
Abstract
At present, more than one cell death pathways have been found, one of which is ferroptosis. Ferroptosis was discovered in 2012 and described as an iron-dependent and lipid peroxidation-driven regulated cell death pathway. In the past few years, ferroptosis has been shown to induce tumor cell death, providing new ideas for tumor treatment. In this article, we summarize the latest advances in ferroptosis-induced tumor therapy at the intersection of tumor biology, molecular biology, redox biology, and materials chemistry. First, we state the characteristics of ferroptosis in cells, then introduce the key molecular mechanism of ferroptosis, and describes the relationship between ferroptosis and oxidative stress signaling pathways. Finally, we focused on several types of ferroptosis inducers discovered by scholars, and the application of ferroptosis in systemic chemotherapy, radiotherapy, immunotherapy and nanomedicine, in the hope that ferroptosis can exert its potential in the treatment of tumors.
Similar content being viewed by others


Introduction
Cancer is the second leading cause of death in the world, causing approximately 10 million deaths each year. The treatment of cancer is currently one of the most researched topics. Eliminating cancer cells in the human body without affecting other healthy cells is the main concept of cancer treatment. Since the discovery of regulated cell death in the 1960s, people have realized that cell death is controllable and diverse (Fig. 1) [1]. RCD refers to the regulation of a series of specialized molecular mechanisms in pharmacology, molecular biology and genetics in the process of cell death [2, 3]. Caspase-dependent apoptosis has long been considered the only form of RCD [4], making anticancer drugs induce apoptosis of cells as one of the most important methods to kill cancer cells. However, in recent years, it has been discovered that cancer cells are resistant to drugs and have certain resistance to apoptosis [5, 6]. Therefore, targeting other forms of non-apoptotic cell death has become a new treatment approach to eliminate cancer cells and reduce the drug resistance of cancer cells.

The progression of cell death
In this series of non-apoptotic forms of RCD, ferroptosis was discovered in 2012 and described as a lipid peroxidation driven and iron-dependent RCD [7]. Although the term "ferroptosis" is a compound created after screening of small molecules that can inhibit the growth of RAS mutant cancer cells, in the past few years, ferroptosis has been shown to be closely related to the death of cancer cells [7, 8]. For example, the most classic p53 cancer suppressor gene can inhibit the expression of cystine/glutamate antiporter, thereby regulating ferroptosis [9]. In particular, cancer cells that are resistant to conventional therapies or have a high tendency to metastasize may be particularly susceptible to ferroptosis [10]. In addition, ferroptosis has recently been shown to be related to cancer immunotherapy, in which T cells and INFγ promote the sensitivity of cancer cells to ferroptosis [11]. In recent years, with the development of nanotechnology, the application of nanomedicine in cancer treatment has increased accordingly. Due to the unique physicochemical (high targeting efficiency, strong water solubility, low side effects) and some special properties (e.g., magnetic property, photothermal effect, electrochemical property, etc.), nanomaterials can kill cancer cells efficiently. And it is also found that nanomaterials can induce ferroptosis [12]. In this review, we first introduce the regulatory mechanism of ferroptosis and various death inducers, then elaborate on the current application status and possibilities of ferroptosis in cancer treatments, and finally express the expectations for its potential for clinical transformation.
The basic characteristics of ferroptosis
The occurrence of ferroptosis is always accompanied by a series of variations in cellular, molecular, and genetic levels, which shares similarities and differences with other cell death modalities. Therefore, it is necessary to summarize the characteristics of ferroptosis and distinguish it from other cell death phenotypes. The confirmation of the ferroptosis phenotype mainly depends on the morphological changes at the cellular and subcellular levels and the expression of intracellular ferroptosis-related molecules (such as labile iron, ROS, peroxidized lipids and GSH). In addition, a series of related proteins and genes change when ferroptosis occurs [13]. Next, we will introduce a series of characteristics of morphology, molecular biology and genetics when ferroptosis occurs in cells.
Morphological features
The iron-dead cells show morphological changes at the cellular and ultra-micro level: on the one hand, they lose the integrity of the plasma membrane, the cytoplasm is swollen (oncosis), the mitochondria are smaller than the normal cells, the mitochondrial cristaes shrink or disappear, the outer mitochondrial membrane ruptures, and the membrane density increases. On the other hand, the nuclei in ferroptotic cells remain structural integrity, without condensation or chromatin margination [7, 13]. In some special cases, ferroptosis is also accompanied by the detachment and aggregation of cells, as well as the increase of autophagosomes [14]. It is worth noting that ferroptosis occurs in one cell can quickly spread to neighboring cells [15, 16]. Whereas, apoptotic cells showed cell shrinkage and blebbing, fragmentation and marginalization of chromatin, accompanied by plasma membrane blebbing and the production of apoptotic bodies (Fig. 2). Apoptosis regulators (e.g., BCL-2 family members BAX and BAK) do not affect mitochondrial permeability [7, 17]. H2O2-induced necrosis is characterized by rupture of the plasma membrane and swelling of the cytoplasm and organelles, resulting in broken plasma membrane fragments, which are released and cause cell swelling [7, 18]. Autophagy induced by rapamycin always forms double-membrane enclosed vesicles [13]. However, these morphological features are not observed in ferroptotic cells (Fig. 2).

Morphological features. Transmission electron microscopy of BJeLR cells treated with DMSO (10 h), erastin (37 mM, 10 h), staurosporine (STS, 0.75 mM, 8 h), H2O2 (16 mM, 1 h), and rapamycin (Rap, 100 nM, 24 h). Single white arrowheads, shrunken mitochondria; paired white arrowheads, chromatin condensation; black arrowheads, cytoplasmic and organelle swelling and plasma membrane rupture; black arrow, formation of double-membrane vesicles. A minimum of 10 cells per treatment condition were examined
Biochemical features
Iron accumulation
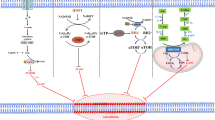
Compared with non-malignant cells, tumor cells have a stronger demand for iron. Researchers have increased the accumulation of iron in cells by increasing the absorption of iron ions by cells, reducing intracellular binding iron, reducing iron outflow, and by using various iron death inducers [19]. Excessive iron in cells can directly generate reactive oxygen species (ROS) through the Fenton reaction; or activate iron-containing enzymes (such as lipoxygenase ALOX or prolyl hydroxylase EGLN) to promote lipid peroxidation [20, 21]. It has also been discovered that free iron ions can also generate ROS through mitochondria. (Fig. 3) By inhibiting iron-related genes such as transferrin or using the DFO, it can effectively reduce intracellular free iron and effectively inhibit ferroptosis. At present, in various experiments, it has been found that a variety of metal elements can cause Fenton reaction, but it is still unclear why only iron can generate ROS through Fenton reaction in cells, and induce ferroptosis of cells [7].

The regulatory mechanisms of ferroptosis
Lipid peroxidation
Lipid peroxidation is a reaction driven by free radicals, which mainly affects the polyunsaturated fatty acids (PUFAs) in the cell membrane. PUFAs are most prone to peroxidation, which leads to the destruction of lipid bilayers and affects membrane function [22,23,24]. The products of lipid peroxidation include the initial lipid hydroperoxides (LOOHs) and subsequent reactive aldehydes (e.g., malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE)), which will increase during ferroptosis. Different lipoxygenases, especially ALOXs, have an up-and-down related role in mediating lipid peroxidation to produce the hydroperoxides, thereby promoting ferroptosis [24, 25]. Various cell membrane lipids (e.g., phosphatidylcholine, phosphatidylethanolamine (PE) and cardiolipin) may be oxidized [25]. Several membrane electron transfer proteins, especially NADPH oxidase (NOX), contribute to the production of ROS for lipid peroxidation in ferroptosis. In other cases, mitochondria participate in the induction of ferroptosis through processes such as the electron transport chain, the tricarboxylic acid cycle, the breakdown of glutamine, and the synthesis of lipids. Although mitochondria undergo strong changes in the process of ferroptosis, cardiolipin peroxidation has not been found in ferroptosis, and the role of mitochondria themselves in the process of ferroptosis is also controversial [26,27,28]. In different types of cancer cells, the occurrence and development of lipid peroxidation may be different.
Genetic features
As early as 2012, the ferroptosis activator erastin was identified because it can selectively trigger cell death in cancer cells harbouring mutant but not wild- type RAS. The cell death induced by erastin activates RAS-RAF-MEK-ERK pathway [29, 30]. Subsequent research identified some proteins and genes that can be considered as biomarkers of ferroptosis. It has recently been shown that KRAS is involved in the regulation of multiple metabolic pathways such as ROS production, glutamine metabolism and the TCA cycle, making tumor cells in a delicate high-energy state. In this state, once the oxidation-antioxidant balance is broken (excessive ROS production or reduced GSH), ferroptosis is prone to occur [31, 32]. Genes such as prostaglandin endoperoxide synthase 2 (PTGS2/COX2), Acyl-CoA synthase long-chain family member 4 (ACSL4), and nuclear factor erythrocyte-like 2 (NRF2/ NFE2L2) etc. PTGS2 does not use prostaglandins as a substrate for lipid peroxidation, but can oxidize lysophospholipids. PTGS2 is generally considered to be a biomarker of ferroptosis, but it is not a driving factor [33]. ACSL4 is involved in the synthesis of fatty acids and is considered to be a specific biomarker and driving factor of ferroptosis. (Fig. 3) [22, 25] The activation of NRF2 can inhibit the occurrence of ferroptosis, and excessive activation may promote ferroptosis. In addition, some traditional tumor suppressor genes have also been found to be related to ferroptosis [34]. For example, p53-mediated SLC7A11 transcriptional inhibition promotes ferroptosis in cancer cells. But p53-regulated ferroptosis does not depend on the GPX4-ACSL4 pathway, suggesting that may be other ways in the ferroptosis process regulated by p53 [9]. For example, p21 is responsible for encoding a 21kd protein of RAS family, it can prevent p53-induced ferroptosis by adjusting ROS levels [35]. Subsequent research identified a complex signaling pathway that regulates ferroptosis by generating excess ROS through iron accumulation and lipid peroxidation. This network is of special significance as a regulatory pathway for RCD in oncology.
Regulation mechanism of ferroptosis
Iron metabolism
The iron in the circulatory system is mainly Fe3+ [36], which is present in serotransferrin- mediated or lactotransferrin- mediated iron, and is combined with endocytosis through the cell membrane transferrin receptor 1 (TFR1) into the cell to form endosomes. In endosomes, the STEAP3 metalloreductase reduces Fe3+ to Fe2+, and then releases Fe2+ from endosomes into the cytoplasm through solute carrier family 11 member 2 (SLC11A2/DMT1), and excess iron is stored in ferritin [37]. In the case of balanced iron metabolism, very little iron enters and leaves the cell every day. When iron accumulates too much, a small iron pool (the labile iron pool, LIP) containing Fe2+ is formed in the cell, which can directly catalyze the formation of ROS through the Fenton reaction. In addition, iron and iron derivatives (heme or iron-sulfur [Fe-S] clusters) [38], which is vital to the activity of enzymes that can catalyze the production of ROS (eg., nicotinamide adenine dinucleotide phosphate hydride (NADPH) oxidases (NOXs), LOXs, and mitochondrial electron transport complexes), which can promote the production of ROS, and further promote lipid peroxidation induce ferroptosis [39].
Non-canonical ferroptosis induction refers to it caused by increasing LIP, such as increased expression of TFR1, decreased expression of ferritin, decreased expression of iron transporter, or excessive activation of heme oxygenase 1 (HMOX1) [40]. In cells with mutations in the RAS gene, the expression of TFR1 increases, while the expression of ferritin, which stores iron, decreases, increasing the Fe2+ content of LIP in the cell [30, 41]. Iron response element binding protein 2 (IREB2) gene which is the main transcription factor gene silencing can increase the expression of ferritin heavy and light chains and reduce the iron content in cells, indicating that IREB2 can indirectly regulate iron in the cell [7, 39]. HMOX1 has antioxidant activity, and excessive activation of HMOX1 can catalyze the degradation of heme into ferrous iron, biliverdin and carbon monoxide, and the increase of Fe2+ can induce ferroptosis [34, 42]. When free Fe2+ increases, the oxidative stress in the mitochondria will also increase, and the production of ROS will increase, which promotes lipid peroxidation and induces ferroptosis [39] Iron chelating agents inhibit ferroptosis by limiting iron overload, and increasing exogenous iron can promote ferroptosis [43]. For example, using iron such as iron chloride, hemoglobin, hemin or ferrous ammonium sulfate to overload the LIP of cells, inducing the non-canonical ferroptosis. But what is reassuring is that LIP only accounts for a small part of the total iron of the cell. The vast majority of iron is stored in ferritin or used by the cell in the metalloprotein.
Excess iron in cells is stored in ferritin, which is composed of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1), which absorb iron ions in the form of multimers and store them in the cytoplasm. Among them, FTH1 contains a ferrous oxidase center, which can quickly convert Fe2+ to Fe3+ and store it in ferritin. Lysosomes degrade ferritin through nuclear receptor coactivator 4 (NCOA4) to increase free iron levels (this process is called ferritin autophagy), inhibit NCOA4-mediated ferritin autophagy, which can increase iron storage and limit ferroptosis happen [44,45,46]. Finally, solute carrier family 11 member 3 (SLC11A3, also known as ferroportin) can oxidize Fe2+ to Fe3+, and then iron-efflux protein solute carrier family 40 member 1 (SLC40A1/ferroportin1/FPN) squeezes Fe3+ into the extracellular space [37, 46]. Alternatively, prominin 2 (a member of the prominin family of pentaspan membrane glycoproteins, PROM2) exports iron by forming ferritin-containing exosomes in epithelial and breast cancer cells, leading to ferroptosis resistance [47].
Epidemiological evidence suggests that high dietary iron intake increases the risk of several cancers, such as hepatocellular carcinoma (HCC) and breast cancer [2]. Through the understanding of iron metabolism, it is found that the changes in total iron levels in the human body are mainly caused by Fe3+; while the occurrence of ferroptosis is mainly induced by the increase of intracellular Fe2+. Increasing iron intake or reducing iron output will make cancer cells sensitive to oxidative damage and ferroptosis. Ferroptosis-mediated cancer targeted therapy has limitless prospect.
Lipid peroxidation
During ferroptosis, polyunsaturated fatty acids (PUFAs), especially arachidonic acid (AA) and adrenic acid (AdA), are most prone to peroxidation to produce lipid hydroperoxides (LOOHs) and subsequent reactive aldehydes (such as 4-HNEs or MDAs) cause damage to the lipid bilayer and affect membrane function [22,23,24,25]. The biosynthesis and remodeling of PUFAs in cell membranes are mainly regulated by ACSL4 and lysophosphatidylcholine acyltransferase 3 (LPCAT3) [47]. ACSL4 catalyzes the combination of free AA or AdA and CoA to form derivatives AA-CoA or AdA-CoA, and LPCAT3 then promotes their esterification to membrane phosphatidylethanolamine to form AA-PE or AdA-PE. If ACSL4 is knocked down, AA will be converted into acylated AA; or LPCAT3 will be silenced, which catalyzes the insertion of acylated AA into PLs, making cells resistant to ferroptosis. The up-regulation of ACSL4 is considered to be a biomarker and contributor to ferroptosis [23, 48]. ACSL3 converts monounsaturated fatty acids (MUFAs) into acyl-CoA esters to incorporate membrane phospholipids, thereby protecting cells from ferroptosis [49]. At the same time, studies have found that peroxisome (PEX)-mediated plasmalogen biosynthesis provides another source of PUFA for lipid peroxidation during ferroptosis, indicating that PEX may promote lipid peroxidation related to ferroptosis [50].
The peroxidation process is mainly divided into two categories: enzymatic lipid peroxidation and non-enzymatic lipid peroxidation [51]. Enzymatic lipid peroxidation is mainly mediated by the activity of the arachidonate lipoxygenase (ALOX) family in a controlled manner. The mammalian ALOX family consists of six members (ALOXE3, ALOX5, ALOX12, ALOX12B, ALOX15 and ALOX15B). Different lipoxygenases mediate lipid peroxidation to produce hydroperoxides(AA-PE-OOH or AdA-PE-OOH) has a context-dependent role to promote ferroptosis. For example, ALOX5, ALOXE3, ALOX15, and ALOX15B are responsible for ferroptosis in human cell lines derived from various cancer types (BJeLR, HT-1080 or PANC1 cells), while ALOX15 and ALOX12 mediate the derivation of non-small-cell lung cancer (NSCLC) p53 induced ferroptosis in H1299 cells, in which ALOX12 induced ferroptosis through TP53-mediated down-regulation of SLC7A11 [20, 24, 52, 53]. Current studies have found that inhibiting or knocking down lipoxygenase can inhibit ferroptosis in certain cell types. It is still unknown whether other oxygenases (such as cyclooxygenase and peroxygenase) also play a similar role in lipid peroxidation [33].
Non-enzymatic lipid peroxidation is a free radical-driven chain reaction. Reactive oxygen species (ROS) initiate the oxidation of PUFAs, which mainly involve hydrogen reactions. In the presence of Fe2+, the Fenton reaction generates hydroxyl radicals (a highly mobile, water-soluble ROS that can trigger lipid peroxidation) [51]. The hydroxyl radicals extract hydrogen from PUFAs to form carbon-centric lipid radicals(L.) [54]. Molecular oxygen (O2) reacts quickly with lipid radicals to produce lipid peroxy radicals (LOO.). Subsequently, LOO. acts as a catalyst to extract hydrogen from PUFAs to form lipid hydroperoxides (LOOH) and new LOO.. And LOOH can be converted into alkoxy radicals (LO.), which reacts with adjacent PUFAs to initiate another chain reaction. Lipophilic antioxidants can reduce ROS by reducing reactions or combine with peroxides generated by the auto-oxidation reaction by releasing hydrogen atoms, interrupting the chain reaction. Or iron chelating agents such as DFO, which can be complexed iron interrupts the reaction [51].
Although the exact mechanism of lipid peroxidation leading to ferroptosis in cells is unknown, current studies have found that lipid peroxides have toxic effects on cancer cells through two mechanisms. Molecularly, lipid peroxides are further broken down into active substances, which can consume amino acids, nucleic acids and proteins, driving cells to ferroptotic death [54]. In addition, lipid hydroperoxides may be broken down into reactive toxic aldehydes, such as 4-HNEs or MDAs, which by crosslinking may inactivate proteins involved in essential cellular processes to promote ferroptosis [55]. Structurally, extensive lipid peroxidation leads to biofilm thinning and increasing curvature, leading to a vicious cycle of lipid peroxidation, and ultimately leading to membrane instability and the formation of lipid pores (similar to the proteinaceous pores observed in necroptosis and pyroptosis) and micelle formation [40, 56,57,58,59]. Or continuous large-scale oxidation and consumption of PUFAs may change the fluidity and structure of the membrane, and increase the permeability of the membrane, leading to the loss of membrane integrity, and ferroptosis of cancer cells [60].
Oxidation
ROS is a group of molecules containing partially reduced oxygen, which can cause cancer cells to die by destroying biological molecules such as DNA/RNA, proteins, and lipids [54]. ROS involved in ferroptosis can be produced from various sources, and the accumulation of oxidation products (especially phospholipid hydroperoxide) is considered to be a sign of ferroptosis [59]. ROS is a by-product of aerobic metabolism. In the presence of mitochondrial superoxide dismutase (SOD), the electron transport chain on the inner mitochondrial membrane produces H2O2, which then diffuses from the mitochondria to the cytoplasm. The rate depends on the mitochondrial transmembrane potential. At a high iron concentration that is conducive to the Fenton reaction, H2O2 forms highly reactive oxygen radicals to promote lipid peroxidation [28, 52, 61]. In order to deal with the excessive production of ROS, there is a complete anti-oxidative stress system in the cell.
Antioxidant mechanism
xCT
Amino acids cannot diffuse directly into cells, they must be transported across the cell membrane with the help of specific transport proteins. The amino acid antiporter system Xc− is one of transporters and consists of two core components: the light chain SLC7A11 (xCT) and the heavy chain SLC3A2 (4F2hc). It introduces extracellular oxidized form of cysteine and cystine to exchange intracellular glutamate. After cystine enters the cell, it is reduced to cysteine, and cysteine is involved in the synthesis of GSH (a major endogenous antioxidant) [7]. Under the conditions of extracellular oxidation, the exchange of cystine and glutamate is the most upstream event of ferroptosis. The inhibition of the SLC7A11 pathway is the most critical upstream mechanism for inducing ferroptosis [34, 62]. Its expression or activity is regulated by many factors. For example, under the positive regulation of Nrf2 and the negative regulation of cancer suppressor genes (such as TP53, BAP1 and BECN1) [63, 64], they form a complex network to control the level of GSH in ferroptosis. Small molecule compounds or drugs (eg., erastin, sorafenib and sulfasalazine) inhibit SLC7A11 or reduce glutamate and cause glutathione depletion to trigger ferroptosis [49, 65]. AMPK is the main regulator of ATP homeostasis (Fig. 2). Its mediated BECN1 phosphorylation promotes ferroptosis by inhibiting SLC7A11 activity, while mediated Acetyl CoA carboxylase(ACACA) phosphorylation inhibits ferroptosis by inhibiting fatty acid biosynthesis, indicating that the energy state may be affect the lipid biosynthesis and peroxidation during the ferroptosis [61, 66]. When the system Xc− is inhibited, the trans-sulfuration pathway (methionine through which cysteine is provided from cystathionine for glutathione synthesis) regulates ferroptosis in certain cells, which is regulated by the aminoacyl tRNA synthetase family (such as CARS1). Up-regulation of the transsulfur pathway can make cells insensitive to ferroptosis induced by erastin [67]. Recently, the deubiquitinating enzyme ubiquitin aldehyde binding 1 (OTUB1), a member of the ovarian cancer (OTU) family, has been identified as an important factor in stabilizing SLC7A11. The inactivation of OTUB1 makes cancer cells sensitive to ferroptosis. In addition, OTUB1 is overexpressed in cancer cells, making OTUB1 a potential target for ferroptosis-mediated cancer treatment [68].
GSH
GSH is the main antioxidant in mammalian cells and can be used as a cofactor of selenium-dependent GPX4 to reduce lipid hydroperoxides [69]. Ferroptosis can be triggered by GSH consumption (such as the consumption of GSH by erastin indirectly inactivates GPX4) or direct reduction of GSH synthesis (such as the inhibition by buthionine sulfoximine (BSO)), which leads to ROS accumulation and subsequent lipid peroxidation [70, 71]. It should be noted that, researchers have found that lack of cystine can inhibit cell growth, while lipophilic antioxidants and iron chelators agents can inhibit this type of cell death [39]. Multidrug resistance protein 1 (MRP1) is an adenosine triphosphate (ATP) binding cassette family transporter that can export certain types of chemotherapeutic drugs. Cancer cells with high MRP1 expression show a multidrug resistance phenotype. Recently, MRP1 has been identified as a negative regulator of intracellular GSH levels, and high MRP1 expression can effectively sensitize cancer cells to ferroptosis inducers that target GSH metabolism [72]. The study not only provides a potential strategy to eradicate drug-resistant cancers, but also explains from another perspective why some aggressive malignancies are sensitive to ferroptosis (Fig. 3).
GPX4
The classic pathway induces ferroptosis through peroxidative damage to the membrane, and the main detoxification mechanism is achieved through the catalytic detoxification of GPX4. Early experiments found that mice that knock out or silence the GPX4 gene cannot survive, proving that GPX4 is an essential gene for survival. As a central downstream regulator of ferroptosis, GPX4 uses two molecules of GSH as electron donors to reduce toxic phospholipid hydroperoxides to non-toxic phospholipids, even if they has been inserted into the membrane or lipoproteins to combat lipid peroxidation [25, 33]. The relationship between GPX4 expression and patient survival outcome depends on the cancer type. For example, the high expression level of GPX4 is negatively correlated with the prognosis of breast cancer patients [73], but has good survival outcomes for pancreatic cancer patients [74]. This may be related to the KRAS mutation in patients with pancreatic cancer. In PDAC cells, mutant KRAS transcription activates NRF2, and NRF2 up-regulates xCT, thereby regulating GPX4 expression [32]. The expression and activity of GPX4 in ferroptosis depend on the presence of GSH and selenium. When synthesizing GPX4, the nascent polypeptide chain combines selenium into selenocysteine (Sec), where selenium replaces the sulfur of cysteine, increasing the anti- ferroptosis activity of GPX4 [75, 76]. GPX4 can be inactivated through direct or indirect targeting mechanisms. Such as direct pharmacological (eg., RSL3, altretamine, ML162, ML210, FIN56 or FINO2) or genetic (Cre recombinase method) interventions to GPX4 can induce ferroptosis. In addition, consumption of GSH is an indirect method of inactivating GPX4 [77, 78]. GPX4 depletion also mediates other non-ferroptosis RCDs (e.g., apoptosis, necroptosis and pyroptosis) [78,79,80], indicating that lipid peroxidation is located at the crossroads of several of these pathways, but downstream effectors may be different. Although GPX4 inhibition is an important downstream signal in the process of ferroptosis, ferroptosis unrelated to GPX4 may still occur. For example, TP53-mediated ferroptosis does not need to inhibit GPX4, although TP53 can inhibit SLC7A11 expression [9].
Other oxidative stress pathways
Nrf2
In addition to GPX4, antioxidant proteins such as Nrf2 are also the main regulators of oxidative stress signals, and can inhibit lipid peroxidation. However, excessive activation may induce ferroptosis through unstable iron-catalyzed ROS metabolism mediated by HMOX1 [81,82,83]. Nrf2 also has a dual role in cancer progression, lack of Nrf2 activity can contribute to early cancerigenesis, while high constitutive Nrf2 activity can trigger cancer progression and resistance to treatment [81]. Nrf2 activates the protective genes by transactivating iron metabolism, oxidative defense, and redox signaling [34, 84]. Preclinical studies have shown that Nrf2 signaling is an important defense mechanism against ferroptosis and is involved in the resistance of HCC cells to sorafenib. After erastin and sorafenib are used to inhibit or silence Nrf2 genes, the sensitivity of cells to ferroptosis increases, which emphasizes its key role in antioxidant mechanisms [34, 81, 85]. The contribution of Nrf2 to ferroptosis resistance and the therapeutic potential of Nrf2 inhibitors (such as brusatol and trigonelline) to enhance the treatment of ferroptosis need to be further addressed in preclinical and clinical studies.
HIF
Hypoxia-inducible factor (HIF) plays a central role in the response to oxidative stress, affecting various pathological conditions of tissues and cells, and is a key factor in intracellular metabolism. The main regulator of hypoxia HIF1 is a heterodimeric transcription factor, including an unstable α-subunit (including HIF1A, endothelial PAS domain protein 1 (EPAS1, also known as HIF2A) and HIF3A) and stable β subunit (for example, aromatic hydrocarbon receptor nuclear transport protein (ARNT1/HIF1B)). The expression of HIF1A and EPAS1 are elevated in a variety of cancer types, and is usually associated with a poor prognosis of patients [86, 87]. Under normoxic conditions, HIF1A and EPAS1 are hydroxylated by members of the EGLN family of hypoxia-inducible factors, and then recognized by the E3 ubiquitin ligase VHL for proteasomal degradation. Under hypoxic conditions, the inactivation of hydroxylase causes HIF1A and EPAS1 to accumulate and form heterodimers with ARNT, thereby inducing the transcription of genes involved in hypoxia adaptation and survival [87]. EGLN protein is not only an iron-dependent sensor of oxygen, but also an iron-dependent sensor of cysteine, used to catalyze the hydroxylation of HIF. EGLN is a key target of iron chelator (such as deferoxamine), which can increase the stability of HIF by inhibiting the activity of EGLN to prevent ferroptosis by ischemia–reperfusion injury [86].
In early studies, HIF had a dual role in regulating the ferroptosis of cancer cells. In HT-1080 fibrosarcoma cells, hypoxia-induced HIF1A expression inhibits ferroptosis by increasing the expression of fatty acid-binding proteins 3 and 7 to promote fatty acid uptake and increase lipid storage capacity to avoid subsequent lipid peroxidation [88]. In renal cell carcinoma (RCC), hypoxia induces EPAS1 activation and up-regulates the expression of HILPDA to promote PUFAs production and subsequent lipid peroxidation, and induce ferroptosis [89, 90]. Therefore, effective control of HIF is necessary to maintain lipid homeostasis to regulate ferroptosis. In clinical trials, we can consider using HIF inhibitors to regulate ferroptosis according to the situation.
Inducers of ferroptosis
Since ferroptosis was discovered, many researchers have tried to discover all the activation methods that induce ferroptosis, hoping to target the induction of ferroptosis in cells through different ways to achieve the purpose of suppressing cancers. At present, experiments have shown that various small molecule drugs can initiate ferroptosis in four ways: Class I ferroptosis inducers (FINs) mainly consume intracellular GSH, class II FINs mainly target GPX4 and inactivate its activity, class III FINs mainly consume GPX4 and endogenous antioxidant CoQ10 through the SQS-mevalonate pathway, class IV FINs induce lipid peroxidation by increasing the LIP or oxidizing iron [51]. There are still some small molecule compounds, due to their complex mechanism of action, cannot be specifically judged as one of the above four categories. Now summarized in other ferroptosis inducers (Table 1).
Class I FINs
The concentration of GSH in cancer cells is more than 1000 times that of extracellular cells and 4 times that of normal cells. GSH plays an important role in scavenging superoxide and resisting cell death [7, 91]. So GSH has been considered as cancer’s Achilles’ heel [51] (Fig. 4).

The inducers of ferroptosis
SystemXc- and the transsulfur pathway are the two main sources of cysteine and GSH synthesis. In some cancers, apparent silencing or loss of transsulfase enzymes makes cells more dependent on SLC7A11 regulated cystine uptake pathways [91, 92]. After suppressing the System Xc-, SLC7A11 will be compensated up. These changes can be used as pharmacodynamic biomarkers to identify SLC7A11 inhibition and ferroptosis [65]. Based on this, SLC7A11 is considered a very good anti-cancer drug target.
Among Class I FINs, erastin is the prototype of ferroptosis inducer, which can reduce GSH level by directly inhibiting system Xc-, and has been found to inhibit the growth of cervical cancer and ovarian cancer cells [93]. In addition to system Xc-, erastin also targets mitochondrial voltage-dependent anion channel (VDAC) and RAS genes. Knockdown or silencing of these genes will trigger cell resistance to erastin [30]. In addition to causing ferroptosis in cancer treatment, erastin has also been shown to enhance the chemotherapy effects of traditional anti-cancer drugs (such as doxorubicin, cisplatin, temozolomide, cytarabine, etc.) in certain cancer cell lines [94,95,96]. Although erastin has an inhibitory effect, its poor water solubility and unstable metabolism limit its application in the body. Therefore, scientists have developed erastin derivatives piperazine erastin (PE) and imidazolone erastin [28], which act on cancer cells in a similar manner to erastin. However, PE and IKE show better water solubility and stability than their prototypes in the physiological environment, and effectively inhibit tumor growth in experimental models of fibrosarcoma and diffuse large B cell lymphoma (DLBCL) [33, 97].
In addition, the FDA-approved immunosuppressant sulfasalazine (SAS, trade name Azulfidine, Salazopyrin, Sulazine, etc.) is used as a first-line treatment for rheumatoid arthritis and can also be used as a System Xc- inhibitor. The SAS mode of action is similar to erastins, but the effect is much weaker. It is currently used to treat lymphoma, pancreatic cancer and lung cancer [98,99,100]. However, due to its poor pharmacokinetics, lower potency and metabolic stability, the clinical application of this drug is limited [101, 102]. SAS is also used as a combination therapy to enhance the therapeutic effect of other chemotherapy on glioma [103]. Sorafenib is an anti-cancer drug approved by the FDA for the treatment of HCC, RCC and thyroid cancer. In addition to inhibiting receptor tyrosine kinases, it can also promote cell ferroptosis by inhibiting the function of System Xc-. Sorafenib was determined to be class I FINs [51].
BSO is an ferroptosis inducer that directly blocked the synthesis of GSH, which can inhibit the growth of mouse breast cancers and increase the melphalan chemosensitivity of melanoma and neuroblastoma cells [104,105,106]. With the deepening of research, other inhibitors that inhibit GSH synthesis have also been discovered and verified in different cancer cells. Finally, as a genetic approach to increase the efficacy of GSH depletion, an optimized human cystathionine glyase (CGL), coined cyst(e)inase, was engineered to degrade cysteine and cystine with a higher kinetic rate. This approach impedes the growth of prostate and breast cancer xenografts and increases mouse survival in a chronic lymphocytic leukemia model [51, 107].
Class II and III FINs
Class I FINs are a promising anti-cancer drug for cells with high SLC7A11 expression. However, in other cancer cells, there may be other ways to resist ferroptosis caused by class I FINs. For example, the up-regulation of the heat shock protein (HSPB1) can make cells resistant to the treatment of class I FINs [98]. If inhibiting the synthesis of GSH fails to induce ferroptosis in some cells, it may be due to the existence of the transsulfur pathway that the cells can still survive [105, 108] (Fig. 4). Therefore, for this type of cancer cells, the targeted inactivation of the activity of GPX4 by class II and III FINs can induce ferroptosis of the cells. At present, among many class II FINs, RSL3 can induce ferroptosis by directly targeting GPX4. RSL3 targets enzymes with nucleophilic sites, and directly inactivates GPX4 through the alkylation of selenocysteine [33]. At present, RSL3 is widely used. For example, in mouse models, it can block the activity of GPX4 to promote ferroptosis and thus inhibit the growth of fibrosarcoma [33]. Withaferin A inhibits the growth and recurrence rate of neuroblastoma xenografts [82]. Altretamine is an FDA-approved anti-cancer drug for the treatment of ovarian cancer, which induces ferroptosis through GPX4 inhibition [77]. In class III FINs, the latest iFSP1 small molecule inhibitors developed by American and German scientists can block the function of the ferroptosis inhibitor the protein FSP1 and reduce the content of ubiquinone to increase lipid peroxidation [109, 110]. FIN56 is an ferroptosis inducer derived from CIL56, which was discovered through regulatory analysis of 56 lethal compounds that do not depend on caspase. FIN56 leads to depletion of GPX4 and CoQ10 through the SQS-mevalonate pathway [111].
Class IV FINs
Compared with normal cells, cancer cells have a stronger demand for iron, which makes cancer cells more sensitive to the induction of ferroptosis. Based on the characteristics of cancer cells, class IV FINs mainly promote the synthesis of lipid peroxides by increasing intracellular LIP or iron oxide to induce ferroptosis, and may provide new opportunities for cancer treatment [112]. In addition to make GPX4 inactivation, withaferin A also increases LIP through HMOX1-mediated heme degradation, thereby inducing ferroptosis in neuroblastoma [82]. Heme and ferrous ammonium sulfate induce intracellular iron accumulation, which leads to ferroptosis of neuroblastoma cells [82]. BAY87-2243, a known inhibitor of IκBα, can up-regulate the expression of HMOX1 and enrichment of iron ions in an NFκB-dependent manner, and induce ferroptosis [83]. FINO2 is a class of organic peroxides, which share many characteristics with artemisinin (for example, iron is required to induce cell death, and ROS is produced). The peroxide containing 1,2-dioxolane structure has been identified as ferroptosis inducers, which are more effective than artemisinins in some cancer cell lines. The ferroptosis caused by FINO2 is due to the combined effects of the direct oxidation of unstable iron and the inactivation of GPX4 (Fig. 4). Compared with non-malignant cells in the same tissue, FINO2 is more effective in malignant cells. In addition, in vitro experiments have shown that FINO2 can bypass chemoresistance-related pathways (eg., p53 mutations, BCL-2 overexpression) [55]. The combination of the kinase tyrosine inhibitor lapatinib and the lysosomal drug silamexine that destabilizes the lysosomes synergistically induces ferroptosis by disrupting iron transport in breast cancer cells [113]. The use of iron oxide nanoparticles to promote iron overload has been shown to induce ferroptosis and inhibit cancer growth in nutrient-deficient cancer cells [114]. In addition, recently FDA approved polyethylene glycol-coated ultra-small nanoparticles Coined Cornel dots(C'dots) can absorb and integrate extracellular iron and transport it into the cell, causing iron overload and inducing cell death [114].
Ferroptosis and cancer therapy
Although breakthroughs have been made in the field of cancer therapy, cancers are still the second leading cause of death in the world. At present, the main treatment approach is to use anti-cancer drugs to trigger the apoptotic death of cancer cells. However, due to the inherent and acquired resistance of cancer cells to apoptosis, the therapeutic effect is limited. Drug resistance is still the main limiting factor for the cure of cancer patients. Inducing ferroptosis of cancer cells is one of the best ways to avoid drug resistance [51, 131]. It can be through the use of exogenous molecules or drugs, or regulation of extracellular physiological conditions (eg., high concentration of extracellular glutamate) blocking system xCT to induce ferroptosis extensively, and it can also target cytogenesis according to the difference between cancer cells and normal cells. At present, several small molecules and FDA-approved clinical drugs activate ferroptosis in cancer cells, and the efficacy of ferroptosis inducers to inhibit cancers in various experimental models. At the same time, there are also a variety of treatments that can be effectively induced ferroptosis in experiments, emphasizing the potential as a new type of anti-cancer therapy [51, 129, 131]. Next, we will introduce how some current treatments use ferroptosis to treat cancer (Table 2).
Ferroptosis and chemotherapy
Chemotherapy is one of the main treatment methods for malignant cancers, but in the course of cancer chemotherapy, various mechanisms have led to cancer multidurg resistance (MDR), and resistance to chemotherapy drugs has become the main reason of chemotherapy failure in cancer patients [132]. In recent years, more and more studies have been conducted on how to effectively overcome cancer MDR. With the ferroptosis into the eyes of researchers, there is hope for overcoming cancer chemotherapy resistance [132, 133]. It is currently known that inhibiting xCT and GPX4 can effectively enhance the sensitivity of tumors (eg., pancreatic ductal carcinoma, NSCLC and osteosarcoma) to gemcitabine and cisplatin [118, 119]. In addition, there are several other drugs that are already in clinical use or have strong clinical transformation potential that can promote ferroptosis.
Sorafenib
Sorafenib is a clinically approved multi-kinase inhibitor for the treatment of advanced cancers [65]. Studies have shown that in the treatment of HCC, RCC, lung cancer or pancreatic cancer, the anti-cancer activity of sorafenib mainly depends on inducing ferroptosis by inhibiting the activity of the system xc-, rather than relying on the inhibition of its kinase [34, 65, 134]. However, in certain cancer cell lines, drug resistance has been observed in sorafenib-mediated cancer therapy. In the study of drug-resistant cancer cells, it is found that the target gene of metallothioneins-1G (MT-1G) is a biomarker and contributing factor of sorafenib resistance [34, 135]. Therefore, inhibiting the MT-1G pathway during sorafenib treatment can reduce the risk of chemotherapy resistance and improve the therapeutic effect [130].
Artemisinin
In addition to its therapeutic value in the treatment of malaria, artemisinin also has a killing effect on a variety of cancers. In addition to inducing cell apoptosis, artemisinin (especially artesunate and dihydroartemisinin) can also increase the level of intracellular free iron by promoting ferritin autophagy, thereby triggering ferroptosis in cancer cells [122,123,124,125]. Iron supplements, such as holotransferrin, can enhance the anti-cancer properties of artemisinin [136]. This is because cancer cells have more heme, which favors the cancer targeting specificity of artemisinins in a similar manner as in the case of malaria [137]. In clinical trials, artemisinin has been proved to be effective in treating acute myeloid leukemia, [123] and head and neck squamous cell carcinoma (HNSCC) [122].
Cyst(e)inase
Cyst(e)inase is an engineered human enzyme that can effectively degrade cysteine and cystine (cyst(e)ine) in serum. Subsequent depletion of extracellular cystine leads to the death of prostate cancer and chronic lymphocytic leukemia cells in vitro and in vivo [107]. Cyst(e)inase-mediated depletion of cyst(e)ine can induce ferroptosis in pancreatic without causing obvious toxicities, suggesting acceptable safety and tolerability [116]. The strategy of using cystinase to regulate extracellular cystine levels can provide new therapeutic opportunities for ferroptosis-based anticancer therapies, especially with drugs that induce ROS (eg., doxorubicin, gemcitabine, paclitaxel, 5-fluorouracil, Bortezomib).
Statins
Statins (eg., fluvastatin, lovastatin and simvastatin) are a class of drugs used to hypotensive by inhibiting HMG-CoA reductase (HMGCR). By inhibiting the mevalonate pathway, statins can reduce selenoproteins (such as GPX4) and CoQ10 biosynthesis, thereby enhancing ferroptosis [111, 129]. Data from clinical trials indicate that atorvastatin and fluvastatin may have anti-proliferative effects in cancers overexpressing HMGCR [138, 139]. A deeper understanding of the ferroptosis pathway regulated by cholesterol may help to better use statins in future clinical studies (Fig. 3).
Ferroptosis and radiotherapy
Radiotherapy (RT), as one of the effective cancer treatments, uses ionizing radiation (IR) from a radioactive source to cause DNA damage and cause cell apoptosis. And now studies have found that RT can directly induce ferroptosis of cancer cells [120]. Ataxia telangiectasia-mutated (ATM) is a key protein kinase in the process of DNA damage repair. The ATM-mediated down-regulation of SLC7A11 caused by RT is the cause of ferroptosis in cancer cells [140]. When the SLC7A11 is overexpressed, it can promote RT resistance by inhibiting ferroptosis [126]. Studies have shown that FINs (such as RSL3, erastin, sorafenib, and sulfasalazine) can synergistically enhance RT efficacy by inhibiting SLC7A11 or inactivating GPX4 in models of glioma, lung cancer, fibrosarcoma, melanoma, breast cancer, and cervical cancer [60, 120, 126, 140,141,142,143,144,145]. In addition to down-regulating SLC7A11, RT also up-regulates ACSL4, thereby increasing lipid synthesis and peroxidation, inducing ferroptosis [126].
These studies have revealed the molecular mechanism between ferroptosis and RT sensitization, provide a theoretical basis for further elucidating the mechanism of ferroptosis in RT sensitization, and have groundbreaking significance for the development of ferroptosis-related drugs with RT sensitization.
Ferroptosis and immunotherapy
Immunotherapy is currently one of the promising treatment methods for anti-cancer. It is achieved by activating the immune system and enhancing its inherent cancer treatment capabilities. Immune checkpoint inhibitors (ICIs) mainly act by activating effective anti-cancer immune responses driven by cytotoxic T cells. The currently approved ICIs target CTLA4, PD-1 and their ligand PD-L1. Judging from ongoing clinical trials, PD-1/L1 inhibitors are mainly be used for combination therapy, with targeted therapy or other immunotherapy. In recent years, it has been found that ferroptosis is closely related to immune regulation. For example, PD-L1 antibodies can promote lipid peroxidation-dependent ferroptosis in cancer cells, PD-L1 antibodies and ferroptosis inducers synergistically inhibit tumor growth in vitro and in vivo [11]. Cytotoxic T cell-driven immunity can induce ferroptosis in cancer cells. In terms of mechanism, CD8 + T cells can down-regulate the expression of SLC3A2 and SLC7A11 by releasing IFNγ, thereby reducing the uptake of cystine and promoting lipid peroxidation in cancer cells [11]. TGFβ1 can promote ferroptosis through SLC7A11 transcription inhibition and ZEB1 activation. However, TGF-β inhibitors and PD-1 antibodies can synergistically generate an immunogenic microenvironment and produce H2O2, thereby promoting the Fenton reaction, triggering ferroptosis of cancer cells. And the cancer antigens released after cell death in turn promote the immunogenicity of the microenvironment [146]. Under certain circumstances, ferroptosis may have a cancer-promoting effect. The release of damage-related molecular patterns (DAMP) in exosomes during death, which ultimately leads to the polarization of macrophages to the M2 phenotype and stimulates tumor growth [147]. These findings bridge the gap between ferroptosis and immunotherapy, laying a theoretical foundation for the synergy of ferroptosis-immunotherapy in the treatment of malignant cancers. At the same time, it provides a direction for further exploring the molecular mechanism of ferroptosis to promote the efficacy of immunotherapy (Fig. 5).

Dual role of ferroptosis in tumor immunity. a CD8+ T cell-mediated IFNG release inhibits SLC7A11 expression in cancer cells through activation of the STAT1 pathway, thereby inducing tumor cell ferroptosis. b Ferroptotic cancer cell-mediated KRASG12D release increases M2 macrophage polarization through activation of the STAT3 pathway, thereby limiting antitumor immunity
Ferroptosis and nanomedicine
The current clinical treatments are far from satisfactory due to various reasons. Combining emerging biological discoveries and traditional treatment methods has become the development trend of effective cancer treatment. There have been studies on ferroptosis-related nano-preparations combined with other treatment methods to enhance the therapeutic effect of cancers. One of the anti-cancer strategies is to use nano-drugs to induce ferroptosis.
The most direct method is to develop iron-containing nanoparticles, which can transport and release iron into cells. At present, in addition to solid iron-based nanocrystals, some other iron-based nanocomposites have been developed, such as amorphous iron (Fe0) nano-metallic glass and metal organic framework (MOF) have been developed to effectively release iron in TME, improving the iron release efficiency [148]. Iron nanoparticles kill cancer cells by increasing iron levels and ROS. At the same time, chemotherapeutic drugs, such as cisplatin prodrugs, can also be modified on iron nanoparticles to form FePt-NP2 to increase the sensitivity of cancer cells to cisplatin [117]. Or add iron to the nanoparticles of chemotherapy drugs, such as depositing Fe3+ and tannic acid (TA) on the nanocrystals of SRF, resulting SRF@FeIIITA (SFT). After entering the cell, it can be destroyed in the lysosomal acidic microenvironment, thereby releasing SRF to inhibit GPX4. At the same time, TA is an acid-activated reducing agent that can reduce Fe3+ to Fe2+ to generate ROS and enhance ferroptosis [69]. In addition, methylene blue (MB) is loaded into this SFT nanoplatform for bioimaging guided photodynamic therapy (PDT) [69].
Another intuitive method is to add FINs to the nano-delivery vehicle. Compared with free drugs, the nano-platform can improve solubility and biocompatibility, and increase accumulation through active or passive targeting, improve the pharmacokinetics of FINs, and improve the therapeutic effect [97]. For example, nanoparticle formulations of withaferin A promoted accumulation due to increased permeability and EPR retention effects and inhibited tumor growth in neuroblastoma [82, 102]. Similarly, the use of IKE nanoparticles resulted in increased accumulation of IKE in DLBCLs, suppressing tumor growth in mice while reducing toxicity [97].
In addition to small molecule drugs, nanocarriers can also be combined with other small molecules, such as amino acids, plasmids and other unconventional drugs. Some researchers have developed arginine-terminated manganese silicate nanobubbles (AMSNs) [149]. Due to the deficiency of arginine succinate synthase (ASS) in cancer, AMSN has high biocompatibility and strongly cancer targeting ability. After entering cells, AMSN releases high-valence manganese ions (Mn3+ and Mn4+), which are reduced to Mn2+ by GSH. The consumption of GSH leads to increased intracellular oxidative stress and inactivation of GPX4. In addition, the magnetic hollow structure enables AMSN to be used as a bioimaging contrast agent and drug delivery carrier [149]. Other researchers have also developed a metal–organic network (MON-p53) encapsulated by p53 plasmids, which can kill cancer cells through a mixed ferroptosis/apoptosis pathway, in which ferroptosis plays a major role. MON-p53 nanoparticles induce ferroptosis by providing unstable iron, and also deliver p53 to cancer cells for gene therapy, effectively inhibiting cancer growth and metastasis [150].
What's more attractive is that nanomaterials can not only serve as carriers, but they can also induce ferroptosis by participating in biochemical reactions and disturbing metabolic balance. In 2016, the first nanoparticle ferroptosis inducer was reported [114]. The FDA-approved ultra-small poly(ethylene glycol) coated silica nanoparticles create Cornel dots (C′ dots), which induce ferroptosis in amino acid starved cancer cells and inhibit cancer growth. Researchers found that the PEGylated C′ dots absorbs extracellular iron and binds to it. When it enters cancer cells, it releases it and increases intracellular iron levels. Accompanied by the production of ROS and the consumption of GSH, it eventually leads to ferroptosis [151].
Researchers have used the tunable physicochemical properties of nanomaterials to study the combined treatment effects of ferroptosis and phototherapy in addition to the classic treatment. These studies mainly focus on the aspects of photodynamic therapy (PDT) and photothermal therapy (PTT), because they are intrinsically related to ROS and O2, and ferroptosis can produce ROS and provide O2 [152, 153]. The researchers prepared a nanosystem assembled by the photosensitizer Ce6 and erastin. The results showed that the Ce6-erastin nanoassembly produced a large amount of toxic reactive oxygen species under localized cancer irradiation, and showed strong anti-cancer activity on xenograft cancers [154]. Studies have also found that the ferroptosis inducer SRF has a synergistic effect with PDT, and the ferroptosis inhibitor DFO can reverse the anti-cancer ability of PDT [155,156,157]. Recently, scientists loaded SRF and SPIO nanoparticles on the mesopores and surface of MPDANPs to form SRF@MPDA-SPIO, which can release Fe3+/ Fe2+ and SRF in cells, which can be used for MR imaging-guided ferroptosis—PTT combination therapy [158]. In addition, there is a high-efficiency nanosystem that combines chemotherapy, ferroptosis and PTT triple therapy for the combined treatment of ER + breast cancer [121]. Other researchers have constructed the nanocomposite FPMF@CpGODN, which is a perfect combination of ferroptosis, PTT, chemotherapeutics and immunotherapy, which has a stronger effect on eliminating primary tumors and preventing tumor recurrence [159]. This concept of combining ferroptosis with nanomaterials, chemotherapeutics, immunotherapy and other treatment methods can provide a new clinical vision for cancer treatment.
Although the existing nanomaterials have produced good ferroptosis effects, there are still many problems to be solved before entering the clinic. The current biosafety of nanomaterials still needs to be improved in many ways, such as biological targeting, biocompatibility, biodegradability and immunogenicity. At the same time, we must continue to develop new types of nanomaterials that can induce ferroptosis and suitable for making the nanomedicines. In general, although nanomaterials can induce ferroptosis of tumor cells and can achieve ideal tumor suppression effects, almost all research data comes from experimental animals, so there are still many preclinical experiments that need to be further carried out.
Conclusions and perspectives
Although much progress has been made in tumor biology and therapeutics, there is still a long way to go to win the fight against cancer. Since ferroptosis was discovered, researchers in the field of biomedicine have been enthusiastic about it. How to identify ferroptosis, how to regulate ferroptosis, how to induce ferroptosis, and how to apply ferroptosis has gradually become a research hotspot among researchers. As the above problems are solved one by one, ferroptosis has received more and more attention in tumor biology and tumor treatment. In various cell or animal cancer models, ferroptosis has a significant anti-cancer effect. Targeted regulation of ferroptosis in tumor cells has become an emerging anti-cancer strategy. Further exploration of unknown key molecules or pathways of ferroptosis can provide new targets and new methods for tumor treatment. For example, the induction of ferroptosis through nanomedicine can not only enrich tumor treatment methods, but also treat malignant tumors in combination with traditional treatment methods. There are still many unknowns and challenges in the basic research and clinical transformation of ferroptosis. However, we believe that with the deepening of basic research on ferroptosis, future research results on induction of ferroptosis can provide more theoretical guidance and programs for tumor treatment.
Availability of data and materials
The data sets supporting the results of this article are included within the article.
References
Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130–43.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
Parisi LR, Morrow LM, Visser MB, Atilla-Gokcumen GE. Turning the spotlight on lipids in non-apoptotic cell death. ACS Chem Biol. 2018;13(3):506–15.
Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254(3):439–59.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–64.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–91.
Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33(5):890-904 e5.
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4.
Wang Y, Sun S, Zhang Z, Shi D. Nanomaterials for cancer precision medicine. Adv Mater. 2018;30(17):e1705660.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–91.
Katikaneni A, Jelcic M, Gerlach GF, Ma Y, Overholtzer M, Niethammer P. Lipid peroxidation regulates long-range wound detection through 5-lipoxygenase in zebrafish. Nat Cell Biol. 2020;22(9):1049–55.
Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22(9):1042–8.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85.
Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169(2):286-300 e16.
Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. 2018;22(12):3826–36.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–75.
Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90.
Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628-41 e26.
Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–43.
Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16(3):278–90.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354-63 e3.
Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–96.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–8.
Mukhopadhyay S, Vander Heiden MG, McCormick F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat Cancer. 2021;2(3):271–83.
Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM, et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Can Res. 2020;80(8):1630–43.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–31.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–84.
Venkatesh D, Stockwell BR, Prives C. p21 can be a barrier to ferroptosis independent of p53. Aging (Albany NY). 2020;12(18):17800–14.
Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93(4):1721–41.
Lane DJ, Merlot AM, Huang ML, Bae DH, Jansson PJ, Sahni S, et al. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim Biophys Acta. 2015;1853(5):1130–44.
Patel SJ, Frey AG, Palenchar DJ, Achar S, Bullough KZ, Vashisht A, et al. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat Chem Biol. 2019;15(9):872–81.
Kim MJ, Yun GJ, Kim SE. Metabolic regulation of ferroptosis in cancer. Biology (Basel). 2021;10(2):83.
Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38(1):27.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–45.
Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6(27):24393–403.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–8.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–32.
Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575-86 e4.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604–9.
Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438.
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26(3):420–32.
Distefano AM, Martin MV, Cordoba JP, Bellido AM, D’Ippolito S, Colman SL, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 2017;216(2):463–76.
Hassannia B, Vandenabeele P, Vanden BT. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–49.
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17(4):948–60.
Imoto S, Kono M, Suzuki T, Shibuya Y, Sawamura T, Mizokoshi Y, et al. Haemin-induced cell death in human monocytic cells is consistent with ferroptosis. Transfus Apher Sci. 2018;57(4):524–31.
Lin LS, Song J, Song L, Ke K, Liu Y, Zhou Z, et al. Simultaneous fenton-like ion delivery and glutathione depletion by MnO2 -based nanoagent to enhance chemodynamic therapy. Angew Chem Int Ed Engl. 2018;57(18):4902–6.
Abrams RP, Carroll WL, Woerpel KA. Five-Membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem Biol. 2016;11(5):1305–12.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–71.
Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16(5):e2006203.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103.
Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482(3):419–25.
Cobler L, Zhang H, Suri P, Park C, Timmerman LA. xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget. 2018;9(64):32280–97.
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–34.
Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. 2017;69(6):423–34.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–92.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr Biol. 2018;28(15):2388-99 e5.
Jang EJ, Kim DH, Lee B, Lee EK, Chung KW, Moon KM, et al. Activation of proinflammatory signaling by 4-hydroxynonenal-Src adducts in aged kidneys. Oncotarget. 2016;7(32):50864–74.
Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510(2):278–83.
Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262-79 e25.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20(7):1692–704.
Shah R, Margison K, Pratt DA. The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem Biol. 2017;12(10):2538–45.
Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 2019;26(6):1544-56 e8.
Zhang X, Sui S, Wang L, Li H, Zhang L, Xu S, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol. 2020;235(4):3425–37.
Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339.
Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409-22 e21.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85.
Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162(2):441–51.
Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237–48.
Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139–48.
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24(1):97-108 e4.
Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43.
Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. 2018;128(8):3341–55.
Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett. 2018;416:124–37.
Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 2020;27(4):436–47.
Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500.
Speer RE, Karuppagounder SS, Basso M, Sleiman SF, Kumar A, Brand D, et al. Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: From ferroptosis to stroke. Free Radic Biol Med. 2013;62:26–36.
Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22.
Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238.
Courtney KD, Ma Y, Diaz de Leon A, Christie A, Xie Z, Woolford L, et al. HIF-2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first-in-class HIF-2 inhibitor, in patients with clear cell renal cell carcinoma. Clin Cancer Res. 2020;26(4):793–803.
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617.
Jeschke J, O’Hagan HM, Zhang W, Vatapalli R, Calmon MF, Danilova L, et al. Frequent inactivation of cysteine dioxygenase type 1 contributes to survival of breast cancer cells and resistance to anthracyclines. Clin Cancer Res. 2013;19(12):3201–11.
Zhao H, Li Q, Wang J, Su X, Ng KM, Qiu T, et al. Frequent epigenetic silencing of the folate-metabolising gene cystathionine-beta-synthase in gastrointestinal cancer. PLoS ONE. 2012;7(11):e49683.
Li Y, Zeng X, Lu D, Yin M, Shan M, Gao Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum Reprod. 2021;36(4):951–64.
Lou JS, Zhao LP, Huang ZH, Chen XY, Xu JT, Tai WC, et al. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine. 2021;80:153370.
Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol Rep. 2015;33(3):1465–74.
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2(4):e1054549.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26(5):623-33 e9.
Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15(10):1633–40.
Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR, et al. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother Pharmacol. 2009;64(3):463–72.
Lo M, Ling V, Low C, Wang YZ, Gout PW. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol. 2010;17(3):9–16.
Azadkhan AK, Truelove SC, Aronson JK. The disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br J Clin Pharmacol. 1982;13(4):523–8.
Robe PA, Martin DH, Nguyen-Khac MT, Artesi M, Deprez M, Albert A, et al. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer. 2009;9:372.
Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget. 2016;7(24):36021–33.
Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27(2):211–22.
Anderson CP, Reynolds CP. Synergistic cytotoxicity of buthionine sulfoximine (BSO) and intensive melphalan (L-PAM) for neuroblastoma cell lines established at relapse after myeloablative therapy. Bone Marrow Transplant. 2002;30(3):135–40.
Ongaro A, Pellati A, De Mattei M, De Terlizzi F, Rossi CR, Campana LG. Enhancement of melphalan activity by buthionine sulfoximine and electroporation in melanoma cells. Anticancer Drugs. 2015;26(3):284–92.
Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23(1):120–7.
Mandal PK, Seiler A, Perisic T, Kolle P, Banjac Canak A, Forster H, et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285(29):22244–53.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–92.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–8.
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–76.
Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307.
Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11(11):977–85.
Nishizawa S, Araki H, Ishikawa Y, Kitazawa S, Hata A, Soga T, et al. Low tumor glutathione level as a sensitivity marker for glutamate-cysteine ligase inhibitors. Oncol Lett. 2018;15(6):8735–43.
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85–9.
Ma P, Xiao H, Yu C, Liu J, Cheng Z, Song H, et al. Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett. 2017;17(2):928–37.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50(2):445–60.
Liu Q, Wang K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol Int. 2019;43(11):1245–56.
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9(12):1673–85.
Xiong H, Wang C, Wang Z, Jiang Z, Zhou J, Yao J. Intracellular cascade activated nanosystem for improving ER+ breast cancer therapy through attacking GSH-mediated metabolic vulnerability. J Control Release. 2019;309:145–57.
Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016;381(1):165–75.
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–69.
Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–32.
Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27(1):242–54.
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30(2):146–62.
Nagpal A, Redvers RP, Ling X, Ayton S, Fuentes M, Tavancheh E, et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 2019;21(1):94.
Mai TT, Hamai A, Hienzsch A, Caneque T, Muller S, Wicinski J, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9(10):1025–33.
Jiang M, Qiao M, Zhao C, Deng J, Li X, Zhou C. Targeting ferroptosis for cancer therapy: exploring novel strategies from its mechanisms and role in cancers. Transl Lung Cancer Res. 2020;9(4):1569–84.
Arlt A, Sebens S, Krebs S, Geismann C, Grossmann M, Kruse ML, et al. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene. 2013;32(40):4825–35.
Elgendy SM, Alyammahi SK, Alhamad DW, Abdin SM, Omar HA. Ferroptosis: an emerging approach for targeting cancer stem cells and drug resistance. Crit Rev Oncol Hematol. 2020;155:103095.
Zhou Y, Shen Y, Chen C, Sui X, Yang J, Wang L, et al. The crosstalk between autophagy and ferroptosis: what can we learn to target drug resistance in cancer? Cancer Biol Med. 2019;16(4):630–46.
Kazan HH, Urfali-Mamatoglu C, Gunduz U. Iron metabolism and drug resistance in cancer. Biometals. 2017;30(5):629–41.
Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, Chauffert B, Galmiche A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34(11):6417–22.
Houessinon A, Francois C, Sauzay C, Louandre C, Mongelard G, Godin C, et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol Cancer. 2016;15(1):38.
Stockwin LH, Han B, Yu SX, Hollingshead MG, ElSohly MA, Gul W, et al. Artemisinin dimer anticancer activity correlates with heme-catalyzed reactive oxygen species generation and endoplasmic reticulum stress induction. Int J Cancer. 2009;125(6):1266–75.
Li Z, Li Q, Wu J, Wang M, Yu J. Artemisinin and its derivatives as a repurposing anticancer agent: what else do we need to do? Molecules. 2016;21(10):1331.
Bjarnadottir O, Romero Q, Bendahl PO, Jirstrom K, Ryden L, Loman N, et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. 2013;138(2):499–508.
Garwood ER, Kumar AS, Baehner FL, Moore DH, Au A, Hylton N, et al. Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res Treat. 2010;119(1):137–44.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6.
Sleire L, Skeie BS, Netland IA, Forde HE, Dodoo E, Selheim F, et al. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene. 2015;34(49):5951–9.
Nagane M, Kanai E, Shibata Y, Shimizu T, Yoshioka C, Maruo T, et al. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE. 2018;13(4):e0195151.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15(2):469–84.
Shibata Y, Yasui H, Higashikawa K, Miyamoto N, Kuge Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE. 2019;14(12):e0225931.
Pan X, Lin Z, Jiang D, Yu Y, Yang D, Zhou H, et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett. 2019;17(3):3001–8.
Zhang F, Li F, Lu GH, Nie W, Zhang L, Lv Y, et al. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano. 2019;13(5):5662–73.
Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16(11):2069–83.
Zheng H, Jiang J, Xu S, Liu W, Xie Q, Cai X, et al. Nanoparticle-induced ferroptosis: detection methods, mechanisms and applications. Nanoscale. 2021;13(4):2266–85.
Wang S, Li F, Qiao R, Hu X, Liao H, Chen L, et al. Arginine-rich manganese silicate nanobubbles as a ferroptosis-inducing agent for tumor-targeted theranostics. ACS Nano. 2018;12(12):12380–92.
Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li C, et al. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. 2017;17(1):284–91.
Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci Transl Med. 2014;6(260):260ra149.
Hu P, Wu T, Fan W, Chen L, Liu Y, Ni D, et al. Near infrared-assisted Fenton reaction for tumor-specific and mitochondrial DNA-targeted photochemotherapy. Biomaterials. 2017;141:86–95.
He Y, Del Valle A, Qian Y, Huang YF. Near infrared light-mediated enhancement of reactive oxygen species generation through electron transfer from graphene oxide to iron hydroxide/oxide. Nanoscale. 2017;9(4):1559–66.
Zhu T, Shi L, Yu C, Dong Y, Qiu F, Shen L, et al. Ferroptosis promotes photodynamic therapy: supramolecular photosensitizer-inducer nanodrug for enhanced cancer treatment. Theranostics. 2019;9(11):3293–307.
Sun X, Cao Z, Mao K, Wu C, Chen H, Wang J, et al. Photodynamic therapy produces enhanced efficacy of antitumor immunotherapy by simultaneously inducing intratumoral release of sorafenib. Biomaterials. 2020;240:119845.
Xu T, Ma Y, Yuan Q, Hu H, Hu X, Qian Z, et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS Nano. 2020;14(3):3414–25.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–7.
Guan Q, Guo R, Huang S, Zhang F, Liu J, Wang Z, et al. Mesoporous polydopamine carrying sorafenib and SPIO nanoparticles for MRI-guided ferroptosis cancer therapy. J Control Release. 2020;320:392–403.
Zhang D, Cui P, Dai Z, Yang B, Yao X, Liu Q, et al. Tumor microenvironment responsive FePt/MoS2 nanocomposites with chemotherapy and photothermal therapy for enhancing cancer immunotherapy. Nanoscale. 2019;11(42):19912–22.
Acknowledgements
We would like to thank the Jilin University for supporting this manuscript.
Funding
Project supported by the National Science Foundation of JiLin Province, China (Grant No.20210101337JC).
Author information
Authors and Affiliations
Contributions
QN drafted this article. YH revised the article, XY and XL contributed to the acquisition and sorting of the data for this article. XF designed this project. All authors participated in the review, editing and revision of the manuscript, and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This review does not involve ethics approval and consent to participate.
Consent for publication
We agree to the publication of this article.
Competing interests
The authors declare no conflict of interest and have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nie, Q., Hu, Y., Yu, X. et al. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int 22, 12 (2022). https://doi.org/10.1186/s12935-021-02366-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02366-0