
Abstract
Despite the significant progress in decreasing the occurrence and mortality of hepatocellular carcinoma (HCC), it remains a public health issue worldwide on the basis of its late presentation and tumor recurrence. To date, apart from surgical interventions, such as surgical resection, liver transplantation and locoregional ablation, current standard antitumor protocols include conventional cytotoxic chemotherapy. However, due to the high chemoresistance nature, most current therapeutic agents show dismal outcomes for this refractory malignancy, leading to disease relapse. Nevertheless, the molecular mechanisms involved in chemotherapy resistance remain systematically ambiguous. Herein, HCC is hierarchically characterized by the formation of primitive cancer stem cells (CSCs), progression of epithelial-mesenchymal transition (EMT), unbalanced autophagy, delivery of extracellular vesicles (EVs), escape of immune surveillance, disruption of ferroptosis, alteration of the tumor microenvironment and multidrug resistance-related signaling pathways that mediate the multiplicity and complexity of chemoresistance. Of note, anecdotal evidence has corroborated that noncoding RNAs (ncRNAs) extensively participate in the critical physiological processes mentioned above. Therefore, understanding the detailed regulatory bases that underlie ncRNA-mediated chemoresistance is expected to yield novel insights into HCC treatment. In the present review, a comprehensive summary of the latest progress in the investigation of chemotherapy resistance concerning ncRNAs will be elucidated to promote tailored individual treatment for HCC patients.
Similar content being viewed by others

Background
As one of the most typical and fatal malignancies worldwide, hepatocellular carcinoma (HCC) accounts for nearly 90% of all liver cancers and originates from primary hepatocytes under carcinogenic factors such as alcohol-related cirrhosis, hepatitis B & C virus and exposure to diverse dietary toxins, which causes over 600,000 deaths annually [1]. In the last several decades, the incidence and mortality of HCC have doubled due to its heterogeneity, with an unfavorable 5-year survival rate of no more than 10% among patients [2]. Despite advancements in the diagnosis and treatment of HCC, curative treatments such as surgical removal, locoregional ablation or liver transplantation are options for a minority of newly diagnosed individuals with relatively favorable prognoses [3]. Unfortunately, the majority of HCC cohorts are asymptomatic, and patients tend to be diagnosed at an advanced stage to receive palliative therapies, including systematic chemotherapy, radiotherapy and transcatheter arterial chemoembolization (TACE). In particular, systematic antitumor agents have played an increasingly promising role in the management of HCC with the development of multiple kinase inhibitors. However, progress in the field of conventional chemotherapy has stalled owing in a large part to chemoresistance that leaves patients with ineffective therapeutic choices and an unsatisfactory prognosis.
Pioneering studies have demonstrated that chemotherapy resistance presents a major hurdle in the current health care system, which in turn contributes to the tumorigenesis and progression of various carcinomas. To enforce strict regulation for advanced HCC, sorafenib is the first oral targeted medication approved by the FDA, and its therapeutic effects are accomplished through the inhibition of several kinases and signaling pathways, including the VEGFR kinase, EGFR kinase, PDGFR kinase, MAPK, NF-κB and STAT3 signaling pathways [4]. Specifically, sorafenib exhibits antitumor activity against HCC by inhibiting angiogenesis, suppressing oncogenic gene mutations and inducing tumor cell apoptosis. In addition, other kinase inhibitors and immune checkpoint inhibitors, such as sunitinib, erlotinib, lenvatinib, regorafenib, nivolumab, pembrolizumab, ipilimumab and tremelimumab, were subsequently established individually or in combination to improve curative efficacy [5, 6]. However, the overall survival (OS) rate of advanced HCC patients remains low, and less than one-third of them benefit from chemotherapy. Drug resistance is apparent within six months of initiating the treatment regimen [7]. Thus, further research is necessary to improve the outcome of such a devastating disease.
Currently, growing attention has been focused on the annotation of specific molecular mechanisms underlying chemoresistance. Accordingly, cancer stem cells (CSCs), epithelial-mesenchymal transition (EMT), autophagy, extracellular vesicles (EVs), immune evasion, ferroptosis, tumor microenvironment (TME) and multidrug resistance (MDR) relevant signaling pathways are considered pivotal determinants for drug resistance in HCC [8,9,10,11,12]. Notably, high-throughput sequencing and bioinformatics analysis have revealed the significance of noncoding RNAs (ncRNAs) in the mentioned physiological processes in a broad spectrum of cancers [13,14,15,16]. As the majority of transcriptional precursor RNAs, over 70% of the whole genome is transcribed into ncRNAs and regulates residual genes by employing epigenetic modification, transcriptional regulation, posttranscriptional modification and targeted protein translation. Nevertheless, until now, the crosstalk between ncRNAs and the molecular bases of chemoresistance has been poorly understood in HCC. Furthermore, strategies aimed at reversing ncRNA-mediated refractoriness are currently underway and represent a promising future that converts traditional agents into potent killers of cancerous cells. The current review provides an update of accessible information regarding ncRNA-mediated molecular mechanisms of drug resistance with a remarkable impact on the effectiveness of HCC therapy.
The biogenesis and function of ncRNAs
ncRNAs refer to the prevalent clusters of RNA transcripts that cannot encode peptides or proteins. According to their classification, ncRNAs are divided into transfer RNAs (tRNAs), microRNAs (miRNAs), small interfering RNAs (siRNAs), circular RNAs (circRNAs), ribosomal RNAs (rRNAs), long noncoding RNAs (lncRNAs) and small nuclear RNAs (snRNAs). Alternatively, considered the “junk” of gene transcription, regulatory ncRNAs fall into three prevailing categories: miRNAs with a transcript length less than 200 nucleotides, lncRNAs with a transcript length exceeding 200 nucleotides and circRNAs with unique circular structures. Functionally, apart from directly operating on host gene expression, ncRNAs consistently tend to regulate cell processes in a coordinated network via internal interplay. Furthermore, as the central members, miRNAs have been demonstrated in numerous studies to be indispensably linked to lncRNAs, circRNAs and messenger RNAs (mRNAs) [17, 18]. Additionally, miRNAs and lncRNAs are biosynthesized via subsequent procedures with RNA polymerase II, and RNase III enzyme, ribonuclease III, among others. which is similar to the transcription process of mRNAs. Moreover, with stable molecular characteristics, circRNAs are mainly generated by the primary mRNA through a back splicing mechanism that is defined to reverse the downstream splicing site and connect to the upstream splicing site to form a closed circular molecule, which endows circRNAs with resistance to RNase R. Specifically, lncRNAs and circRNAs can act as competitive RNA or RNA sponges to bind miRNAs and restrict the regulation of mRNA targets. Nonetheless, the biogenesis and function of ncRNAs are complicated processes that require in-depth studies of regulatory details. Above all, given the distinct roles of ncRNAs in the regulation of HCC progression, we further describe the potential interaction between ncRNAs and chemoresistance-related features of HCC in the following sections.
The involvement of ncRNAs in liver cancer stem cells (LCSCs)
Cancer stem cells (CSCs) represent a small proportion of pluripotent cancer cells that have the capacity for self-renewal, infinite proliferation and tumorigenesis. Collectively, CD13, CD24, CD44, CD90, CD133 and EpCAM are well-accepted stemness markers used to distinguish CSCs from noncancer stem cells. Historically, the manifest characteristic of CSCs confers high chemoresistance to numerous malignancies. Therapy-induced CSC senescence allows for elevated efficacy of traditional agents and prolonged survival rates. Currently, compelling lines of evidence have confirmed the importance of liver cancer stem cells (LCSCs) in mediating the refractoriness of HCC. A brief description of the characteristics of LCSCs and their correlations with ncRNA-related resistance is introduced as follows.
Critical regulatory elements of LCSCs
Originally, LCSCs were derived from transformed liver progenitor cells, which differentiate into liver parenchymal cells, including hepatocytes and cholangiocytes. Mechanistically, Nanog, Oct4, SALL4 and Sox2 are the dominant genes modulating the stemness of LCSCs. For example, as an essential downstream effector of CD24-associated hepatocarcinogenesis, Nanog can maintain the pluripotency of LCSCs dependent on the STAT3 signaling pathway [19]. Consistently, in hepatic stem cell-like HCC cells, the upregulated transcription level of SALL4 is characterized by the activation of CD44 and EpCAM with a highly carcinogenic and metastatic nature, probably via the Wnt/β-catenin pathway [20]. In particular, the transcription factors Sox2 and Oct4 work in a synergistic network to maintain the self-renewal and stemness of pluripotent stem cells [21]. Recently, the TME has been reported to play an increasing role in preserving stem cell traits, which comprise a complicated system of cells other than cancer cells. Hypoxia is a representative feature of the TME, and hypoxia increases the transcription of ubiquitin-specific protease 22 (USP22) by activating hypoxia-inducible factor-1α (HIF-1α), a key component in stemness maintenance, thereby causing HIF-1α/USP22 positive feedback to repress TP53 expression and stabilize cancer stemness in HCC [22]. Moreover, an increase in stem cell-like properties is also induced by several cytokines, and the inflammatory factors IL6, IL-8, MCP-1, TGF-β, CCL2 and CXCL11 are positively correlated with LCSC markers [23,24,25]. Li et al. further demonstrated that IL6 plays an important role in the conversion of non-LCSCs to LCSCs via STAT3 signaling, which contributes to chemoresistance in HCC [26]. Similarly, CXCL11, a chemokine overexpressed in HCC, can facilitate cancer stemness by stimulating downstream CXCR3/ERK1/2 signaling [25]. Other components, such as immune cells, cancer-associated fibroblasts (CAFs) and the extracellular matrix, are also closely linked with stemness traits [27]. Ultimately, dysregulated changes in DNA methylation are implicated in liver cancer stemness. Noticeable upregulation of YTHDF2-dependent m6A methylation in the OCT4 mRNA 5′-UTR has been reported in Hep3B and Huh7 cells to support CSC properties [28]. Taken together, epigenetic modifications of stemness-mediated genes, dysregulated levels of relevant signaling pathways and alterations of TME-associated elements are considerable contributors to LCSCs.
Crosstalk between ncRNAs and LCSC-mediated chemoresistance in HCC
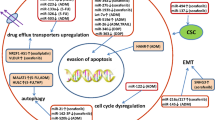
Plenty of research has been conducted exploring the role of ncRNAs in LCSC-induced chemoresistance. First, data from a range of investigations have suggested that liver cancer cells with CSCs are resistant to common therapies through several regulatory mechanisms, which consist of activated prosurvival signaling pathways, repressed apoptotic pathways, the release of TME-related factors, and enhanced drug efflux and metabolism [26, 29, 30]. Shortly thereafter, as another method of epigenetic modification, aberrant expression profiling of ncRNAs showed that ncRNAs can enhance stemness maintenance and confer chemoresistance to LCSCs. As illustrated in Fig. 1, ncRNAs can be classified into oncogenes or tumor suppressors depending on where they are expressed and what they regulate. MiR-33a is downregulated in Lgr5+ HCC-CSCs, and restoring its expression with ectopic mimics could sensitize HCC to doxorubicin by directly targeting the drug efflux-related protein ABCA1 [31]. In the HCC Huh7 cell line, miR-137 functions as a tumor suppressor to inhibit the formation of LSCS traits and reverse sorafenib resistance by degrading adenine nucleotide translocator 2 (ANT2) [32]. Regarding tumor promotors, miR-452 is responsible for stem-like characteristics and chemoresistance by inhibiting SOX7 via the Wnt/β-catenin signaling pathway in HCC [33]. Guan et al. [34] found that miR-130b overexpression not only facilitates CD133+ liver-initial cell growth but also increases resistance to a certain degree by silencing TP53INP1. Meanwhile, lncRNA-H19 is enriched in CD90+ liver cancer cells, and antisense H19 oligonucleotide transfection induces a remarkable increase in the ratio of MDR1 promoter methylation, which gives rise to doxorubicin accumulation [35, 36]. LncRNA-HOTAIR is of great significance in the malignant transformation from normal liver stem cells to LCSCs via miR-10b-induced EMT activation, which is another factor in drug resistance [37]. Simultaneously, lncRNA-CUDR is positively involved in the malignant transformation of LCSCs through the downstream cascade of HULC/β-catenin signaling [38], which sheds light on the reversal of chemoresistance in HCC. As a newly identified class of ncRNAs, circRNAs with covalently closed single-stranded RNA loop structures play a potential role in the regulation of CSCs. A microarray assay was performed to determine the circRNA profile in the SK‑Hep‑1 cell line, in which hsa_circ_0067531 was significantly decreased in CD90+ cells and restrained the sphere-forming ability of LCSCs via the PI3K/Akt pathway [39]. Moreover, circMEG3 inversely correlates with the expression of Cbf5 and shortens the telomere lifespan of LCSCs through METTL3 in a HULC-dependent manner [40]. In particular, as the most generalizable interrelation of the circRNA-miRNA axis, circ-FOXP1 is a driver for the carcinogenesis and progression of LCSCs that concurrently sponges miR-421 and miR-875-3p, leading to an increased level of the target gene SOX9 [41]. Likewise, circ-MALAT1 can act as both a miRNA sponge and an mRNA brake to stabilize the stemness of LCSCs, including miR-6887-3p and PAX5 mRNA [42]. Overall, current and ongoing studies have underscored the significance of ncRNAs in LCSC-induced chemoresistance and provided prospects for HCC therapy.

Aberrant expression profiling of ncRNAs enhance stemness maintenance and confer chemoresistance to LCSCs. Aberrant ncRNAs expression could influence the stemness and resistance of LCSCs probably via Wnt/β-catenin signaling, PI3K/Akt signaling, cell cycle arrest, cell proliferation and apoptosis
Significance of ncRNAs in EMT-related chemoresistance of HCC
Molecular bases of EMT-related chemoresistance in HCC
Epithelial-mesenchymal transition (EMT), as the name implies, represents a morphogenetic process whereby epithelial cells shed cell polarity and adhesion and acquire properties of migration and invasiveness to become mesenchymal cells. Abnormal EMT triggered by cytokines, growth factors or the TME is characterized by detachment of cell contact, changes in the cytoskeletal network and enhanced proteolytic activity, consequently leading to tumorigenesis and drug resistance. In terms of HCC, as a submember of the TGF-β family, BMP4 was reported by Shen et al. to accelerate mesenchymal marker-induced oxaliplatin resistance via MEK/ERK/ELK1 signaling activation [43]. As the critical downstream element of hepatocyte growth factor (HGF), MET is a substantial contributor to EMT-associated resistance in HCC mainly through autophagy and the canonical PI3K/Akt axis, whether in vivo or in vitro [44, 45]. More importantly, as a ubiquitous feature of solid tumors, hypoxia exhibits a positive correlation with EMT-induced cisplatin refractoriness via the PI3K/Akt/HIF-1α pathway, such that suppression of the aforementioned axis will undoubtedly enhance the therapeutic outcomes of hypoxic chemotherapy in HCC [46]. Intriguingly, a potential link between EMT and CSCs has been recently described: coexpression of Oct4/Nanog in LCSCs accelerates EMT via upregulation of the STAT3/Snail pathway [47], which reveals another regulatory base of EMT-involved resistance. As described above, EMT is closely related to HCC chemoresistance through different molecular machinery.
Interplay between ncRNAs and EMT-associated resistance in HCC
Over a dozen observations have revealed that ncRNA-mediated EMT may also regulate therapeutic refractoriness, thus paving a new avenue for the identification of modulated mechanisms and suggesting a latent paradigm for HCC treatment. The profiles of ncRNA expression are tissue- and cell type dependent-, as shown in Table 1. Concerning miRNAs, miR-212-3p is viewed as a tumor inhibitor in the Huh7 cell line, and its overexpression effectively counteracts paclitaxel resistance by targeting ZEB2, a key factor in EMT [48]. Analogously, miR-106a has been reported to act as a link between PDGF-D and Twist1, and restoration of miR-106a levels reverses EMT-mediated gemcitabine resistance in HCC [49]. In line with these findings, Wang et al. [50] emphasized the ability of miR-9 to enhance the sensitivity of HCC to cisplatin by regulating EIF5A2-related EMT. In contrast, a new cluster of miR-216a/217 was found to be positively associated with the EMT phenotype and sorafenib resistance through the TGF-β and PI3K/Akt signaling pathways by targeting PTEN and SMAD7, respectively [51]. Subsequently, with respect to lncRNAs, a previous study posited the positive effect of lincRNA-ROR on doxorubicin resistance by interacting with the mesenchymal cell marker, Twist1 [52]. In addition, lncRNA-H19 can either directly function by targeting the MDR1 gene [36] or indirectly sponge miR-675 [53] to aggravate EMT-mediated chemoresistance in HCC. Strikingly, lncRNA-POIR, a competing endogenous RNA (ceRNA), was determined to function as a molecular sponge to silence miR-182-5p [54], which was identified as a negative mediator of EMT-associated sorafenib insensitivity. Similarly, EMT-mediated sorafenib resistance may also be induced by lncRNA-SNHG3 via modulation of the miR-128/CD151 pathway in HCC [55]. Nevertheless, the importance of anti-EMT lncRNAs in the chemoresistance of HCC remains to be further explored. Current evidence shows that the antitumor lncRNA-AOC4P and lincRNA-p21 suppress EMT-induced HCC metastasis via degradation of vimentin [56] and inhibition of Notch signaling [57], suggesting a reasonable clue for the potential therapeutic target of HCC chemoresistance. Ultimately, the mechanisms underlying circRNAs and EMT-related resistance have been clarified in several studies [58, 59]. Specifically, circ_0003998 is elevated in doxorubicin-resistant HCC cells and serves as a ceRNA to downregulate miR-218-5p, which is negatively associated with the content of the EMT marker EIF5A2. Consistently, circFoxo3 is overexpressed in HCC samples and promotes adriamycin resistance via the miR-199a-5p/ABCC1 axis, contributing to EMT progression. Altogether, we summarize the basic intercorrelation between ncRNAs and EMT-involved resistance, and further efforts are required to probe deeper mechanisms and address refractoriness in HCC.
Participation of ncRNAs in autophagy-involved resistance of HCC
The molecular machinery of autophagy-mediated chemoresistance in HCC
Autophagy is characterized as an evolutionarily conserved procedure in which damaged proteins, aberrant organelles and excessive cytoplasmic components are degraded via lysosomal or vacuole-dependent pathways. On the basis of delivery modes, there are three main forms of autophagy: microautophagy, macroautophagy and chaperone-mediated autophagy. As the most studied form, macroautophagy usually occurs constitutively at a basal level and can be further induced by various metabolic stresses. For brevity, macroautophagy henceforth referred to as autophagy, involves the formation of autophagosomes in the cytoplasm, fusion with lysosomes or vacuoles and degradation of autolysosomes by proteases or nucleases, which is regulated via autophagic essential genes (ATGs, Beclin1, ULK1, LC3 complex, etc.) and signaling pathways to sustain the microenvironment. More importantly, dysfunctional autophagy may be a necessary regulator not only in the development of different cancers but also in determining the efficacy of antitumor chemotherapy. For HCC, on the one hand, basal autophagy is reported to remove damaged mitochondria and mutated cells to maintain genomic stability in the dysplastic phase of hepatocytes; on the other hand, dysregulated autophagy continues to sustain HCC cell survival when a solid tumor is established [60]. Considering the paradoxical role of autophagy in HCC, therapy choice varies depending on the density of hepatocytes. Currently, with a growing number of studies performed on the relationship between autophagy and chemoresistance, we coincidentally determined the positive effect of autophagy on preventing HCC cells from traditional therapies through multiple mechanisms. Accordingly, as a representative feature of the TME, oxidative stress may also act as a driver of autophagy-induced chemoresistance via the Egr-1/LC3 [61] and FOXO3a/Beclin-1 [62] axes. As another component of the TME, tumor-associated macrophages (TAMs) induce autophagy, and inhibition of ATG5 expression and LC3 conversion enhances the efficacy of oxaliplatin in TACE [63]. Interestingly, the stemness-related marker CD13 was reported by Wang et al. [12] to promote HCC resistance via the P38/Hsp27/CREB/ATG7 pathway, which uncovers the intercross between autophagy and LCSCs. In general, induced autophagy contributes to chemoresistance to a certain degree through diverse machinery.
Regulatory mechanism of ncRNAs in autophagy-mediated HCC resistance
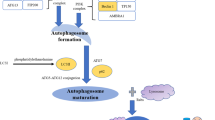
As described previously, induced autophagy exerts a protective effect from current chemotherapeutic agents in established HCC, whether in cells or samples. During the past few decades, growing enthusiasm has been geared toward investigating the characteristics and activities of ncRNAs in autophagy. According to the density of ncRNAs, they are classified as either promoting or inhibiting autophagy and are depicted in Fig. 2. Li et al. [64] found that the miR-26a/b level was downregulated and accompanied by induced autophagy in doxorubicin-resistant HCC cells. In turn, restoration of ULK1 expression inhibited ULK1-initiated autophagy and promoted cell apoptosis by doxorubicin. In particular, the tumor suppressor miR-375 plays a significant role in autophagy inhibition in that it can inhibit the formation of autophagosome-fused lysosomes and prolong the residence duration of sorafenib [65]. Mechanistically, miR-375 may also promote the sensitivity of HCC cells to sorafenib in a manner dependent on ATG14 regulation [66]. Moreover, insight into the role of miR-233 in doxorubicin-initiated autophagy uncovered the resistance mechanism by which it suppresses FOXO3-mediated autophagy in HCC [67]. Moreover, miR-101 is reported to block tumor progression in vivo [68] and prohibit the formation of autophagosomes by reducing ATG4D expression and mTOR signaling in vitro [69], showing that the presence of miR-101 attenuates autophagy and chemoresistance in HCC. Consistently, hsa-miR-30a and its subtype hsa-miR-30a-5p are inversely related to autophagy via the ATG5 and Beclin1 axes, thereby countering anoikis [70] and drug resistance [71] in HCC. Conversely, pro-autophagic miR-423-5p is upregulated in sorafenib-treated HCC cells and samples, and ectopic mimics facilitate autophagosome and vacuole formation, implying its potential to predict the response of patients to sorafenib [72]. Subsequently, lncRNAs have been shown to mediate the interplay between autophagy and resistance. Strikingly, the existing lncRNAs are all pro-autophagic in HCC. By comparison with normal SK-HEP-1 cells, Huang et al. found that the content of lncRNA-KCNQ1OT1 was statistically higher in cisplatin-resistant cells. The authors further determined that knockdown of lncRNA-KCNQ1OT1 overcomes autophagy-mediated resistance by leveraging the miR-338-3p/Beclin1 axis [73]. Upstream of hsa-miR-30a-5p, lncRNA-HNF1A-AS1 functions as an autophagy promoter to induce hepatocarcinogenesis via ATG5 regulation [74], which may be a promising therapeutic target in the reversal of drug resistance. Similarly, lncRNA-NEAT1 and lncRNA-HANR competitively bind with corresponding miR-204 and miR-29b, thus stimulating autophagy-initiated resistance via ATG3 [75] and ATG9A [76], respectively. Finally, lncRNA-MALAT1 induced by HIF-2α is upregulated in 5-FU-resistant Bel-7402 cells, and transfection of MALAT1 siRNA or miR-216b mimics leads to reduced LC3-II levels and increased p62 accumulation, implying the significance of the HIF-2α/MALAT1/miR-216b axis in autophagy-related chemoresistance of HCC [77]. However, to the best of our knowledge, there exists no literature on the role of circRNAs in autophagy-related resistance, which provides possibilities for subsequent studies. Aside from the aforementioned molecular machinery of ncRNAs in nonselective autophagy-mediated resistance of HCC, it is urgent to explore detailed mechanisms of ncRNA-mediated refractoriness underlying selective autophagy, such as mitophagy, reticulophagy, pexophagy and lipophagy.

Regulatory mechanism of ncRNAs in autophagy-mediated HCC resistance. ncRNAs may regulate the refractoriness with regard to the autophagy related genes, including ULK1, Beclin1, ATG3, ATG4D, ATG5, ATG7, ATH9A, ATG14, LC3 and p62
ncRNA-containing extracellular vesicles and chemoresistance in HCC
Extracellular vesicles (EVs) are defined as a heterogeneous category of membrane-bound vesicles that encompass lipids, proteins, mRNAs, miRNAs, lncRNAs, circRNAs and transcription factors and are classified as exosomes (diameter from 40 to 100 nm), microvesicles (MVs, diameter from 100 nm to 1 μm) and apoptotic bodies (diameter from 1 to 4 μm). EVs are generated and released upon cell activation or apoptosis with various components depending on the original cellular and the stimulus for their formation. As the main form of biological communication, EV delivery of ncRNAs in liver cancer has been thoroughly reviewed. Herein, as the third molecular regulatory tool, we summarize the functions of these exosomes and MV-containing biomolecules in the regulation of progression and therapeutic efficacy in HCC, and the potential machinery is clarified as follows in Table 2.
Specifically, the expression of exosomal ncRNAs is significantly associated with the development and chemoresistance of HCC. Accordingly, serum exosomes from HCC patients show a notably lower level of miR-9-3p. By bioinformatics analysis, the authors identified the target gene HBGF-5, through which miR-9-3p reduced cell viability and ERK1/2 signaling [78]. Meanwhile, in HCV-related HCC, activated mast cells can transfer exosomal miR-490 to cancer cells and restrain the EGFR/Akt/ERK1/2 pathway to shuttle HCC metastasis [79]. Furthermore, Dou et al. [80] demonstrated the loss of exosomal miR-320a in activated CAFs and the promotion of malignant phenotype transition via the ERK1/2 axis, establishing additional insight into the exploration of exosomes and the TME. Conspicuously, miR-122 is closely related to a superior prognosis of HCC, and exosome-mediated miR-122 transfer to HepG2 cells increases sensitivity to sorafenib by targeting CCNG1, ADAM10 and IGF1R [81]. Interestingly, as an important host risk factor for HCC, HCV RNA accumulation is promoted by miR-122 in hepatitis C patients. Decreased intrahepatic contents of miR-122 were detected in HCC patients, indicating a potential role of miR-122 during the development of HCC. Reportedly, a clinical trial demonstrated that the miR-122 antagonist Miravirsen was developed for the management of HCV, and phase I and II studies displayed its dose-dependent pharmacology with mild side effects [82], which is of great importance for the evaluation of anti-HCC therapy. Regarding protumor exosomal miRNAs, a recent study reported that exosome-containing miR-1247-3p from HCC cells could facilitate remote lung metastasis and conversion of CAFs through β1-integrin-NFκB signaling [83]. Similarly, a cluster of miR-21 and miR-10b secreted from exosomes was induced in the acidic milieu and promoted HCC cell proliferation and metastasis [84]. In addition, it is hypothesized that circulating miR-32-5p induces MDR in HCC via exosomes through the PTEN/PI3K/Akt axis [85], which is related to angiogenesis, proliferation, EMT and autophagy.
On the other hand, exosomal lncRNAs possess unique expression profiles characterizing the progression and therapeutic resistance of HCC. To be concrete, along with exosomal miR-21, lncRNA-ATB secreted from exosomes is positively related to the TNM stage and unfavorable prognosis of patients, which provides considerable value in the therapeutic prediction of HCC [86]. In parallel, serum levels of lncRNA-ENSG00000258332.1 and LincRNA-00635 [87] from exosomes are remarkably higher in HCC patients, which correlates with lymph node metastasis, portal vein tumor emboli, TNM stage and OS. Tash et al. [88] found that exosomal lncRNA-RP11-513I15.6 enhances the level of aggressiveness and metastasis in HCC via its competitive endogenous exosomal miR-1262/RAB11A network. Additionally, a previous study reported that CD90+ liver cancer cell-derived exosomes could facilitate angiogenesis in the TME, and lncRNA profiles revealed that lncRNA-H19 is enriched in exosomes and acts as a possible mediator of angiogenic effects [35]. For circRNAs, an increased level of exosomal circ-UHRF1 indicates unfavorable outcomes in patients with HCC. Moreover, Ke et al. [89] demonstrated the delivery of circ-UHRF1 via exosomes to NK cells and the induction of NK-cell exhaustion by sponging miR-499c-5p, leading to refractoriness to anti-PD1 immunotherapy.
Regarding MVs, several studies have elucidated that MV-mediated transfer of lncRNAs represents a distinct mechanism to modulate the TME, tumor proliferation and chemoresistance in HCC. Specifically, MV-containing lncRNA-TUC339 has been implicated in hepatocarcinogenesis via intercellular signaling mediators [90], which makes such extracellular lncRNA a promising marker for HCC. In addition, Patel et al. [91] reported the enrichment of lincRNA-ROR within MVs derived from HCC cells. Incubation of MV-containing lincRNA-ROR and sorafenib suppresses chemotherapy-induced cell apoptosis by interacting with TGF-β, which is also a critical characteristic of LCSCs.
In this chapter, we systematically summarize the implications of EV-containing ncRNAs in the progression and drug sensitivity of HCC. Nevertheless, further efforts are warranted to elucidate the molecular bases underlying the importance of circRNAs, especially EV-related circRNAs.
Intercorrelation between ncRNAs and immune evasion in HCC
Recently, immunotherapy has become a research hotspot in terms of investigations into immunotherapeutic agents that confer a notable prognostic benefit by altering the immune microenvironment to develop an effective response against several malignancies. Indeed, its combination with traditional agents may change the treatment landscape. However, as the main immune organ, the liver plays an overarching role in host defense and is characterized by a strong immunosuppressive microenvironment and high immune evasion, which creates a vital impediment to an efficacious immune response. Considering the density of immune cells, chemokines and cytokines, the immune microenvironment may elicit dual activating and suppressive roles in tumor progression or tumor eradication. Specifically, liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs), regulatory T cells (Tregs) [92], dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs) [93], natural killer cells (NKs) [94], immune checkpoints PD-1/PD-L1 [95], CTLA4 [96], lymphocyte activation gene 3 (LAG-3), glypican-3 (GPC-3), galectin 9 [97], immune-related cytokines [98] IL1, TNF-α, IFN-γ, IL-4, IL-5, IL-8 and IL10 are significant factors engaging in the maintenance of immune balance, which provides some hints for HCC immunotherapy. In this framework, a thorough understanding of the immune microenvironment and ncRNA-mediated immune evasion will assist to counterbalance immunosuppression in HCC treatment.
Biofunction of ncRNAs in HCC immunotherapy
Currently, numerous studies have highlighted the immunomodulatory effects of ncRNAs that are involved in immune cells, immune checkpoint molecules, chemokines, cytokines, immunosuppressive signaling pathways, etc. According to Fig. 3, preclinical studies have provided intuitive evidence that miRNAs participate in HCC immune escape from NK-cell activity. As a novel targeting miRNA of MICB, miR-889 is overexpressed in HCC cell lines and confers resistance to NK-mediated cytotoxicity [99], and a specific histone deacetylase inhibitor can enhance the susceptibility of HCC to NK-based immune strategies. Similar to miR-889, miR-561-5p is overexpressed in HCC tissue with high metastatic potential and accelerates pulmonary metastasis by regulating CX3CR1+ NKs via the CX3CL1/STAT3 signaling axis [100]. Comparatively, in HBV and HVC-related HCC, miR-152 is reported to be positively correlated with the degree of NK cytolysis against tumor cells, probably by facilitating the degradation of the immunotolerance molecule HLA-G [101]. Coincidently, Abdelrahman et al. [102] also found that miR-182, an immunoinhibitory miRNA, could counter the imbalance between the activating receptor NKG2D and the inhibitory receptor NKG2A, which ultimately augments NK lytic activity in HCC immunotherapy. As another immune cell, the efficacy of γδ T-cell-based immunotherapy was amplified by the forced overexpression of miR-382 [103], which directly inhibited the antiapoptotic member c-FLIP and triggered caspase-8 mediated apoptosis. In addition, the intercross between miRNAs and peptide GPC-3-related vaccine therapy is described as follows. As an oncofetal glycoprotein, GPC-3 is overexpressed and associated with a poor prognosis in HCC. A microarray test revealed that miR-527 is an upstream effector of GPC-3, and transfection of miR-527 suppressed the invasiveness of HCC [104]. Consistently, by targeting GPC-3, miR-520c-3p can play an inducible role in HCC cell apoptosis [105], therefore inhibiting proliferation and invasion. A recent functional screening also revealed a suppressive effect of miR-1271 on GPC-3 by binding to the 3′-UTR [106], which constitutes a promising avenue of anti-HCC therapy. Thus, the clarified significance of miRNAs in GPC-3 will pave a new way for the improvement of vaccine therapy. In addition, Zhang et al. [107] found a negative relationship between miR-4717 and PD-1 in that HBV-induced release of PD-1 was putatively abolished by miR-4717, which increased the production of TNF-α and IFN-γ and blocked transformation of hepatocarcinogenesis resulting from HBV infection. As a key signaling pathway regulating immune suppression, the STAT3 axis is responsible for the transcription of miR-146a [108]. Importantly, inhibition of miR-146a not only modifies the STAT3-associated cytokine profile but also attenuates HCC-induced NK dysfunction and enhances the antitumor activity of lymphocytes. More impressively, a liposome-based miR-34 mimic, MRX34, has been investigated in phase I clinical trial for the treatment of HCC patients. MiR-34 plays an important tumor-suppressive role by downregulating the immune checkpoint PD-L1 and multiple oncogenes, including CD44, MET, CDK4/6 and NOTCH. Preclinical studies have demonstrated the therapeutic effects of MRX34, which include reduced tumor volume and enhanced survival in orthotopic mouse models of HCC [109]. An update on MRX34 during the ASCO Annual Meeting 2016 clarified its manageable toxicity profile and evident antitumor activity in liver cancer. However, the clinical trial has been at a standstill due to several immune-associated severe adverse effects among the patients, which remains to be further evaluated and investigated in the field of miRNAs-related therapy.

Biofunction of ncRNAs in HCC immunotherapy. ncRNAs are involved in the immune evasion that compromises NKs, KCs, TAMs, CD4 + T, CD8 + T, γδ T cells, GPC-3, PD-1, PD-L1, CTLA4, chemokines and cytokines
In the context of lncRNAs, lncRNA-FTX [110] upregulates the M1/M2 KC ratio and inhibits the NAFLD conversion to HCC. However, oncogenic lncRNA-SNHG20 promotes the aforementioned progression by inducing M2 polarization of KCs via STAT6 activation [111]. A functional enrichment analysis evaluated the critical role of lncRNA-GIHCG in HCC progression with negative regulators in the infiltration of DCs, NKs, CD4+ and CD8+ T cells [112]. Moreover, another genome-wide association study showed that LINC01149 competitively sponges miR-128-3p and releases highly soluble MICA, thus inducing NK exhaustion and immune evasion [113]. Moreover, immunosuppressive TAMs are positively correlated with the expression of HMGA2, which is mediated by the lncRNA-CCAT1/miR-Let7b axis [114]. Induced TAMs may also further increase the level of lncRNA-H19 and trigger the downstream miR-193b/MAPK1 pathway to promote HCC aggressiveness [115]. In addition, as a typical immunogenicity marker, TIM3-mediated CD8+ T cell activity is determined by lncRNA-Tim3 [116] and lncRNA-NEAT1 [117]. LncRNA-TIM3 exacerbates CD8+ T-cell exhaustion to compromise antitumor immunity by binding to TIM3, and repression of lncRNA-NEAT1 amplifies the anticancer effect of CD8+ T-cells against HCC via the miR-155/Tim-3 axis. In terms of immune checkpoints, lncRNA-MIAT and lncRNA-MIR155HG are remarkably related to PD-1, PD-L1 and CTLA-4, which are engaged in the immune escape process of HCC [118, 119]. Next, lncRNA-PCED1B-AS1 is highly expressed in HCC cells and exosomal lncRNA-PCED1B-AS1 elevates PD-Ls-related immunosuppression by sponging hsa‑miR‑194‑5p [120]. In regard to cancer vaccines, by epigenetically activating the oncogene GPC-3 through PCAF recruitment [121], lncRNA-GPC3-AS1 was found to enhance HCC cell proliferation and migration. Of note, lncRNA-HOXA-AS2 promotes GPC-3-related proliferation and EMT in HCC via the miR-520c-3p/GPC3 interaction [122]. For ACT therapy, Wang et al. found that lncRNA-NNT-AS1 reduced the level of CD4+ TILs via activation of the TGF-β pathway in HCC [123].
Regarding circRNAs, several substantial studies have also provided comparable findings. CircARSP91 can enhance innate immune surveillance by strengthening NK cytotoxicity by upgrading ULBP1 in HCC immunity [124]. Likewise, circ_0007456 further assists in avoiding immune escape by magnifying NK-mediated immunomodulation through the miR-6852-3p/ICAM-1 axis [125]. As indicated before, Ke et al. [89] found an immunosuppressive effect of circ-UHRF1 in inducing NK exhaustion and inhibiting IFN-γ and TNF-α secretion, thus causing anti-PD-1 resistance in HCC via miR-449c-5p/TIM3, which reveals an interplay between exosomes and immune evasion through circRNAs. In addition, circTRIM33-12 exerts its antitumor role in activating NKG2D in γδ T cells, CD8+ T cells and NKs by guaranteeing TET1 from miR-191 [126]. In particular, circ_0064428 is reported to be inversely related to CD8+ TILs [127], which potentially reflects the sensitivity and specificity of ACT-based immunotherapy in HCC. Overall, data from initial immunotherapy trials indicate that the specificity of ncRNAs is of paramount importance.
Biological nexus between ncRNA and ferroptosis in HCC
Ferroptosis, a newly coined form of regulated cell death, is defined as an iron-dependent process that is characterized by the accumulation of lipid peroxides and reactive oxygen species (ROS) under oxidative conditions. As a nonapoptotic cell death process, ferroptosis shares different molecular mechanisms with apoptosis, which provides a new perspective for cancer therapeutics. Historically, traditional anticancer drugs have promoted cell death, likely via caspase-dependent apoptosis. However, tumoricidal efficacy is far from satisfactory due to apoptosis resistance, which occurs frequently in various cancer cell types. Currently, with the fast-growing investigations of ferroptosis in cancer, it is gradually recognized as an adaptive feature to effectively eliminate malignant cells, which may circumvent the limitations of HCC therapy.
Mechanistically, ferroptosis is triggered by iron accumulation and polyunsaturated fatty acid (PUFA) and cysteine metabolism. Multiple classic pathways and a series of molecules are important causative factors in ferroptosis regulation. Normally, intracellular iron maintains an exquisite balance via iron transport systems. Either decreased iron export or increased iron import will sensitize cancerous cells to oxidative stress. As a vital factor in ferroptosis induction, the labile iron level will be elevated with transferrin-mediated iron import or ferritinophagy which is mediated by NCOA4, a specific autophagy cargo that transfers ferritin to lysosomes for the release of free iron [128]. Cystine and glutamate are important amino acids for ferroptosis induction, both of which need to be exchanged across the cell membrane with the specific transporter system Xc−, a disulfide-linked heterodimer that consists of SLC3A2 and SLC7A11 [129]. Under the oxidative conditions of cancer cells, the exchange of cystine with glutamate ranks as the most upstream event in ferroptosis, which could be triggered by the inhibition of system Xc−. Reportedly, several genes such as GLS1, GLS2, p53 and BAP1, have been identified to regulate SLC7A11 [130, 131], thus influencing cystine uptake and ferroptosis. As the major intracellular antioxidant, glutathione (GSH) is generated from cysteine, glutamate and glycine under the catalysis of glutathione synthetase (GSS). Ferroptosis can be initiated with the depletion of GSH, which can be achieved either by the inhibition of GSH biosynthesis or by the blockage of cystine acquisition via system Xc−. Interestingly, the CSC marker CD44 is reported to stabilize system Xc−, suggesting a potential link between CSCs and ferroptosis that contributes to cancer treatment [132]. Subsequently, ferroptosis is accompanied by iron-dependent ROS accumulation and PUFA depletion. PUFAs are more susceptible to lipid peroxidation, which results in a variety of products that drive cells to ferroptotic death [133]. As a dominant downstream molecule, GPX4 combats lipid peroxides by reducing GSH to leave oxidized GSH as a byproduct, and silencing of GPX4 facilitates ferroptosis [134]. Incidentally, the Ras/Raf/MEK and p62-Keap1-Nrf2 pathways are reported to be decisive targets for ferroptosis in HCC [135, 136]. In addition, several posttranslational modifications of the mentioned genes also play considerable roles in ferroptosis, including methylation, acetylation, phosphorylation and ubiquitination [137]. Therefore, based on the above signatures, the following tactics could be established to promote ferroptosis in HCC.
In HCC, several studies have pinpointed the significance of ncRNAs in ferroptosis regulation. Accordingly, as an activation subunit of Nrf2, GABPB1 is downregulated by lncRNA GABPB1-AS1 at the translational level, leading to the suppression of PRDX5-mediated antioxidant capacity and elevated ferroptosis in HCC [138]. Liu et al. [139] demonstrated a ferritinophagy-related circRNA in which cIARS positively regulates sorafenib-mediated ferroptosis by preventing ALKBH5-induced autophagy inhibition. A microarray study revealed that circ0097009 is involved in the regulation of SLC7A11, a key mediator in ferroptosis, by binding to miR-1261 in HCC [140]. Based on the current research on the interrelation between ncRNAs and ferroptosis depicted in Fig. 4, we boldly speculate that ncRNAs could regulate HCC progression by mediating ferroptosis, thereby proposing a new molecular mechanism for chemoresistance reversal.

Biological interrelation between ncRNAs and ferroptosis in HCC. Phosphatidylethanolamines (PEs) containing arachidonic acid (AA) and adrenic acid (AdA) are domination substrates that are involved in oxidation and ferroptosis. With the help of fatty acid transport protein (FATP) and fatty acid translocase (FAT), AA and AdA are absorbed via cellular membrane. long-chain acyl-CoA synthetase member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are responsible for the esterification and incorporation of PUFAs. Iron-dependent arachidonate lipoxygenases (ALOXs) and Fenton chemistry participate in the synthesis of phospholipid hydroperoxides (PE-AA-OOH/PE-AdA-OOH). GPX4 combats with lipid peroxidation through degrading toxic PE-AA-OOH/PE-AdA-OOH to nontoxic phospholipid alcohols (PE-AA-OH/PE-AdA-OH)
Conclusions
In conclusion, we have summarized a substantial amount of available information generated by the current broadly active investigations in the field of ncRNA-related chemoresistance in HCC. Remarkably, a variety of these pathological characteristics are intertwined and are involved in the lack of an effective response to common therapies in HCC. This review aims to improve the understanding of the dynamic and complex processes of drug refractoriness, which is indispensable to define novel markers with considerable sensitivity and selectivity that will be useful to precisely predict prognosis. In this regard, the detection of ncRNAs in the biomodulation of the above features, in both fundamental and applied aspects, will prove essential in motivating future innovations for HCC treatment.
Availability of data and materials
Not applicable.
Abbreviations
- ncRNAs:
-
Noncoding RNAs
- HCC:
-
Hepatocellular carcinoma
- CSCs:
-
Cancer stem cells
- EMT:
-
Epithelial-mesenchymal transition
- EVs:
-
Extracellular vesicles
- TME:
-
Tumor microenvironment
- MDR:
-
Multidrug resistance
- TACE:
-
Transcatheter arterial chemoembolization
- OS:
-
Overall survival
- tRNAs:
-
Transfer RNAs
- miRNAs:
-
MicroRNAs
- siRNAs:
-
Small interfering RNAs
- circRNAs:
-
Circular RNAs
- rRNAs:
-
Ribosomal RNAs
- lncRNAs:
-
Long noncoding RNAs
- snRNAs:
-
Small nuclear RNAs
- mRNAs:
-
Messenger RNAs
- LCSCs:
-
Liver cancer stem cells
- USP22:
-
Ubiquitin-specific protease 22
- HIF-1α:
-
Hypoxia-inducible factor-1α
- CAFs:
-
Cancer-associated fibroblasts
- HGF:
-
Hepatocyte growth factor
- ceRNA:
-
Competing endogenous RNA
- TAMs:
-
Tumor-associated macrophages
- MVs:
-
Microvesicles; LSECs
- LSECs:
-
Liver sinusoidal endothelial cells
- PD-L1:
-
Programmed cell death ligand-1
- KCs:
-
Kupffer cells
- Tregs:
-
Regulatory T cells
- DCs:
-
Dendritic cells
- MDSCs:
-
Myeloid-derived suppressor cells
- NK:
-
Natural killer
- LAG-3:
-
Lymphocyte activation gene 3
- AOSC:
-
American Society of Clinical Oncology
- GPC-3:
-
Glypican-3
- ROS:
-
Reactive oxygen species
- PUFAs:
-
Polyunsaturated fatty acids
- GSH:
-
Glutathione
- GSS:
-
Glutathione synthetase
- Glu:
-
Glutamate
- Gly:
-
Glycine
- GCL:
-
Glutamate-cysteine ligase
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NAD+:
-
Nicotinamide adenine dinucleotide phosphate
- AA:
-
Arachidonic acid
- AdA:
-
Adrenic acid
- ACSL4:
-
Long-chain acyl-CoA synthetase member 4
- LPCAT3:
-
Lysophosphatidylcholine acyltransferase 3
- PE:
-
Phosphatidylethanolamines
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. Cancer J Clin. 2018;68(1):7–30.
Malato Y, Willenbring H. The MAP3K TAK1: a chock block to liver cancer formation. Hepatology. 2010;52(4):1506–9.
Sala M, Llovet JM, Vilana R, Bianchi L, Sole M, Ayuso C, Bru C, Bruix J. Barcelona clinic liver cancer G: Initial response to percutaneous ablation predicts survival in patients with hepatocellular carcinoma. Hepatology. 2004;40(6):1352–60.
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–109.
Kudo M. Immune checkpoint blockade in hepatocellular carcinoma. Liver Cancer. 2015;4(4):201–7.
Zhang B, Finn RS. Personalized clinical trials in hepatocellular carcinoma based on biomarker selection. Liver Cancer. 2016;5(3):221–32.
El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134(6):1752–63.
Herraez E, Lozano E, Macias RI, Vaquero J, Bujanda L, Banales JM, Marin JJ, Briz O. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology. 2013;58(3):1065–73.
Leung MS, Chan KK, Dai WJ, Wong CY, Au KY, Wong PY, Wong CC, Lee TK, Ng IO, Kao WJ, et al. Anti-tumour effects of PIM kinase inhibition on progression and chemoresistance of hepatocellular carcinoma. J Pathol. 2020;252(1):65–76.
Bhagyaraj E, Ahuja N, Kumar S, Tiwari D, Gupta S, Nanduri R, Gupta P. TGF-beta induced chemoresistance in liver cancer is modulated by xenobiotic nuclear receptor PXR. Cell Cycle. 2019;18(24):3589–602.
Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55(4):838–45.
Zhao Y, Wu H, Xing X, Ma Y, Ji S, Xu X, Zhao X, Wang S, Jiang W, Fang C, et al. CD13 induces autophagy to promote hepatocellular carcinoma cell chemoresistance through the P38/Hsp27/CREB/ATG7 pathway. J Pharmacol Exp Ther. 2020;374(3):512–20.
Luo Y, Zheng S, Wu Q, Wu J, Zhou R, Wang C, Wu Z, Rong X, Huang N, Sun L, et al. Long noncoding RNA (lncRNA) EIF3J-DT induces chemoresistance of gastric cancer via autophagy activation. Autophagy. 2021. https://doi.org/10.1080/15548627.2021.1901204.
Zhou Y, Hu Y, Yang M, Jat P, Li K, Lombardo Y, Xiong D, Coombes RC, Raguz S, Yague E. The miR-106b ~ 25 cluster promotes bypass of doxorubicin-induced senescence and increase in motility and invasion by targeting the E-cadherin transcriptional activator EP300. Cell Death Differ. 2014;21(3):462–74.
Gao W, Guo H, Niu M, Zheng X, Zhang Y, Xue X, Bo Y, Guan X, Li Z, Guo Y, et al. circPARD3 drives malignant progression and chemoresistance of laryngeal squamous cell carcinoma by inhibiting autophagy through the PRKCI-Akt-mTOR pathway. Mol Cancer. 2020;19(1):166.
Chaudhary AK, Mondal G, Kumar V, Kattel K, Mahato RI. Chemosensitization and inhibition of pancreatic cancer stem cell proliferation by overexpression of microRNA-205. Cancer Lett. 2017;402:1–8.
Shen XY, Zheng LL, Huang J, Kong HF, Chang YJ, Wang F, Xin H. CircTRNC18 inhibits trophoblast cell migration and epithelial-mesenchymal transition by regulating miR-762/Grhl2 pathway in pre-eclampsia. RNA Biol. 2019;16(11):1565–73.
Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB, Yin DD, Kong R, Xia R, Lu KH, Li JH, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92.
Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9(1):50–63.
Zeng SS, Yamashita T, Kondo M, Nio K, Hayashi T, Hara Y, Nomura Y, Yoshida M, Hayashi T, Oishi N, et al. The transcription factor SALL4 regulates stemness of EpCAM-positive hepatocellular carcinoma. J Hepatol. 2014;60(1):127–34.
Schmidt R, Plath K. The roles of the reprogramming factors Oct4, Sox2 and Klf4 in resetting the somatic cell epigenome during induced pluripotent stem cell generation. Genome Biol. 2012;13(10):251.
Ling S, Shan Q, Zhan Q, Ye Q, Liu P, Xu S, He X, Ma J, Xiang J, Jiang G, et al. USP22 promotes hypoxia-induced hepatocellular carcinoma stemness by a HIF1alpha/USP22 positive feedback loop upon TP53 inactivation. Gut. 2020;69(7):1322–34.
Jiang J, Ye F, Yang X, Zong C, Gao L, Yang Y, Zhao Q, Han Z, Wei L. Peri-tumor associated fibroblasts promote intrahepatic metastasis of hepatocellular carcinoma by recruiting cancer stem cells. Cancer Lett. 2017;404:19–28.
Liu ZH, Bai MJ, Li JQ, Yan SH, Ye XW, Bu JG, Chen CY. Correlation between inflammatory cytokines and liver cancer stem cell markers in DEN-induced liver cancer rats. Eur Rev Med Pharmacol Sci. 2021;25(2):710–21.
Zhang Y, Zhao W, Li S, Lv M, Yang X, Li M, Zhang Z. CXCL11 promotes self-renewal and tumorigenicity of alpha2delta1(+) liver tumor-initiating cells through CXCR3/ERK1/2 signaling. Cancer Lett. 2019;449:163–71.
Li Y, Chen G, Han Z, Cheng H, Qiao L, Li Y. Erratum: IL-6/STAT3 signaling contributes to sorafenib resistance in hepatocellular carcinoma through targeting cancer stem cells [Corrigendum]. OncoTargets Ther. 2020;13:12253–4.
Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, Chai S, Lin CH, Tsang SY, Ma S, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016;15(6):1175–89.
Zhang C, Huang S, Zhuang H, Ruan S, Zhou Z, Huang K, Ji F, Ma Z, Hou B, He X. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene. 2020;39(23):4507–18.
Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133 + HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27(12):1749–58.
Cheung ST, Cheung PF, Cheng CK, Wong NC, Fan ST. Granulin-epithelin precursor and ATP-dependent binding cassette (ABC)B5 regulate liver cancer cell chemoresistance. Gastroenterology. 2011;140(1):344–55.
Hou H, Kang Y, Li Y, Zeng Y, Ding G, Shang J. miR-33a expression sensitizes Lgr5 + HCC-CSCs to doxorubicin via ABCA1. Neoplasma. 2017;64(1):81–91.
Lu AQ, Lv B, Qiu F, Wang XY, Cao XH. Upregulation of miR-137 reverses sorafenib resistance and cancer-initiating cell phenotypes by degrading ANT2 in hepatocellular carcinoma. Oncol Rep. 2017;37(4):2071–8.
Zheng Z, Liu J, Yang Z, Wu L, Xie H, Jiang C, Lin B, Chen T, Xing C, Liu Z, et al. MicroRNA-452 promotes stem-like cells of hepatocellular carcinoma by inhibiting Sox7 involving Wnt/beta-catenin signaling pathway. Oncotarget. 2016;7(19):28000–12.
Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, Ng I, Man K, Wong N, To KF, et al. miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell. 2010;7(6):694–707.
Conigliaro A, Costa V, Lo Dico A, Saieva L, Buccheri S, Dieli F, Manno M, Raccosta S, Mancone C, Tripodi M, et al. CD90 + liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol Cancer. 2015;14:155.
Tsang WP, Kwok TT. Riboregulator H19 induction of MDR1-associated drug resistance in human hepatocellular carcinoma cells. Oncogene. 2007;26(33):4877–81.
Ye P, Wang T, Liu WH, Li XC, Tang LJ, Tian FZ. Enhancing HOTAIR/MiR-10b drives normal liver stem cells toward a tendency to malignant transformation through inducing epithelial- to-mesenchymal transition. Rejuvenation Res. 2015;18(4):332–40.
Gui X, Li H, Li T, Pu H, Lu D. Long noncoding RNA CUDR regulates HULC and beta-catenin to govern human liver stem cell malignant differentiation. Mol Ther J Am Soc Gene Ther. 2015;23(12):1843–53.
Zhang K, Che S, Su Z, Zheng S, Zhang H, Yang S, Li W, Liu J. CD90 promotes cell migration, viability and sphereforming ability of hepatocellular carcinoma cells. Int J Mol Med. 2018;41(2):946–54.
Jiang X, Xing L, Chen Y, Qin R, Song S, Lu Y, Xie S, Wang L, Pu H, Gui X, et al. CircMEG3 inhibits telomerase activity by reducing Cbf5 in human liver cancer stem cells. Mol Ther Nucleic Acids. 2021;23:310–23.
Wang W, Li Y, Li X, Liu B, Han S, Li X, Zhang B, Li J, Sun S. Circular RNA circ-FOXP1 induced by SOX9 promotes hepatocellular carcinoma progression via sponging miR-875-3p and miR-421. Biomed Pharmacother. 2020;121:109517.
Chen L, Kong R, Wu C, Wang S, Liu Z, Liu S, Li S, Chen T, Mao C, Liu S. Circ-MALAT1 functions as both an mRNA translation brake and a microRNA sponge to promote self-renewal of hepatocellular cancer stem cells. Adv Sci. 2020;7(4):1900949.
Ma J, Zeng S, Zhang Y, Deng G, Qu Y, Guo C, Yin L, Han Y, Cai C, Li Y, et al. BMP4 promotes oxaliplatin resistance by an induction of epithelial-mesenchymal transition via MEK1/ERK/ELK1 signaling in hepatocellular carcinoma. Cancer Lett. 2017;411:117–29.
Fu R, Jiang S, Li J, Chen H, Zhang X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med Oncol. 2020;37(4):24.
Huang X, Gan G, Wang X, Xu T, Xie W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy. 2019;15(7):1258–79.
Jiao M, Nan KJ. Activation of PI3 kinase/Akt/HIF-1alpha pathway contributes to hypoxia-induced epithelial-mesenchymal transition and chemoresistance in hepatocellular carcinoma. Int J Oncol. 2012;40(2):461–8.
Yin X, Zhang BH, Zheng SS, Gao DM, Qiu SJ, Wu WZ, Ren ZG. Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling. J Hematol Oncol. 2015;8:23.
Yang J, Cui R, Liu Y. MicroRNA-212-3p inhibits paclitaxel resistance through regulating epithelial-mesenchymal transition, migration and invasion by targeting ZEB2 in human hepatocellular carcinoma. Oncol Lett. 2020;20(4):23.
Wang R, Li Y, Hou Y, Yang Q, Chen S, Wang X, Wang Z, Yang Y, Chen C, Wang Z, et al. The PDGF-D/miR-106a/Twist1 pathway orchestrates epithelial-mesenchymal transition in gemcitabine resistance hepatoma cells. Oncotarget. 2015;6(9):7000–10.
Bao Y, Zhang Y, Lu Y, Guo H, Dong Z, Chen Q, Zhang X, Shen W, Chen W, Wang X. Overexpression of microRNA-9 enhances cisplatin sensitivity in hepatocellular carcinoma by regulating EIF5A2-mediated epithelial-mesenchymal transition. Int J Biol Sci. 2020;16(5):827–37.
Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58(2):629–41.
Zhang Y, Wu W, Sun Q, Ye L, Zhou D, Wang W. lincROR facilitates hepatocellular carcinoma resistance to doxorubicin by regulating TWIST1mediated epithelialmesenchymal transition. Mol Med Rep. 2021. https://doi.org/10.3892/mmr.2021.11979.
Xu Y, Liu Y, Li Z, Li H, Li X, Yan L, Mao J, Shen J, Chen W, Xue F. Long noncoding RNA H19 is involved in sorafenib resistance in hepatocellular carcinoma by upregulating miR675. Oncol Rep. 2020;44(1):165–73.
Chen BW, Zhou Y, Wei T, Wen L, Zhang YB, Shen SC, Zhang J, Ma T, Chen W, Ni L, et al. lncRNA-POIR promotes epithelial-mesenchymal transition and suppresses sorafenib sensitivity simultaneously in hepatocellular carcinoma by sponging miR-182-5p. J Cell Biochem. 2021;122(1):130–42.
Zhang PF, Wang F, Wu J, Wu Y, Huang W, Liu D, Huang XY, Zhang XM, Ke AW. LncRNA SNHG3 induces EMT and sorafenib resistance by modulating the miR-128/CD151 pathway in hepatocellular carcinoma. J Cell Physiol. 2019;234(3):2788–94.
Wang TH, Lin YS, Chen Y, Yeh CT, Huang YL, Hsieh TH, Shieh TM, Hsueh C, Chen TC. Long non-coding RNA AOC4P suppresses hepatocellular carcinoma metastasis by enhancing vimentin degradation and inhibiting epithelial-mesenchymal transition. Oncotarget. 2015;6(27):23342–57.
Jia M, Jiang L, Wang YD, Huang JZ, Yu M, Xue HZ. lincRNA-p21 inhibits invasion and metastasis of hepatocellular carcinoma through Notch signaling-induced epithelial-mesenchymal transition. Hepatol Res. 2016;46(11):1137–44.
Li X, He J, Ren X, Zhao H, Zhao H. Circ_0003998 enhances doxorubicin resistance in hepatocellular carcinoma by regulating miR-218-5p/EIF5A2 pathway. Diagn Pathol. 2020;15(1):141.
Huang W, Huang F, Feng C. CircFoxo3 promotes adriamycin resistance through regulation of miR-199a-5p/ATP binding cassette subfamily C member 1 axis in hepatocellular carcinoma. OncoTargets Ther. 2020;13:5113–22.
Sun K, Guo XL, Zhao QD, Jing YY, Kou XR, Xie XQ, Zhou Y, Cai N, Gao L, Zhao X, et al. Paradoxical role of autophagy in the dysplastic and tumor-forming stages of hepatocarcinoma development in rats. Cell Death Dis. 2013;4:e501.
Peng WX, Xiong EM, Ge L, Wan YY, Zhang CL, Du FY, Xu M, Bhat RA, Jin J, Gong AH. Egr-1 promotes hypoxia-induced autophagy to enhance chemo-resistance of hepatocellular carcinoma cells. Exp Cell Res. 2016;340(1):62–70.
Liang C, Dong Z, Cai X, Shen J, Xu Y, Zhang M, Li H, Yu W, Chen W. Hypoxia induces sorafenib resistance mediated by autophagy via activating FOXO3a in hepatocellular carcinoma. Cell Death Dis. 2020;11(11):1017.
Fu XT, Song K, Zhou J, Shi YH, Liu WR, Shi GM, Gao Q, Wang XY, Ding ZB, Fan J. Tumor-associated macrophages modulate resistance to oxaliplatin via inducing autophagy in hepatocellular carcinoma. Cancer Cell Int. 2019;19:71.
Jin F, Wang Y, Li M, Zhu Y, Liang H, Wang C, Wang F, Zhang CY, Zen K, Li L. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017;8(1):e2540.
Zhao P, Li M, Wang Y, Chen Y, He C, Zhang X, Yang T, Lu Y, You J, Lee RJ, et al. Enhancing anti-tumor efficiency in hepatocellular carcinoma through the autophagy inhibition by miR-375/sorafenib in lipid-coated calcium carbonate nanoparticles. Acta Biomater. 2018;72:248–55.
Yang S, Wang M, Yang L, Li Y, Ma Y, Peng X, Li X, Li B, Jin H, Li H. MicroRNA-375 targets ATG14 to inhibit autophagy and sensitize hepatocellular carcinoma cells to sorafenib. OncoTargets Ther. 2020;13:3557–70.
Zhou Y, Chen E, Tang Y, Mao J, Shen J, Zheng X, Xie S, Zhang S, Wu Y, Liu H, et al. miR-223 overexpression inhibits doxorubicin-induced autophagy by targeting FOXO3a and reverses chemoresistance in hepatocellular carcinoma cells. Cell Death Dis. 2019;10(11):843.
Zheng F, Liao YJ, Cai MY, Liu TH, Chen SP, Wu PH, Wu L, Bian XW, Guan XY, Zeng YX, et al. Systemic delivery of microRNA-101 potently inhibits hepatocellular carcinoma in vivo by repressing multiple targets. PLoS Genet. 2015;11(2):e1004873.
Xu Y, An Y, Wang Y, Zhang C, Zhang H, Huang C, Jiang H, Wang X, Li X. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep. 2013;29(5):2019–24.
Fu XT, Shi YH, Zhou J, Peng YF, Liu WR, Shi GM, Gao Q, Wang XY, Song K, Fan J, et al. MicroRNA-30a suppresses autophagy-mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett. 2018;412:108–17.
Chen MY, Yadav VK, Chu YC, Ong JR, Huang TY, Lee KF, Lee KH, Yeh CT, Lee WH. Hydroxychloroquine (HCQ) modulates autophagy and oxidative dna damage stress in hepatocellular carcinoma to overcome sorafenib resistance via TLR9/SOD1/hsa-miR-30a-5p/Beclin-1 axis. Cancers. 2021. https://doi.org/10.3390/cancers13133227.
Stiuso P, Potenza N, Lombardi A, Ferrandino I, Monaco A, Zappavigna S, Vanacore D, Mosca N, Castiello F, Porto S, et al. MicroRNA-423-5p promotes autophagy in cancer cells and is increased in serum from hepatocarcinoma patients treated with sorafenib. Mol Ther Nucleic Acids. 2015;4:e233.
Zhong W, Dai Q, Huang Q. Effect of lncRNA KCNQ1OT1 on autophagy and drug resistance of hepatocellular carcinoma cells by targeting miR-338-3. Cell Mol Biol. 2020;66(3):191–6.
Liu Z, Wei X, Zhang A, Li C, Bai J, Dong J. Long non-coding RNA HNF1A-AS1 functioned as an oncogene and autophagy promoter in hepatocellular carcinoma through sponging hsa-miR-30b-5p. Biochem Biophys Res Commun. 2016;473(4):1268–75.
Li X, Zhou Y, Yang L, Ma Y, Peng X, Yang S, Li H, Liu J. LncRNA NEAT1 promotes autophagy via regulating miR-204/ATG3 and enhanced cell resistance to sorafenib in hepatocellular carcinoma. J Cell Physiol. 2020;235(4):3402–13.
Shi Y, Yang X, Xue X, Sun D, Cai P, Song Q, Zhang B, Qin L. HANR enhances autophagy-associated sorafenib resistance through miR-29b/ATG9A axis in hepatocellular carcinoma. OncoTargets Ther. 2020;13:2127–37.
Yuan P, Cao W, Zang Q, Li G, Guo X, Fan J. The HIF-2alpha-MALAT1-miR-216b axis regulates multi-drug resistance of hepatocellular carcinoma cells via modulating autophagy. Biochem Biophys Res Commun. 2016;478(3):1067–73.
Tang J, Li Y, Liu K, Zhu Q, Yang WH, Xiong LK, Guo DL. Exosomal miR-9-3p suppresses HBGF-5 expression and is a functional biomarker in hepatocellular carcinoma. Minerva Med. 2018;109(1):15–23.
Xiong L, Zhen S, Yu Q, Gong Z. HCV-E2 inhibits hepatocellular carcinoma metastasis by stimulating mast cells to secrete exosomal shuttle microRNAs. Oncol Lett. 2017;14(2):2141–6.
Zhang Z, Li X, Sun W, Yue S, Yang J, Li J, Ma B, Wang J, Yang X, Pu M, et al. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017;397:33–42.
Lou G, Song X, Yang F, Wu S, Wang J, Chen Z, Liu Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J Hematol Oncol. 2015;8:122.
van der Ree MH, van der Meer AJ, de Bruijne J, Maan R, van Vliet A, Welzel TM, Zeuzem S, Lawitz EJ, Rodriguez-Torres M, Kupcova V, et al. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antiviral Res. 2014;111:53–9.
Fang T, Lv H, Lv G, Li T, Wang C, Han Q, Yu L, Su B, Guo L, Huang S, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9(1):191.
Tian XP, Wang CY, Jin XH, Li M, Wang FW, Huang WJ, Yun JP, Xu RH, Cai QQ, Xie D. Acidic microenvironment up-regulates exosomal miR-21 and miR-10b in early-stage hepatocellular carcinoma to promote cancer cell proliferation and metastasis. Theranostics. 2019;9(7):1965–79.
Fu X, Liu M, Qu S, Ma J, Zhang Y, Shi T, Wen H, Yang Y, Wang S, Wang J, et al. Exosomal microRNA-32-5p induces multidrug resistance in hepatocellular carcinoma via the PI3K/Akt pathway. J Exp Clin Cancer Res. 2018;37(1):52.
Lee YR, Kim G, Tak WY, Jang SY, Kweon YO, Park JG, Lee HW, Han YS, Chun JM, Park SY, et al. Circulating exosomal noncoding RNAs as prognostic biomarkers in human hepatocellular carcinoma. Int J Cancer. 2019;144(6):1444–52.
Xu H, Chen Y, Dong X, Wang X. Serum exosomal long noncoding RNAs ENSG00000258332.1 and LINC00635 for the diagnosis and prognosis of hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev. 2018;27(6):710–6.
Abd El Gwad A, Matboli M, El-Tawdi A, Habib EK, Shehata H, Ibrahim D, Tash F. Role of exosomal competing endogenous RNA in patients with hepatocellular carcinoma. J Cell Biochem. 2018;119(10):8600–10.
Zhang PF, Gao C, Huang XY, Lu JC, Guo XJ, Shi GM, Cai JB, Ke AW. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19(1):110.
Kogure T, Yan IK, Lin WL, Patel T. Extracellular vesicle-mediated transfer of a novel long noncoding RNA TUC339: a mechanism of intercellular signaling in human hepatocellular cancer. Genes Cancer. 2013;4(7–8):261–72.
Takahashi K, Yan IK, Kogure T, Haga H, Patel T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio. 2014;4:458–67.
Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65(6):2457–64.
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–43.
Nan J, Xing YF, Hu B, Tang JX, Dong HM, He YM, Ruan DY, Ye QJ, Cai JR, Ma XK, et al. Endoplasmic reticulum stress induced LOX-1(+) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. 2018;154(1):144–55.
Kudo M. Scientific rationale for combined immunotherapy with PD-1/PD-L1 antibodies and VEGF inhibitors in advanced hepatocellular carcinoma. Cancers. 2020. https://doi.org/10.3390/cancers12051089.
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–65.
Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, Liu J, Shi L, Liu C, Wang G, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342–51.
Nishida N, Kudo M. Immunological microenvironment of hepatocellular carcinoma and its clinical implication. Oncology. 2017;92(Suppl 1):40–9.
Xie H, Zhang Q, Zhou H, Zhou J, Zhang J, Jiang Y, Wang J, Meng X, Zeng L, Jiang X. microRNA-889 is downregulated by histone deacetylase inhibitors and confers resistance to natural killer cytotoxicity in hepatocellular carcinoma cells. Cytotechnology. 2018;70(2):513–21.
Chen EB, Zhou ZJ, Xiao K, Zhu GQ, Yang Y, Wang B, Zhou SL, Chen Q, Yin D, Wang Z, et al. The miR-561-5p/CX3CL1 signaling axis regulates pulmonary metastasis in hepatocellular carcinoma involving CX3CR1(+) natural killer cells infiltration. Theranostics. 2019;9(16):4779–94.
Bian X, Si Y, Zhang M, Wei R, Yang X, Ren H, Zheng G, Wang C, Zhang Y. Down-expression of miR-152 lead to impaired anti-tumor effect of NK via upregulation of HLA-G. Tumour Biol. 2016;37(3):3749–56.
Abdelrahman MM, Fawzy IO, Bassiouni AA, Gomaa AI, Esmat G, Waked I, Abdelaziz AI. Enhancing NK cell cytotoxicity by miR-182 in hepatocellular carcinoma. Hum Immunol. 2016;77(8):667–73.
Chen Z, Zheng Z, Feng L, Huo Z, Huang L, Fu M, Chen Q, Ke Y, Yang J, Hou B. Overexpression of miR-382 sensitizes hepatocellular carcinoma cells to gammadelta T cells by inhibiting the expression of c-FLIP. Mol Ther Oncolytics. 2020;18:467–75.
Nomura K, Kitanaka A, Iwama H, Tani J, Nomura T, Nakahara M, Ohura K, Tadokoro T, Fujita K, Mimura S, et al. Association between microRNA-527 and glypican-3 in hepatocellular carcinoma. Oncol Lett. 2021;21(3):229.
Miao HL, Lei CJ, Qiu ZD, Liu ZK, Li R, Bao ST, Li MY. MicroRNA-520c-3p inhibits hepatocellular carcinoma cell proliferation and invasion through induction of cell apoptosis by targeting glypican-3. Hepatol Res. 2014;44(3):338–48.
Maurel M, Jalvy S, Ladeiro Y, Combe C, Vachet L, Sagliocco F, Bioulac-Sage P, Pitard V, Jacquemin-Sablon H, Zucman-Rossi J, et al. A functional screening identifies five microRNAs controlling glypican-3: role of miR-1271 down-regulation in hepatocellular carcinoma. Hepatology. 2013;57(1):195–204.
Zhang G, Li N, Li Z, Zhu Q, Li F, Yang C, Han Q, Lv Y, Zhou Z, Liu Z. microRNA-4717 differentially interacts with its polymorphic target in the PD1 3′ untranslated region: a mechanism for regulating PD-1 expression and function in HBV-associated liver diseases. Oncotarget. 2015;6(22):18933–44.
Sun X, Zhang J, Hou Z, Han Q, Zhang C, Tian Z. miR-146a is directly regulated by STAT3 in human hepatocellular carcinoma cells and involved in anti-tumor immune suppression. Cell Cycle. 2015;14(2):243–52.
Bader AG. miR-34—a microRNA replacement therapy is headed to the clinic. Front Genet. 2012;3:120.
Wu H, Zhong Z, Wang A, Yuan C, Ning K, Hu H, Wang C, Yin X. LncRNA FTX represses the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma via regulating the M1/M2 polarization of Kupffer cells. Cancer Cell Int. 2020;20:266.
Wang B, Li X, Hu W, Zhou Y, Din Y. Silencing of lncRNA SNHG20 delays the progression of nonalcoholic fatty liver disease to hepatocellular carcinoma via regulating liver Kupffer cells polarization. IUBMB Life. 2019;71(12):1952–61.
Xiao S, Huang S, Yang J. Overexpression of GIHCG is associated with a poor prognosis and immune infiltration in hepatocellular carcinoma. OncoTargets Ther. 2020;13:11607–19.
Zhong R, Tian J, Fu M, Ma S, Liu L, Li J, Shen N, Ke J, Yang Y, Gong Y, et al. LINC01149 variant modulates MICA expression that facilitates hepatitis B virus spontaneous recovery but increases hepatocellular carcinoma risk. Oncogene. 2020;39(9):1944–56.
Deng L, Huang S, Chen B, Tang Y, Huang F, Li D, Tang D. Tumor-linked macrophages promote HCC development by mediating the CCAT1/Let-7b/HMGA2 signaling pathway. OncoTargets Ther. 2020;13:12829–43.
Ye Y, Guo J, Xiao P, Ning J, Zhang R, Liu P, Yu W, Xu L, Zhao Y, Yu J. Macrophages-induced long noncoding RNA H19 up-regulation triggers and activates the miR-193b/MAPK1 axis and promotes cell aggressiveness in hepatocellular carcinoma. Cancer Lett. 2020;469:310–22.
Ji J, Yin Y, Ju H, Xu X, Liu W, Fu Q, Hu J, Zhang X, Sun B. Long non-coding RNA Lnc-Tim3 exacerbates CD8 T cell exhaustion via binding to Tim-3 and inducing nuclear translocation of Bat3 in HCC. Cell Death Dis. 2018;9(5):478.
Yan K, Fu Y, Zhu N, Wang Z, Hong JL, Li Y, Li WJ, Zhang HB, Song JH. Repression of lncRNA NEAT1 enhances the antitumor activity of CD8(+)T cells against hepatocellular carcinoma via regulating miR-155/Tim-3. Int J Biochem Cell Biol. 2019;110:1–8.
Peng L, Chen Z, Chen Y, Wang X, Tang N. MIR155HG is a prognostic biomarker and associated with immune infiltration and immune checkpoint molecules expression in multiple cancers. Cancer Med. 2019;8(17):7161–73.
Peng L, Chen Y, Ou Q, Wang X, Tang N. LncRNA MIAT correlates with immune infiltrates and drug reactions in hepatocellular carcinoma. Int Immunopharmacol. 2020;89(Pt A):107071.
Fan F, Chen K, Lu X, Li A, Liu C, Wu B. Dual targeting of PD-L1 and PD-L2 by PCED1B-AS1 via sponging hsa-miR-194-5p induces immunosuppression in hepatocellular carcinoma. Hepatol Int. 2021;15(2):444–58.
Zhu XT, Yuan JH, Zhu TT, Li YY, Cheng XY. Long noncoding RNA glypican 3 (GPC3) antisense transcript 1 promotes hepatocellular carcinoma progression via epigenetically activating GPC3. FEBS J. 2016;283(20):3739–54.
Zhang Y, Xu J, Zhang S, An J, Zhang J, Huang J, Jin Y. HOXA-AS2 promotes proliferation and induces epithelial-mesenchymal transition via the miR-520c-3p/GPC3 axis in hepatocellular carcinoma. Cell Physiol Biochem. 2018;50(6):2124–38.
Wang Y, Yang L, Dong X, Yang X, Zhang X, Liu Z, Zhao X, Wen T. Overexpression of NNT-AS1 activates TGF-beta signaling to decrease tumor CD4 lymphocyte infiltration in hepatocellular carcinoma. Biomed Res Int. 2020;2020:8216541.
Ma Y, Zhang C, Zhang B, Yu H, Yu Q. circRNA of AR-suppressed PABPC1 91 bp enhances the cytotoxicity of natural killer cells against hepatocellular carcinoma via upregulating UL16 binding protein 1. Oncol Lett. 2019;17(1):388–97.
Shi M, Li ZY, Zhang LM, Wu XY, Xiang SH, Wang YG, Zhang YQ. Hsa_circ_0007456 regulates the natural killer cell-mediated cytotoxicity toward hepatocellular carcinoma via the miR-6852-3p/ICAM-1 axis. Cell Death Dis. 2021;12(1):94.
Zhang PF, Wei CY, Huang XY, Peng R, Yang X, Lu JC, Zhang C, Gao C, Cai JB, Gao PT, et al. Circular RNA circTRIM33-12 acts as the sponge of MicroRNA-191 to suppress hepatocellular carcinoma progression. Mol Cancer. 2019;18(1):105.
Weng Q, Chen M, Li M, Zheng YF, Shao G, Fan W, Xu XM, Ji J. Global microarray profiling identified hsa_circ_0064428 as a potential immune-associated prognosis biomarker for hepatocellular carcinoma. J Med Genet. 2019;56(1):32–8.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–32.
Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, Pearson HB, Fisher OM, Read M, Guerra GR, et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. 2017;8:14844.
Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30(8):918–30.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–92.
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113(34):E4966–75.
Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, Rodriguez Martinez M, Lopez G, Mattioli M, Realubit R, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162(2):441–51.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–84.
Nie J, Lin B, Zhou M, Wu L, Zheng T. Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2018;144(12):2329–37.
Wei X, Yi X, Zhu XH, Jiang DS. Posttranslational modifications in ferroptosis. Oxid Med Cell Longev. 2020;2020:8832043.
Qi W, Li Z, Xia L, Dai J, Zhang Q, Wu C, Xu S. LncRNA GABPB1-AS1 and GABPB1 regulate oxidative stress during erastin-induced ferroptosis in HepG2 hepatocellular carcinoma cells. Sci Rep. 2019;9(1):16185.
Liu Z, Wang Q, Wang X, Xu Z, Wei X, Li J. Circular RNA cIARS regulates ferroptosis in HCC cells through interacting with RNA binding protein ALKBH5. Cell Death Discov. 2020;6:72.
Lyu N, Zeng Y, Kong Y, Chen Q, Deng H, Ou S, Bai Y, Tang H, Wang X, Zhao M. Ferroptosis is involved in the progression of hepatocellular carcinoma through the circ0097009/miR-1261/SLC7A11 axis. Ann Transl Med. 2021;9(8):675.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82072685).
Author information
Authors and Affiliations
Contributions
Conception and design: QH and GC. Compilation of literature: QH, PG, ZB, HY and JY. Article writing and editing: QH and PG. Figure and table organization: QH, PG and GC. Supervision: YW. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, Q., Guo, P., Bo, Z. et al. Noncoding RNA-mediated molecular bases of chemotherapy resistance in hepatocellular carcinoma. Cancer Cell Int 22, 249 (2022). https://doi.org/10.1186/s12935-022-02643-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-022-02643-6