cancer cell line MDA-MB-231 was obtained from the Chinese Academy of Medical Sciences (ATCC®HTB-26™, Beijing, China). The cells were cultured in L15 medium containing 20% fetal bovine serum at 37 °C and in normal air. Lentiviral particles with SDC1 overexpression or shRNA expression and their negative controls were obtained from GenePharma (Shanghai, China). MDA-MB-231 cells were incubated in a 24-well plate for 24 h and then transduced with the shRNA-expressing lentivirus, SDC1 overexpression, or their control empty lentivirus for 48 h. Puromycin (LABLEAD Biotech, Beijing, China) was used to select cells for two weeks. Then the stably transfected SDC1 knockdown (5′-GGAGCAGGACTTCACCTTTGA-3ʹ) cell lines (LV3-NC and LV3-SDC1), overexpression (6382, NM_001006946.2, Additional file 1: Table S1) cell lines (LV5-NC and LV5-SDC1) based on MBA-MD-231 and their control empty cells were cultured in RPMI-1640 (AQ11875, Beijing Aoqing Biotechnology, China) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37 °C in an incubator with 5% CO2.
Cell Counting Kit-8 (CCK-8)
The cells were seeded in the 96-well plates (5000 cells/well) and cultured for 24 h, 48 h, 72 h. Then, 10 μL of CCK-8 reagent (CK04-500 T, DOJINDOi) was added to the 100 μL culture medium. After 4 h incubation in a 37 °C incubator, the absorbance at 450 nm was measured using an EPOCH2 Microwell Plate Spectrophotometer (BioTek, Winooski, VT).
Transwell assay
The cells (4 × 104 cells/well) were plated in a transwell chamber with 8 μM pore size in 200 μL serum-free RPMI-1640, and the lower chamber was filled with 600 μL DMEM with 10% PBS. After 24 h incubation, the cells on the upper side of the chamber were removed and the migrated cells were fixed with 4% paraformaldehyde and stained with crystal violet. Number of migrated cells were measured using Axio Imager A2 microscope (Carl Zeiss Microscopy GmbH).
Quantitative real-time PCR (RT-qPCR)
TRIzol™ Reagent (15596026, ThermoFisher Scientific) was used for total RNA isolation. Reverse transcription into cDNA was done using PrimeScript™ RT Master Mix (Perfect Real Time) Kit (RR036A, TAKARA, Kusatsu, Japan). RT-qPCR was performed by Fast SYBR® Green Master Mix (4385612, ThermoFisher Scientific) on QuantStudio™ 7 Flex Real-Time PCR System (4485701, ThermoFisher Scientific). The relative level was calculated using the 2−△△Ct method, and expressed as the ratio of GAPDH. The primer sequences are listed in Additional file 2: Table S2.
Western blot
The protein extraction was carried out by RIPA lysis buffer (AQ521, Beijing AOqing Biotechology, China) containing ProtLytic Protease Inhibitor Cocktail (P001, New Cell & Molecular Biotech, Shanghai, China). The cell lysates were then resolved by NuPAGE™ (4–12%) Bis–Tris PAGE (NP0322BOX, ThermoFisher Scientific) and transferred onto PVDF membranes. After blocking, the membranes were incubated overnight with primary antibodies against SDC1 (ab128936, 1:1000; Abcam, Cambridge, UK), Smad2 (5339, 1:1000; CST, Danvers, MA, USA), Phospho-Smad2 (18338, 1:1000; CST), anti-smad3 (ab40854, 1:1000; Abcam), Phospho-Smad3 (9520, 1:1000; Abcam), E-Cadherin (3195, 1:1000; CST), N-Cadherin (13116, 1:1000; CST), Vimentin (5741, 1:1000; CST), TGF beta 1 (ab215715, 1:1000; Abcam) and HRP-conjugated GAPDH Monoclonal antibody (HRP-60004, 1:10000; ProteinTech) at 4 °C. Then the membranes were washed three times and incubated with HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H + L) (SA00001-2, 1:10000; ProteinTech, Chicago, IL, USA) for 1 h at 24 °C. The NcmECL Ultra Kit (P10300, New Cell and Molecular Biotech) was added, the proteins were visualized by Amersham Imager 680 (Beijing LABAID Science and Technology. LTD, China) and were evaluated using ImageJ 1.8.0.
Statistics
R3.6.3 Statistical Software and SPSS 23.0 were used to perform statistical analyses. The difference in gene expression among clinical samples was analyzed by an independent t-test. The R “survival” package and Kaplan–Meier method in SPSS were used for survival analysis. We used the survival package to perform a multivariate Cox analysis to establish an immunity-related gene prognosis model. The area under the curve was calculated after creating the ROC curve [33] using the survival ROC package to test the performance of the prognotic model. R was used to draw the charts for the bioinformatic analysis. A P < 0.05 was considered statistically significant.
Results
Obtaining differential genes and construction of an immunity-related gene prognosis model
We extracted 56,753 genes from the TCGA database, including 942 upregulated and 590 downregulated genes (Fig. 1A, C). A total of 377 immunity-related DEGs were extracted after intersection with the ImmPort database, including 203 upregulated and 174 downregulated genes (Fig. 1B, D). Enrichment analysis showed that the immune inflammatory pathway was one of the most significantly enriched pathways (Table 1). T cell receptor complex, leukocyte-mediated immunity, and immune effector process pathways had high enrichment scores (Fig. 1E). Among them, we found that 16 immunity-related DEGs were significantly associated with overall survival (OS) in breast cancer patients (Fig. 1F) and their average differences are shown in Table 2. To better predict the prognosis of patients with breast cancer, an immunity-related gene prognosis model was constructed by multivariate Cox regression analysis. The formula was as follows: [Expression level of ULBP2 * 0.1160] + [Expression level of BCL3 *(− 0.0166)] + [Expression level of IL18 * (− 0.0696)] + [Expression level of IGHE* 0.0602] + [Expression level of SEMA6D* 0.1086] + [Expression level of FGF7 * 0.0919] + [Expression Level of IL22RA1 * (-0.7509)] + [Expression level of NPR3 * 0.0445] + [Expression level of SDC1 * 0.0014] + [Expression level of TRDV1 * (-0.2995)].

A Heatmap and C volcano plot showing differentially expressed genes between breast cancer tissue and normal tissue in the TCGA dataset. Red dots represent differentially up-regulated genes, green dots represent differentially downregulated genes and black dots represent no differentially expressed genes. B Heatmap and D volcano plot showed different immune-related genes between breast cancer tissue and normal tissue in the TCGA dataset. Red dots represent differentially up-regulated genes, green dots represent differentially downregulated genes and black dots represent no differentially expressed genes. E The most significant genomes pathways by gene functional enrichment in immune-related genes. F Sixteen immunity-related differentially expressed genes significantly associated with overall survival in the TCGA datasets
Correlation between immunity-related gene model and clinical features
The model was able to distinguish breast cancer patients into high- and low-risk of death groups, according to the survival outcome of patients (Fig. 2A, B). The area under the curve of the model was 0.674 (Fig. 2C), which indicated that the model could predict the prognosis of breast cancer patients to an extent. Univariate and multivariate Cox analyses showed that the immunity-related gene prognosis model was a good prognostic factor, independent of age, pathological stage, tumor stage, lymph node metastasis status, and distant metastasis status (Fig. 2D, E). The relationship of each sample between immune cell infiltration and the risk value was calculated by the model in the TCGA database (Fig. 2F–K). Patients with high-risk scores had significantly lower immune cell infiltration including B, CD4T+, CD8T+, macrophage, neutrophil, and dendritic cells.

A The survival outcome and B the overall survival between high risk and low-risk patients distinguished by immunity-related gene model. C The prognostic value of the model by receiver operating characteristic (ROC) curve. Univariate analysis (D) and multivariate analysis (E) of immunity-related gene prognosis model and distant metastasis in the METABRIC datasets. The relationships between B cell infiltration (F), CD4+ T cell infiltration (G), CD8+ T cell infiltration (H), dendritic infiltration (I), macrophage infiltration (J), neutrophil infiltration (K) and the risk value by the immunity-related gene prognosis model
SDC1 association with survival and triple negative breast cancer patients
We employed our model to analyze the correlation between immune-related genes and survival of patients with breast cancer, using data on the clinical features and survival outcomes of 1053 and 1093 patients in the TCGA and METABRIC database, respectively. We found SDC1 was significantly associated with overall survival (Fig. 3A), and lymph node metastasis (Fig. 3C) in TCGA database. We also found that SDC1 was also significantly associated with disease-free survival (DFS) (Fig. 3B). In addition, SDC1 was significantly higher in TNBC patients and HER2-positive patients (Fig. 3D, E). SDC1 was also found to be higher in patients ≤ 60 years old than in patients > 60 years old (Fig. 3F).

A The overall survival and B disease-free survival in METABRIC datasets between patients with high expression and low expression of SDC1. C SDC1 expression in patients with or without lymph node metastasis in the TCGA dataset. In the METABRIC dataset, D SDC1 expression in patients with TNBC and in other patients, E SDC1 expression in HER2-positive patients and HER2-negative patients, F SDC1 expression in patients aged ≤ 60 years old and > 60 years old
SDC1 association with survival and recurrence in TNBC patients
We analyzed the 282 TNBC patients treated at PUMCH who had clinical features and survival outcomes. We observed SDC1 expression in tumor cell, PNMDC, and CAF by immunohistochemistry (IHC) (Fig. 4A), and the characteristics of these patients are shown in Table 3. The proportion of patients with positive expression of SDC1 in tumor cells was higher than those patients in PNMDC (Fig. 4B, p = 0.003). The patients with positive expression of SDC1 in tumor cells had significantly lower DFS compared to patients with negative SDC1 expression in tumor cells (Fig. 4C, p < 0.001). The patients with negative expression of SDC1 in CAF had significantly lower DFS compared with the patients with positive SDC1 expression in CAFs (Fig. 4D, p = 0.02). In univariate Cox proportional hazards regression analysis, tumor size, positive lymph nodes, positive expression of SDC1 in tumor cells, and negative expression of SDC1 in CAF were significantly associated with distant recurrence and in multivariate Cox proportional hazards regression analysis, positive lymph nodes, negative expression of SDC1 in CAF were significantly associated with distant recurrence (Table 4).

A Hematoxylin and eosin (HE) staining and immunohistochemistry (IHC) of SDC1 expression in TNBC patients. SDC1-negative expression on tumor cell by (a) HE staining and b IHC (original magnification, × 200). SDC1 1 + expression on tumor cells by (c) HE staining and d IHC (original magnification, × 200). SDC1 2 + expression on tumor cells by (e) HE staining and f IHC (original magnification, × 200). SDC1 3 + expression on tumor cells by (g) HE staining and h IHC (original magnification, × 200). SDC1-negative expression in cancer-associated fibroblasts (CAFs) by (i) HE staining and j IHC (original magnification, × 200). SDC1-positive expression on CAF by (k) HE staining and l IHC (original magnification, × 200). B The percentage of TNBC patients with the expression of SDC1 in tumor cells and in paracancerous normal mammary duct cells (PNMDCs). Disease-free survival between SDC1-positive patients and SDC1-negative patients in tumor cells C and in CAFs D in the TNBC cohort
SDC1 is associated with tumor stromal infiltrating lymphocytes in TNBC patients
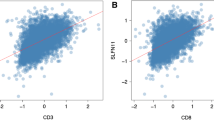
TIL distribution was confirmed by HE, and CD4+TILs, CD3+TILs, CD8+TILs and CD19 +TILs were also observed by IHC in 282 TNBC samples (Fig. 5A, C). The patients with positive expression of SDC1 in tumor cells were associated with lower TIL distribution (Fig. 5B, p = 0.021). CD3 T, CD4 T, CD8 T, and CD19 T cell distributions were lower in patients with positive SDC1 expression than in patients with negative SDC1 expression in tumor cells, albeit there was no significant difference (Fig. 5B). TIL distribution was lower in patients with negative SDC1 expression than patients with positive expression of SDC1 in CAF, although there was no significant difference (Fig. 5D, p = 0.060). CD3 T cells, CD8 T cells were lower in patients with positive expression of SDC1 than those with negative SDC1 expression in CAF (Fig. 5D, p = 0.055, p = 0.140). CD4 T and CD19 T cell counts were higher in patients with positive expression of SDC1 than those with negative expression in CAFs (Fig. 5D, p = 0.730, p = 0.008).

A SDC1 expression in tumor cells associated with TILs in the TNBC cohort. a, b Patients with SDC1-positive expression and a low percentage of CD3+ TILs. c, d Patients with SDC1-positive expression and a low percentage of CD4+ TILs. e, f Patients with SDC1-positive expression and a low percentage of CD8+ TILs. g, h Patients with SDC1-positive expression and a low percentage of CD19+ TILs. B Frequencies of TILs between patients with SDC1-negative expression and SDC1-positive expression in tumor cells in the TNBC cohort. a Frequencies of TILs, b CD3+ TILs, c CD4+ TILs, d CD8+ TILs, e CD19+ TILs associated with SDC1 expression in tumor cells. C SDC1 expression in CAFs associated with TILs in the TNBC cohort. a, b Patients with SDC1-positive expression and a low percentage of CD3+ TILs. c, d Patients with SDC1-positive expression and a high percentage of CD4+ TILs. e, f Patients with SDC1-positive expression and a low percentage of CD8+ TILs. g, h The patients with SDC1-positive expression and a high percentage of CD19+ TILs. D Frequencies of TILs between patients with SDC1-negative expression and SDC1-positive expression in CAF in the TNBC cohort. a Frequencies of TILs, b CD3+ TILs, c CD4+ TILs, d CD8+ TILs, e CD19+ TILs associated with SDC1 expression in CAFs
SDC1 knockdown inhibited the viability in MDA-MB-231 cells
To further explore the mechanism of SDC1 in breast cancer, especially in TNBC, we conducted cell experiments to evaluate the effects of SDC1 on breast cancer cell proliferation and migration. First, we constructed the stable knockdown and overexpression cell lines and verified their expression effects. A significant decrease to 15.4% in gene expression of SDC1 in MDA-MB-231 cell line was observed after SDC1 knockdown (LV3-SDC1) compared with the NC control (LV3-NC, Fig. 6A, B), while SDC1 was increased by 302.6 times after SDC1 overexpression (LV5-SDC1) compared to the NC control (LV5-NC, Fig. 6A, B).

The effect of SDC1 on breast cancer cell proliferation and migration. A Relative SDC1 mRNA expression after knockdown and overexpression. B Relative SDC1 protein expression after knockdown and overexpression. A CCK-8 assay evaluated the effects of SDC1 on cell proliferation after the C knockdown and D overexpression of SDC1. E Transwell assay determined that SDC1 knockdown promoted the invasion of MDA-MB-231 cells. F The protein levels of EMT-related proteins. G Protein expression in the TGF-β1/Smad signaling pathway. Results with P < 0.05 were considered statistically significant. An unpaired t-test was used for data analysis *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
The CCK-8 assay was utilized to evaluate the effects of SDC1 on cell proliferation. The cell proliferation decreased significantly after the knockdown of SDC1 (Fig. 6C), meanwhile the cell viability increased after SDC1 overexpression (Fig. 6D).
SDC1 knockdown promoted the invasion of MDA-MB-231 cells
To understand the potential roles of SDC1 in mediating MDA-MB-231 cell motility, a transwell assay was performed (Fig. 6E). The number of cells passing through the aperture was significantly higher in the siSDC1 group (LV3-SDC1) compared with the MDA-MB-231 and LV3-NC groups after 24 h incubation. The 24 h migration rate was decreased in the SDC1 overexpression group (LV5-SDC1) compared with the LV5-NC group.
SDC1 affected epithelial–mesenchymal transition (EMT) via regulating the TGF-β1/Smad pathway signaling pathway
Protein expression levels of the epithelial marker E-cadherin was reduced, while those of the interstitial markers N-cadherin and vimentin were significantly increased by SDC1 knockdown (Fig. 6F). In the meantime, the overexpression of SDC1 increased the protein level of E-cadherin and reduced the protein expression of N-cadherin and vimentin. The results showed that SDC1 knockdown or overexpression affected the expression of epithelial and interstitial markers, modulating EMT in MDA-MB-231 cells.
Given the correlation between the EMT process and the TGF-β1 pathway, western blotting was used to detect the effect of SDC1 knockdown or overexpression on the TGF-β1/Smad pathway. After the knockdown of SDC1, the expression of TGF-β1 was significantly decreased, while phospho-Smad3 and phospho-Smad2 expression increased. In contrast, overexpression of SDC1 caused the opposite results (Fig. 6G).
Discussion
Although early-stage breast cancer can be successfully treated through surgery, chemotherapy, or other treatment, more than 30% of patients will eventually progress to an advanced stage [34]. Advanced breast cancer has poor prognosis, and the long-term survival rate is less than 5% [35]. The 5-year OS for patients with TNBC is 72%, with a life expectancy of only 3.55 years [36]. Immune checkpoint inhibitors have been successfully used to treat cancer [37], and immunotherapy has become the preferred treatment for TNBC patients with PD-L1 positive tumors [38]. Therefore, it is crucial to explore new therapeutic targets and tumor markers related to TNBC immunotherapy.
Although the importance of immunity-related genes in cancer therapy has been confirmed [39,40,41,42], there has been no comprehensive analysis of the clinical significance and molecular mechanisms of immunity-related genes in breast cancer patients. Here, we found that SDC1 was highly expressed in TNBC patients at protein and transcriptome levels and could be used as a marker of poor prognosis. We also found that negative expression of SDC1 in CAF was positively related to poor prognosis in TNBC patients. TILs were regulated differently by SDC1 in tumor cells and CAFs. Furthermore, we identified that SDC1 reduction promoted the progression of EMT through TGF-β1/Smad pathway in vitro. Finally, based on the above findings, we propose that an SDC1-associated related immune signature as a predictor and a potential target in TNBC.
The expression and immunoregulatory function of SDC1 in cancers are tumor-specific and their details remain controversial. SDC1 ectodomains attenuate allergic lung inflammation via suppression of CC chemokine-mediated Th2 cell recruitment to the lung [43]. SDC1 suppression in TNBC cell line downregulates IL-6, IL-8 [44, 45]. Heparan sulfate chain shedding from SDC-1 facilitates the release of vascular endothelial growth factor C (VEGF-C) into the medium of hepatocarcinoma cells [46]. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation [47]. VEGF induces T-cell apoptosis during extravasation by leading to FASLG on endothelial cells [48]. SDC1 knockdown in SUM-149 cells promotes Th17 cell expansion via upregulation of IL-23 [49]. CD4+ T cells differentiate into Th17 cells through TGFB, while IL6 and TH17 maintain proliferation by IL23 [50]. IL6 trans-signaling enhances T-cell transmigration on tumor vessels [51].
In our TNBC cohort, we found that the expression of SDC1 was significantly higher in tumor cells than in PNMDCs. The positive expression of SDC1 in tumor cells was significantly related to a poor prognosis and lower frequencies of TILs. Negative expression of SDC1 in CAF was significantly related to a poor prognosis and lower frequencies of CD19+TILs. We speculated that SDC1 in TNBC cells suppressed T-lymphocyte infiltration by upregulating IL6, IL8, and VEGF, while T cell infiltration was inhibited by downregulation of IL-23. Immune escape may be the reason why TNBC patients with positive expression of SDC1 in tumor cells had a poor prognosis.
CAFs participate in tumor occurrence, development, and drug resistance [52]. Activation of CAFs remodel the extracellular matrix and promotes cancer cell invasion and metastasis [53,54,55]. Highly invasive human breast cancer cells MDA-MB-231 can enhance SDC1 expression in senescent fibroblasts via the paracrine action of TGF-β [56]. Senescent cells were characterized by exhaustion of SDC1 expression [57]. Fusion of dendritic cells and CAFs produce TNF-α and IL-6 [58]. IL-6 is expressed approximately 100-fold higher in CAFs compared to normal fibroblasts [59]. TNF stimulates T-cell extravasation on tumor vessels [60] and IL6 trans-signaling enhances T-cell transmigration on tumor vessels [51]. Activation of CAF induces CXCL16, which in turn recruits T cells [61]. We believe that senescent CAFs with high expression of SDC1 weaken breast cancer cell invasion and metastasis and decrease cytokine secretion; according, patients in our TNBC cohort with negative expression SDC1 in CAFs had poor prognosis and low frequencies of TILs.
SDC1 has been known to play an important role in the invasion, migration and EMT in cancer cells by regulating ERK/Snail signaling [62,63,64]. As shown in our study, the cell viability and migration of MDA-MB-231 cells were significantly affected after SDC1 knockdown. Western blot assays showed that SDC1 reduction promoted the progression of EMT through the TGF-β1/Smad pathway. These results demonstrated that SDC1 may regulate epithelial-mesenchymal plasticity in TNBC cells and open up the possibility of mechanistic studies to elucidate the mechanisms involved.
In summary, we constructed an immunity-related gene prognosis model that reliably predicted the prognosis of patients with breast cancer. Among the immunity-related genes, SDC1 was identified as being closely related to the prognosis of breast cancer. The patients with positive expression of SDC1 in tumor cells had poor prognosis and low frequencies of TILs, while patients with negative expression of SDC1 in CAFs had poor prognosis and high frequencies of TILs. In MDA-MB-231, SDC1 knockout or overexpression affected cell proliferation and migration. SDC1 overexpression in MDA-MB-231 cells upregulated the expression of E-cadherin and TGF-β1, downregulated the expression of p-SMAD2 and p-SMAD3, and affected the EMT process to regulate migration. We believe that TNBC cells and senescent CAFs with positive expression of SDC1 probably inhibit T cells by regulating cytokines, resulting in immune escape. In our research, the associations between SDC1 in tumor cells and CAFs with TILs and related cytokines need to be further confirmed in in vivo and in vitro studies.
Conclusion
We identified SDC1 as a potential immune-therapeutic target in TNBC through bioinformatics analysis, and the high expression of SDC1 was significantly associated with a poor prognosis.
Additionally, SDC1 was also closely related with TILs in tumor cells and CAFs and may be involved in the regulation of TILs by various means. Furthermore, we demonstrated that SDC1 regulated EMT through the TGF-β1/Smad pathway. Targeting SDC1 to regulate tumor cells, CAFs, and TILs should be a promising research direction for TNBC treatment.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22.
Goldhirsch A, Wood WC, Coates AS, et al. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011;22(8):1736–47.
Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389(10087):2430–42.
Metzger-Filho O, Tutt A, de Azambuja E, et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol. 2012;30(15):1879–87.
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–90.
Fallahpour S, Navaneelan T, De P, Borgo A. Breast cancer survival by molecular subtype: a population-based analysis of cancer registry data. CMAJ Open. 2017;5(3):E734–9.
Malorni L, Shetty PB, De Angelis C, et al. Clinical and biologic features of triple-negative breast cancers in a large cohort of patients with long-term follow-up. Breast Cancer Res Treat. 2012;136(3):795–804.
Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 2013;18(2):123–33.
Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–9.
Couch FJ, Hart SN, Sharma P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33(4):304–11.
Liu Z, Li M, Jiang Z, Wang X. A comprehensive immunologic portrait of triple-negative breast cancer. Transl Oncol. 2018;11(2):311–29.
Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67.
Loi S, Sirtaine N, Piette F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02–98. J Clin Oncol. 2013;31(7):860–7.
Dieci MV, Mathieu MC, Guarneri V, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann Oncol. 2015;26(8):1698–704.
Adams S, Gray RJ, Demaria S, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32(27):2959–66.
Ibrahim EM, Al-Foheidi ME, Al-Mansour MM, Kazkaz GA. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: a meta-analysis. Breast Cancer Res Treat. 2014;148(3):467–76.
Denkert C, von Minckwitz G, Darb-Esfahani S, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018;19(1):40–50.
Salgado R, Denkert C, Campbell C, et al. Tumor-infiltrating lymphocytes and associations with pathological complete response and event-free survival in HER2-positive early-stage breast cancer treated with lapatinib and trastuzumab: a secondary analysis of the NeoALTTO trial. JAMA Oncol. 2015;1(4):448–54.
Hao J, Yu H, Zhang T, An R, Xue Y. Prognostic impact of tumor-infiltrating lymphocytes in high grade serous ovarian cancer: a systematic review and meta-analysis. Ther Adv Med Oncol. 2020;12:1758835920967241.
Cillo AR, Kurten CHL, Tabib T, et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity. 2020;52(1):183–99 e9.
Ali HR, Provenzano E, Dawson SJ, et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann Oncol. 2014;25(8):1536–43.
Cabrita R, Lauss M, Sanna A, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–5.
Berntsson J, Nodin B, Eberhard J, Micke P, Jirstrom K. Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int J Cancer. 2016;139(5):1129–39.
Garaud S, Buisseret L, Solinas C, et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight. 2019;5:e129641.
Shi JY, Gao Q, Wang ZC, et al. Margin-infiltrating CD20(+) B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin Cancer Res. 2013;19(21):5994–6005.
Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–34.
Bhattacharya S, Andorf S, Gomes L, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58(2–3):234–9.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Li T, Fan J, Wang B, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–10.
Hendry S, Salgado R, Gevaert T, et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the International Immunooncology Biomarkers Working Group: Part 1: assessing the host immune response, TILs in invasive breast carcinoma and ductal carcinoma in situ, metastatic tumor deposits and areas for further research. Adv Anat Pathol. 2017;24(5):235–51.
Specht E, Kaemmerer D, Sänger J, Wirtz RM, Schulz S, Lupp A. Comparison of immunoreactive score, HER2/neu score and H score for the immunohistochemical evaluation of somatostatin receptors in bronchopulmonary neuroendocrine neoplasms. Histopathology. 2015;67(3):368–77.
Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56(2):337–44.
Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med. 2013;274(2):113–26.
Greenberg PA, Hortobagyi GN, Smith TL, Ziegler LD, Frye DK, Buzdar AU. Long-term follow-up of patients with complete remission following combination chemotherapy for metastatic breast cancer. J Clin Oncol. 1996;14(8):2197–205.
James M, Dixit A, Robinson B, Frampton C, Davey V. Outcomes for patients with non-metastatic triple-negative breast cancer in New Zealand. Clin Oncol. 2019;31(1):17–24.
Nathan MR, Schmid P. The emerging world of breast cancer immunotherapy. Breast. 2018;37:200–6.
Schmid P, Adams S, Rugo HS, et al. Atezolizumab and Nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–21.
Li B, Severson E, Pignon JC, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174.
Gingras I, Azim HA Jr, Ignatiadis M, Sotiriou C. Immunology and breast cancer: toward a new way of understanding breast cancer and developing novel therapeutic strategies. Clin Adv Hematol Oncol. 2015;13(6):372–82.
Li J, Liu C, Chen Y, et al. Tumor characterization in breast cancer identifies immune-relevant gene signatures associated with prognosis. Front Genet. 2019;10:1119.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5.
Xu J, Park PW, Kheradmand F, Corry DB. Endogenous attenuation of allergic lung inflammation by syndecan-1. J Immunol. 2005;174(9):5758–65.
Ibrahim SA, Gadalla R, El-Ghonaimy EA, et al. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol Cancer. 2017;16(1):57.
Valla S, Hassan N, Vitale DL, et al. Syndecan-1 depletion has a differential impact on hyaluronic acid metabolism and tumor cell behavior in luminal and triple-negative breast cancer cells. Int J Mol Sci. 2021;22(11):5874.
Yu S, Lv H, Zhang H, et al. Heparanase-1-induced shedding of heparan sulfate from syndecan-1 in hepatocarcinoma cell facilitates lymphatic endothelial cell proliferation via VEGF-C/ERK pathway. Biochem Biophys Res Commun. 2017;485(2):432–9.
Huang H, Langenkamp E, Georganaki M, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J Off Publ Feder Am Soc Exp Biol. 2015;29(1):227–38.
Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–30.
Saleh ME, Gadalla R, Hassan H, et al. The immunomodulatory role of tumor syndecan-1 (CD138) on ex vivo tumor microenvironmental CD4+ T cell polarization in inflammatory and non-inflammatory breast cancer patients. PLoS ONE. 2019;14(5):e0217550.
Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24(6):677–88.
Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. 2014;26(1):38–47.
Barcellos-Hoff MH, Lyden D, Wang TC. The evolution of the cancer niche during multistage carcinogenesis. Nat Rev Cancer. 2013;13(7):511–8.
Buchsbaum RJ, Oh SY. Breast cancer-associated fibroblasts: where we are and where we need to go. Cancers. 2016;8(2):19.
Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014;16:321–46.
Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res MCR. 2012;10(11):1403–18.
Liakou E, Mavrogonatou E, Pratsinis H, et al. Ionizing radiation-mediated premature senescence and paracrine interactions with cancer cells enhance the expression of syndecan 1 in human breast stromal fibroblasts: the role of TGF-β. Aging. 2016;8(8):1650–69.
Kang D, Jung SH, Lee GH, et al. Sulfated syndecan 1 is critical to preventing cellular senescence by modulating fibroblast growth factor receptor endocytosis. FASEB J Off Publ Feder Am Soc Exp Biol. 2020;34(8):10316–28.
Qian L, Tang Z, Yin S, et al. Fusion of dendritic cells and cancer-associated fibroblasts for activation of anti-tumor cytotoxic T lymphocytes. J Biomed Nanotechnol. 2018;14(10):1826–35.
Hugo HJ, Lebret S, Tomaskovic-Crook E, et al. Contribution of fibroblast and mast cell (afferent) and tumor (efferent) IL-6 effects within the tumor microenvironment. Cancer Microenviron Off J Int Cancer Microenviron Soc. 2012;5(1):83–93.
Zhao X, Rong L, Zhao X, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Investig. 2012;122(11):4094–104.
Allaoui R, Bergenfelz C, Mohlin S, et al. Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat Commun. 2016;7:13050.
Wang X, He J, Zhao X, Qi T, Zhang T, Kong C. Syndecan-1 suppresses epithelial-mesenchymal transition and migration in human oral cancer cells. Oncol Rep. 2018;39(4):1835–42.
Liu Z, Jin H, Yang S, et al. SDC1 knockdown induces epithelial-mesenchymal transition and invasion of gallbladder cancer cells via the ERK/Snail pathway. J Int Med Res. 2020;48(8):300060520947883.
Kumar-Singh A, Parniewska MM, Giotopoulou N, et al. Nuclear syndecan-1 regulates epithelial-mesenchymal plasticity in tumor cells. Biology. 2021;10(6):521.
Acknowledgements
The authors would like to thank the TCGA, GO, and METABRIC databases and the Department of Pathology of Peking Union Medical College Hospital for the availability of the data. The authors would also like to thank all of the patients who participated in this study.
Funding
This study was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-C&T-B-017). This study was supported by the National High Level Hospital Clinical Research Funding (2022-PUMCH-B-039).
Author information
Authors and Affiliations
Contributions
YZ collected and analyzed clinical and gene data. Experiments were performed by SMZ, ZLY, and DG. XYR, XC. YLX, YDZ, FM, SJS collected and evaluated the TNBC cohort. YZ and FYL conducted statistical analysis. QS supported quality management and directed the team. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study involving patients was approved by the Institutional Reviewer Board of the Peking Union Medical College Hospital. The patients provided individual written informed consent to participate in this study.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. SDC1 overexpression.
Additional file 2
: Table S2. List of primers for RT-qPCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhong, Y., Li, F., Zhang, S. et al. Syndecan-1 as an immunogene in Triple-negative breast cancer: regulation tumor-infiltrating lymphocyte in the tumor microenviroment and EMT by TGFb1/Smad pathway. Cancer Cell Int 23, 76 (2023). https://doi.org/10.1186/s12935-023-02917-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-02917-7