
Abstract
Thyroid carcinoma is a common malignant tumor of endocrine system and head and neck. Recurrence, metastasis and high malignant expression after routine treatment are serious clinical problems, so it is of great significance to explore its mechanism and find action targets. Epithelial–mesenchymal transition (EMT) is associated with tumor malignancy and invasion. One key change in tumour EMT is low expression of E-cadherin. Therefore, this article reviews the expression of E-cadherin in thyroid cancers (TC), discuss the potential mechanisms involved, and outline opportunities to exploit E-cadherin on regulating the occurrence of EMT as a critical factor in cancer therapeutics.
Similar content being viewed by others


Introduction
Thyroid cancer (TC) is one of the common head and neck tumors, which incidence rate increasing year by year. According to statistics, thyroid cancer is the fourth most common cancer in women, accounting for 5.7% of female malignant tumors [1]. The pathological classification included differentiated thyroid carcinoma (DTC) and undifferentiated thyroid carcinoma (UTC). Both of them can have lymph node metastasis, lung metastasis and are more likely to have distant and early bone metastasis, which seriously affect the prognosis and survival of patient [2]. Therefore, it is of great significance to explore the regulatory mechanism related to the metastasis of thyroid cancer. In the related studies of tumors, it has been shown that the malignant expression, metastasis and invasiveness of tumors are all related to epithelial–mesenchymal transition (EMT) [3].
Epithelial–mesenchymal transition (EMT) means that epithelial cells, stimulated by certain cancer-promoting factors, lose epithelial characteristics and their stable cell structure is destroyed and their cytoskeleton recombines, which leads to the weakening or disappearance of intercellular connections, eventually causing cells to express interstitial strong interstimic properties [4]. EMT in malignant tumor is a necessary process of metastasis and invasion, participating in the growth and development of cells and regulating the adhesion of cells [5,6,7,8]. The hallmarks of EMT are low expression of the epithelial marker E-cadherin, and high expression of the mesenchymal markers N-cadherin and Vimentin [4].
E-cadherin, also known as epithelial-cell adhesion molecule cadherin-1 (Cadherin1, Cdh1), is one of the classical adhesion proteins in the calcium adhesion superfamily, mainly expressed in epithelial cells [8, 9]. E-cadherin maintains cell adhesion and epithelial structural integrity, becoming a key marker for regulating the occurrence of EMT [5, 7, 10]. Based on its physiological characteristics, the increased invasiveness of epithelial tumors is related to the low expression of E-cadherin [8]. For example, in colorectal cancer, gastric cancer, breast cancer and other cancers, E-cadherin has been identified as a tumor suppressor gene affecting epithelial mesenchymal transformation, and is closely related to TNM stage, lymph node metastasis, extracapsular invasion and low disease-free survival rate [11, 12]. Moreover, in recent years, there are more and more studies on the inhibition of epithelial mesenchymal transformation and metastasis of thyroid cancer by regulating the expression of E-cadherin [13, 14]. It has been found that E-cadherin in TC is regulated by many signal pathways such as Wnt, PI3K/AKT, ERK1/2 and NF-κB. Therefore, the expression and regulation mechanism of E-cadherin in TC are reviewed signaling pathway of regulating E-cadherin, immune microenvironment, RNA and extracellular matrix to provide targets and new ideas for early diagnosis and treatment of thyroid tumors.
Signaling pathway of regulating E-cadherin
Wnt/β-catenin signaling pathway involved in the regulation of E-cadherin
Wnt signal pathway includes classical Wnt pathway (Wnt/β-catenin), non-classical Wnt pathway and Wnt/Ca2+ pathway, among which Wnt/β-catenin signal pathway is the most widely and fully studied pathway [15]. The activity of Wnt/β-catenin signaling pathway depends on the content of β-catenin in the cytoplasm [16]. When the Wnt signal is activated, the Wnt ligand binds to the receptor complex and then inhibits the phosphorylation of β-catenin, resulting in the accumulation of β-catenin in the cytoplasm and nuclear transfer to regulate the downstream target genes to affect the apoptosis and proliferation of the cells [16, 17].
The activation of Wnt/β-catenin signal pathway promotes the proliferation and stem cell characteristics of TC [15]. FOXN3, acting as anti-tumor effect in thyroid tumors, inhibit the occurrence of EMT by down-regulating the expression of β-catenin protein and up-regulating the expression of E-cadherin [18]. However, after activating the Wnt/β-catenin signaling pathway, invasion and migration as well as the occurrence of EMT are promoted, manifested by low expression of E-cadherin [19].
In addition to participating in gene regulation as an intermediary of Wnt signal pathway, β-catenin binds to E-cadherin in the absence of Wnt signal, and forms adhesion junctions (adherin junction, AJ) with α-catenin and γ-catenin connections, thus playing the role of adhesion and maintaining cell-to-cell stability [20, 21]. Abnormal expression of the complex formed by β-catenin and E-cadherin is closely related to the metastasis and recurrence of thyroid tumors [22]. A clinical study has shown that abnormal expression of β-catenin and deletion of E-cadherin on membrane in PTC patients, which may lead to low survival rate of patients [23]. However, when the above abnormal conditions are improved, the tumor grows slowly [23]. The use of Wnt inhibitor (DKK-1) can cause the accumulation of β-catenin in the cytoplasm and promote its formation of a complex with E-cadherin to maintain cell stability and inhibit the occurrence of EMT, thereby reducing cell migration and invasion [24]. On the other hand, the abnormal expression of them is related to the malignant degree of TC. Studies have shown that as the degree of differentiation increases E-cadherin is mainly expressed in the membrane and cytoplasm, while β-catenin is expressed in the cytoplasm, while E-cadherin and β-catenin almost disappear in poorly differentiated TC, which is related to the higher the TNM [25].
PI3K/AKT and MAPK signaling pathway involved in the regulation of E-cadherin
PI3K/AKT is a common intracellular signal pathway. It has been found that PI3K/AKT signal pathway not only participates in cell proliferation and apoptosis, but also plays an important role in tumor growth, metastasis and tumor angiogenesis [26, 27]. It has been found that the activation of P13K/AKT signal pathway is related to the low expression of E-cadherin in thyroid carcinoma [28]. The PI3K/AKT signaling pathway in thyroid carcinoma is closely related to the high expression of oncogenes and the low expression of tumor suppressor genes. For example, the high expression of oncogenes HPIP, CUX2, miR-107 and miR-144-3p in thyroid carcinoma can activate PI3K/AKT signal pathway and down-regulate the expression of E-cadherin to induce the occurrence of EMT [29,30,31,32]. However, the low expression of tumor suppressor genes miR-146b and ING5 in thyroid carcinoma can not inhibit the expression of PI3K/AKT, which leads to the low expression of E-cadherin and induces the occurrence of EMT [33, 34].
From the above, it can be seen that there are many genes activating PI3K/AKT signal pathway in thyroid carcinoma. The mechanism of PI3K/AKT regulating E-cadherin can be summarized as follows. It has been found that activating AKT can destroy the adhesion junction structure (composed of E-cadherin, α-catenin, β-catenin and p130Cas proteins) between tumor cells, showing low expression of related proteins [35]. And the activation of AKT can directly induce the expression of Snail and then down-regulate the expression of E-cadherin to promote EMT [35]. The activation of AKT also exists in the thyroid carcinoma, and its activation can lead to the low expression of E-cadherin to induce EMT [36, 37]. The activation from gene to PI3K/AKT signal pathway seems to be closely related to PTEN and BRAF (V600E). PTEN and BRAF (V600E) are confirmed mutant genes in thyroid carcinoma, and they are closely related to the clinical manifestations of thyroid carcinoma malignancy [36, 38, 39]. PI3K molecules are divided into three categories, among which class I PI3K is closely related to carcinogenesis [40, 41]. The phosphorylation of PIP2 located on the inside of the lipid membrane promotes the phosphorylation of PIP3 and after AKT is recruited to the membrane, they bind to form p-AKT and finally activates the PI3K/AKT signal pathway [42].
PTEN, as a tumor suppressor, can reverse the phosphorylation of PIP3 and the recruitment of AKT to the inner membrane by terminating the signal transduction of PI3K/AKT pathway [26, 43, 44]. For example, inhibition of miR-146b in TC can promote the expression of PTEN, then inhibit AKT activation, and promote the expression of E-cadherin to inhibit EMT [45]. In addition, other genes such as F-box11, HPIP and DTX can regulate E-cadherin through the PI3K/AKT pathway, but the mechanism is still unclear [29, 46, 47]. In addition, some studies have found that the activation of PI3K/AKT signal pathway may be related to M2-like macrophages [48]. It has been found that when M2-like macrophages are co-cultured with thyroid cancer cells, the secretion of IGF-1 and IGF-2 by M2-like macrophages can activate PI3K/AKT signal pathway to reduce the expression of E-cadherin, thus induce the occurrence of EMT [48].
Mitogen-activated protein kinase (MAPK) pathway is a pathway that activates mitogen-activated protein (MAP) through a phosphate cascade signal cascade (Ras/RAF/MEK/ERK), via intracellular cascade proteins that eventually transfer signals into the nucleus to participate in process such as embryonic development, cell differentiation, proliferation and death [49, 50]. It has been found that RET and TRK rearrangement, BRAF and Ras mutations and B-Raf kinase in MAPKs signal pathway in TC lead to abnormal expression of MAPK/ERK, JNK/SAPK and p38/MAPK signal pathway [50, 51]. Silencing Kin17, DPP4 in TC can inhibit ERK1/2, JNK1 and P38, while promote E-cadherin, which may be related to the inhibition of MAPK/ERK, JNK/SAPK and p38/MAP signal pathways [52, 53]. MAPK/ERK signal pathway in TC can inhibit the expression of E-cadherin by promoting the expression of transcription factors Snail and slug [54].
TGF-β involved in the regulation of E-cadherin
In tumors, TGF-β functions are complex and diverse due to different backgrounds and stages of tumors, not only acting as a suppressor to inhibit the occurrence and development of tumor cells in the early stage, but also to inhibit the invasion and metastasis in the late stage [55, 56]. TGF-β pathway includes classical and non-classical pathways, among which the classical TGF-β pathway plays a major role in the occurrence of EMT. Relevant literature has shown that at the later stage of tumor progression TGF-β affects the metastasis of tumor cells by down-regulating the expression of E-cadherin [57, 58]. For example, in thyroid follicular carcinoma membranous mice (ThrbPV/PV mice), the continuous activation of TGF-β/Smad2/3 signal pathway can reduce the expression of E-cadherin, thus promoting the occurrence of EMT [59, 60].
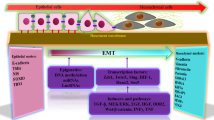
The classical TGF-β signal pathway is called SMAD signal pathway, in which TGF-β ligand specifically binds to T-β RI and T β-RII receptors, transmits the signal into the cell, induces the phosphorylation of Smad2 and Smad3 and forms a complex with Smad4 to aggregate into the nucleus and regulate the expression of target genes, while Smad6 and Smad7 inhibit the generation of classical TGF-β signaling pathways by competitively binding T-βR to Smad2 and Smad3 [56]. It has been found that TGF-β regulates the expression of E-cadherin in TC through a variety of ways, thus participating in the invasion and metastasis of TC. For example, an experiment showed that by down-regulating Smurf, the ubiquitin ligase of Smad3, the TFG-β/SMAD pathway could be affected, thus down-regulating the expression of EMT marker E-cadherin in thyroid cancer [61]. Another study found that E-cadherin was inhibited by affecting the expression of TGF-β/Smad2/3 pathway [62, 63]. It may be related to the regulation of the transcription factor Snail and the tumor suppressor gene BRAFV600E. A study has shown that the BRAFV600E gene mutation in thyroid tumors can down-regulate the expression of E-cadherin and induce the occurrence of EMT, and its abnormal expression in thyroid tumors increases the secretion of TGF-β, which further induces the occurrence of EMT and invasion, manifested as decreased expression of E-cadherin [64, 65]. However, the effect of TGF-β is extremely complex. Unlike the previous study, a clinical study found that the metastasis of thyroid tumors was not related to TGF-β but might be related to the overexpression of BRAFV600E in thyroid tumors and the activation of the transcription factor Snail, which decreased the expression of E-cadherin and ultimately promoted the occurrence of EMT [65]. This further indicates that TGF-β-related pathway can regulate the expression of E-cadherin and affect the occurrence of EMT, and thus affect the metastasis of thyroid cancer (Fig. 1).

Pathway diagram for regulation of E-cadherin in thyroid cancer
E-cadherin is related to the immunity/EMT/metastasis axis
The progression of malignant tumors is associated with inflammatory responses, and inflammation may be a key precipitating factor for the development of EMT during tumor progression [66, 67]. Tumor cells form a chronic inflammatory microenvironment by recruiting immune cells and inflammatory cytokines [68]. In the chronic inflammatory microenvironment created by tumor cells, some immune cells and soluble inflammatory mediators can recruit immunosuppressive cells, damage immune cells or reduce the immunogenicity of cancer cells, thus enabling abnormal cells to escape immune surveillance to form tumors [69]. Immune cells promote the diffusion and migration of tumor cells through matrix remodeling, induction of tumor cell invasion, migration and vascular infiltration [68]. This may be the mechanism of inflammatory reaction involved in tumorigenesis and development. Inflammatory cells continuously produce cytokines that induce EMT, while EMT can promote cancer cells to produce pro-inflammatory cytokines, which support each other and promote tumorigenesis and metastasis [70]. This form that EMT/inflammation axis. It can be seen that there is a bidirectional regulation between EMT and the immune system, and immune escape and immunosuppression play a key role in in tumor microenvironment. So this will be the focus of the next exposition.
Immune cells involved in the regulation of E-cadherin
Innate immune cells involved in the regulation of E-cadherin
There are many kinds of innate immune cells in the immune microenvironment created by tumor tissues, in which macrophages and mast cells play an important role in the invasion and migration of thyroid cancer. As the largest number of immune cells in tumor microenvironment, macrophages can promote the malignant progression of tumor [71, 72]. Existing studies have shown that the polarization of macrophages into M2 macrophages (MA-TAMs) in tumor microenvironment is closely related to the malignant phenotype of tumor [70]. M2 macrophages can stimulate angiogenesis, inhibit T cell activity and release soluble cytokines to make tumor cells aggressive [73, 74]. A large number of studies have shown that M2 macrophages induce EMT by down-regulating the expression of E-cadherin [75,76,77]. In thyroid tumors, M2 macrophages can down-regulate the expression of Ecadherin, promote cell invasion and migration, and induce the occurrence of EMT [78]. There are a large number of inflammatory factors (such as TGF-β, TNF-α and IL) in the polarized environment of macrophages [76, 79]. These inflammatory factors can down-regulate the expression of β-catenin, thus inhibit the activation of Wnt/β-catenin signal pathway, and then up-regulate the expression of Ecadherin and inhibit EMT [77, 80]. In addition, the up-regulation of Wnt1 and Wnt3a is also present in the M2-TAM environment of thyroid cancer [78]. After gene knockout of Wnt1 and Wnt3a, the expression of β-catenin can be significantly decreased, then the Wnt signal pathway can be inhibited to up-regulate E-cadherin to inhibit EMT [78].
In addition, it was found that mast cells could also promote the occurrence of tumor EMT [81,82,83]. Mast cell infiltration also exists in the microenvironment of thyroid carcinoma, and the infiltration density of mast cells is closely related to the invasion of thyroid carcinoma [84]. Soluble factors (such as IL-8, IL-6, TBF- α, CXCR-1) and VEGF secreted by thyroid cancer cells can activate macrophages and recruit macrophages into tumor tissues [84, 85]. On the other hand, the activated macrophages secreted vesicles and mast cell chymase (MCC) could degrade the extracellular matrix and inhibit the expression of E-cadherin by overexpression of transcription factors Slug and Snail, thus inducing the occurrence of EMT [28, 86]. Or activated mast cells regulate the occurrence of EMT by regulating downstream kinases and promote tumor invasion and metastasis [85]. Thus a biphasic regulatory loop is produced in the local part of the tumor. A study shows that mast cells activated by thyroid cancer may induce EMT through IL-8-p-Akt-Slug signal pathway, and transcription factor Slug activation can inhibit the expression of E-cadherin and promote EMT [85]. To sum up, there is a biphasic regulatory loop between thyroid carcinoma and mast cells in the tumor. The cytokines produced by cancer cells activate mast cells, and mast cells promote the expression of transcription factors by activating downstream kinases, thus affecting the occurrence of EMT by regulating Ecadherin.
Adaptive immune cells involved in the regulation of E-cadherin
Many studies have shown that chronic inflammation has a pro-cancer effect in TC [87]. Immunosuppression of tumor infiltrating lymphocytes has been confirmed in advanced cancer. Similarly, this feature also exists in thyroid cancer. Through the immunohistochemical study of the tumor tissues of 74 patients with PTC, it was found that the characteristics of EMT (positive expression of E-cadherin, negative expression of Vimentin) and down-regulation of CD8+ surface markers occurred at the same time, and the characteristic changes were more significant in patients with lymph node metastasis and lymphatic invasion [88]. Through literature search, this process may be related to PD-1, a programmed death protein [89]. PD-1 exists on the surface of activated T cells, which mainly inhibits the activity of immune cells and regulates peripheral immune homeostasis [89]. Its ligand, programmed cell death ligand (PD-L1), as an immune checkpoint, its activation with EMT and its role in inhibiting tumor immune response have been confirmed [90,91,92]. There is also overexpression of PDL1 in thyroid carcinoma. It was found that there was a significant positive correlation between the positive expression rate of PD-L1 and mesenchymal phenotype in patients with PTC (P = 0.012), and the correlation between PD-L1 and EMT was enhanced in patients with recurrent and metastatic disease [88]. The induction of PD-L1 overexpression in 8505C and K1 cells could significantly promote the occurrence of EMT, showing the transformation of pebble-like cells into extensive spindle-like cells [88]. The above results show that the activation of EMT is related to PD-L1. This regulatory process, on the one hand, is due to the fact that the promoter region of PD-L1 gene contains Zeb1 binding sites [93, 94]. Zeb1 is a transcription factor of E-cadherin, which can induce EMT and maintain the stem cell nature of cancer cells to contribute to tumorigenesis [93]. A study found that Zeb1 can relieve the inhibition of PD-L1 in tumor cells, resulting in a decrease in the number of CD8+ T cells and an increase in the percentage of exhausted CD8+ T cells, leading to immunosuppression and metastatic nodules [95]. Another part of the view is that the occurrence of thyroid cancer EMT is related to glycosylation. A large number of studies have confirmed the key role of abnormal glycosylation in malignant tumors [96, 97]. Similarly, there is abnormal glycosylation in thyroid carcinoma, which may be related to FTU8, a fucosyltransferase [98]. FTU8 can mediate core fucosylation and thus participate in tumor invasion and metastasis [99, 100]. Through the study of lung and colon cancer, it was found that the up-regulation of FUT8 could increase the core fucosylation level of E-cadherin and decrease the expression of Ecadherin protein [99, 101]. There are N-glycosylation sites on the surface of PD-1 protein on T cell surface [102]. Through FTU8 gene knockout or drug inhibition of FUT8 activity, it can inhibit the expression of PD-1 on T cell surface, thus promote the activation of T cells and inhibit tumorigenesis [102]. Through the preparation of FUT8 gene knockout and FUT8−/− mouse model, it was found that the activity of FUT8 regulates the total amount of E-cadherin [99]. Therefore, the regulation of EMT by T cells through E-cadherin may be related to PD-1, and this regulation process may be related to transcription factor Zeb1 and glycosylation.
Inflammatory factors involved in the regulation of E-cadherin
Inflammatory cytokines play an important role in maintaining inflammatory microenvironment and tumor progression [103]. Abnormal expression of different types of cytokines can be found in different types of cancer. For example, in breast cancer [104], lung cancer [105] and liver cancer [106], the abnormal expression of inflammatory cytokines is closely related to the progression of tumor migration. There is also abnormal expression of inflammatory cytokines in thyroid carcinoma. The expression of IL-34 was up-regulated in serum and tissue samples of PTC, and the overexpression of IL-34 was significantly correlated with distant metastasis and lymphatic metastasis of tumor [13, 107]. Tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) can induce the malignant development of thyroid cancer cell line, change the cell morphology from fat circle to spindle shape gradually, increase the invasion and migration ability of cells, decrease E-cadherin and increase the expression of N-cadherin and Vimentin at protein level [101, 108]. After stimulated by TNF-α and IFN-γ, E-cadherin was located in the cytoplasm and N-cadherin was located on the cell membrane, and the cancer cells showed interstitial characteristics [101]. A study found that IL-1a can cause ultrastructural changes in thyroid follicular cells, reduce the tightness between thyroid epithelial cells, and lead to the disintegration of intercellular adhesion junction structure (AJ), composed of E-cadherin and α-catenin, β-catenin, γ-catenin [109]. Further study found that the decreased expression of E-cadherin protein in thyroid cancer cell lines stimulated by inflammatory factors may be related to the activation of transcription factors Snail, Slug, Twist1 and Zeb1 [14]. This regulatory process can inhibit the invasion and migration of thyroid cancer cells induced by inflammatory cytokines by using AKT, NF-κB and SATAT3 signaling pathway [14]. Nuclear factor kappa B (NF-κB) is a transcription factor that regulates apoptosis, inflammation and immune response [110]. With studies suggesting that NF-κB may be a key link between inflammation and cancer, it’s overactivation is related to the proliferation, angiogenesis and metastasis of many malignant tumors and can serve as a potent inductor of EMT [110,111,112]. Similarly, the abnormal activation of NF-κB in thyroid carcinoma has been shown to be related to tumor proliferation, invasion and migration [113, 114]. Through the study of NF-κB in other tumors, its regulatory mechanism may be related to the NK-b/Twist axis [115, 116]. After knockout of NF-κB, the proliferation and migration ability of cells stimulated by TNF-α decreased, and the expression of E-cadherin was up-regulated, N-cadherin and Twsit was down-regulated [115]. However, the inhibitory effect of knockout Twist was the same as before [115]. After the thyroid cancer cells were treated with TNF-α, the expression of Twist mRNA was up-regulated and then the expression of E-cadherin was down-regulated [116]. After the use of NF-κB inhibitor, the expression of Twist mRNA and protein was inhibited, and the expression of E-cadherin protein was up-regulated [116]. To sum up, inflammatory cytokines may activate transcription factors through signal pathways, thus down-regulating E-cadherin from promoting the occurrence of EMT.
E-cadherin is related to RNAs
MicroRNAsinvolved in the regulation of E-cadherin
MicroRNAs (miRNAs or miRs) are non-coding small RNA that is transcribed from DNA into a precursor miRNAs (pri-miRNAs) in the nucleus and processed into mature miRNAs for export to the cytoplasm, where they can then degrade or regulate mRNA by targeting the 3′-untranslated region, thereby regulating cell growth, cycle, apoptosis and other biological processes [52, 117]. MiRNA can be used as both a tumor suppressor gene and a tumor promoting gene to regulate the occurrence of tumor EMT in thyroid carcinoma. For example, overexpression of miR-203, MIR-597-3p, MIR-200b, miR-122 and miR-146b-5p significantly increase the expression of E-cadherin and decrease the expression of Ncadherin or Vimentin [118,119,120,121,122]. On the contrary, MiR-144-3p and miR-483-3p in thyroid carcinom, as an oncogene, is overexpressed in PTC to reduce E-cadherin to induce EMT [123, 124].
Although miRNAs is abundant in thyroid carcinoma, its expression is high or low to regulate the expression of Ecadherin. The regulatory mechanism is that miRNA affects target gene transcription by binding to the 3′-UTR of target gene mRNA [52, 117]. For example, in TPC-1 cells, miR-599 as a suppressor gene, up-regulation of its expression can be targeted to bind to the 3'-UTR of Hey2, thus down-regulating its expression [125]. Down-regulation of Hey2 expression inhibits Notch signal pathway, suppresses the expression of transcription factors Snail and Slug, thus up-regulates the expression of E-cadherin and inhibits the occurrence of EMT [125]. MiR-483-3p inhibits the expression of PARD3 by targeting the 3′-UTR of it, and then up-regulates the expression of Snail, Slug, Zeb1 and Twist to inhibit the expression of E-cadherin and finally induce EMT [124]. MiR-31 upregulates the expression of E-cadherin through targeted inhibition of Sox11 to inhibit ERK and AKT signaling pathways, thus inhibiting EMT [126]. In addition, miR199a-5p can directly inhibit the expression of Snail to up-regulate the expression of E-cadherin and induce EMT [127]. Many other situations in which miRNAS regulates E-cadherin are shown in Table 1.
lncRNAs involved in the regulation of E-cadherin
LncRNAs are a type of coding gene that transcribes > 200 nucleotides in length and don’t encode protein, acting as a separate transcriptional unit, an intron of an enhancer (ERNAs) or promoter [134]. Cancer is now thought to occur in association with genetic mutations, and since the coding genome accounts for less than 2% of all sequences, studies suggest that cancer may also be driven by aberrations in the non-coding genome [53]. lncRNAs participate in the regulation of tumor cell proliferation, growth inhibition, invasion and metastasis and EMT [53, 135]. In thyroid tumors, the regulation of EMT by lncRNAs is bidirectional. The overexpression of lncRNAs may lead to the low expression of E-cadherin in thyroid tumors and cancer cells, which can induce EMT, while the low expression of lncRNA may also occur the above process. For example, there are high expressions of lncRNA-HOTAIR, lncRNA-NORAD, lncRNA-linc00673, lncRNA-SLC26A4-AS1, lncRNA-ROR, lncRNA-PVT1, lncRNA-HOTAIR and other lncRNAs in thyroid cancer patients, which are positively correlated with tumor malignant phenotype and lymph node metastasis, while lncRNAs knockout in thyroid cancer cells can up-regulate the expression of E-cadherin protein and inhibit the occurrence of EMT [19, 136,137,138,139,140]. On the contrary, the low expression of lncRNA-CASC2 and lncRNA-linc 00106 in thyroid carcinoma was positively correlated with lymph node metastasis, and up-regulation of lncRNAs could up-regulate the expression of E-cadherin protein and inhibit the occurrence of EMT [141,142,143]. In addition, the expression level of the same lncRNA is also different in different thyroid cancer tumors. For example, the expression level of lncRNA-BANCR in bCPAP is lower than that of CAL-62, WRO and FTC-133 [144]. MALAT1 is highly expressed in PTC, as an oncogene to promote tumor EMT, and as a tumor suppressor gene to inhibit tumorigenesis in poorly differentiated and anaplastic thyroid carcinoma [145].
There are many kinds of lncRNAs, but the mechanism of regulating E-cadherin can be summarized as the following three points. First of all, lncRNAs can play a sponge role on miRNAs to inhibit the expression of miRNAs, thus affecting the expression of downstream transcription factors by regulating related signal pathways to regulate the expression of E-cadherin, and finally regulate the occurrence of EMT. Overexpression of LncRNA-UCA1 can down-regulate the expression of transcription factor Snail by activating Hippo and JNK signal pathways, and up-regulate the expression of E-cadherin to inhibit the occurrence of EMT [146]. In addition, lnc-TUG1, lnc-NORAD and LINC02471 inhibit the expression of E-cadherin by regulating the expression of miRNAs and promoting the expression of transcription factors Snail, Slug and Zeb1, thus promoting the occurrence of EMT [136, 147, 148]. Secondly, lncRNAs can directly regulate the expression of tumor-related genes, and then regulate the expression of E-cadherin. For example, KLF-2-like factor 2 (KLLF2), as a tumor suppressor gene, has low expression in thyroid carcinoma and is associated with lymph node metastasis, malignant histological type and high TNM stage [149]. LIN00673 is highly expressed in thyroid carcinoma and knockout of LIN00673 can enhance the expression of KLF-2 and increase the expression of E-cadherin, thus inhibit the occurrence of EMT [149]. On the contrary, CRABP2 (retinoic acid binding protein 2) is a necessary protein for tumor growth [150]. Overexpression of LINC01816 in thyroid carcinoma can target miR-34c-5p to act as a sponge, and then up-regulate the expression of CRABP2 and inhibit the expression of E-cadherin, thus promoting the occurrence of EMT [151]. Thirdly, LncRNAs can directly regulate the relevant signal pathways to regulate E-cadherin. The overexpression of lnc-BANCR in bCPAP cells can down-regulate the expression of E-cadherin and inhibit EMT by activating Raf/MEK/ERK signal pathway [144]. LncRNA-HOTAIR is highly expressed in thyroid carcinoma [19]. Inhibition of HOTAIR can decrease the expression of Wnt inhibitory factor (WIFI), up-regulate β-catenin, activate Wnt/β-catenin signal pathway, and then down-regulate E-cadherin to promote the occurrence of EMT [19]. On the contrary, knockout of LINC00106 gene can down-regulate E-cadherin and β-catenin, and induce the occurrence of EMT [152]. LINC00313 promotes the methylation of ALX4 promoter by recruiting methylated proteins, while LINC00313 knockout can inhibit the methylation of ALX4 [153]. The accumulation of ALX4 in the cytoplasm can inactivate the AKT/mTOR signal pathway and down-regulate the expression of E-cadherin, thus promoting the occurrence of EMT [153]. In thyroid carcinoma, lncRNA-ZFAS1 and its transcription factor GREB3 are up-regulated [154]. GREB3 activates the expression of lncRNA-ZFAS1 and then targets to inhibit the expression of miR373-3p/MMP-3 to up-regulation of Slug and Snail, and then down-regulation of E-cadherin to promote the occurrence of EMT [154] (Table 2).
E-cadherin is related to extracellular matrix
Matrix metalloproteinases family (MMPs) involved in the regulation of E-cadherin
Extracellular matrix participates in cell movement and apoptosis and provides cytoskeleton support, of which matrix metalloproteinases is the major components [155]. MMPs are zinc-dependent endopeptidases that degrade various protein components of the extracellular matrix (ECM) and are closely related to tumor invasion, metastasis, and angiogenesis [156,157,158]. In the early growth phase of tumor, MMPs inhibitor restrain tumor growth and degrade it [159]. MMPs promote the development of EMT in tumor cells by regulating a variety of regulatory factors [157]. TGF-β can promote EMT in TC, manifested as low expression of E-cadherin, while inhibiting the expression of MMP-9 can inhibit the occurrence [160]. EGFR is related to angiogenesis and tumor growth. MMP-2/9 activates EGFR to regulate E-cadherin through ERK1/2 and AKT/GSK3-β/β-catenin [161]. Twist (an E-cadherin transcription factor) can induce the formation of actin enrichment membrane protrusions that recruit MMP-7, MMP-9 and MMP-14 on the membrane to degrade locally and destroy the cytoskeleton [162]. The above studies may be a powerful explanation for the regulation of E-cadherin by MMPs to promote tumor invasion and migration.
Different cadherins involved in the regulation of E-cadherin
Cadherin is a Ca2+-dependent intercellular adhesion molecule, which acts as a bridge between cells in the extracellular matrix and participates in the regulation of tumor invasion, migration and angiogenesis [163]. E-cadherin (CdH1), as a tumor suppressor of EMT, has a significant correlation with malignant phenotype, invasion and migration of tumor cells [164]. In addition, cadherin also includes CDH5 (VE-Cadherin), cadherin 6 (CDH6), cadherin 17 (CDH17), N-cadherin and so on, which play a similar role to E-cadherin in EMT, or regulate E-cadherin through some pathways.
Abnormal expression of N-cadherin in thyroid tumors significantly inhibits expression of E-cadherin, the conversion of E-cadherin to N-cadherin is the key factor to promote the development of malignant tumors [6]. A study has shown that E-cadherin expression can be up-regulated by inhibiting the expression of N-cadherin in thyroid tumors, possibly by promoting the expression of transcription factors Twist, Snail, and Slug to promote the occurrence of EMT and invasive migration [165]. Studies have shown that CDH16 appears to be another marker of EMT, regardless of E-cadherin negative or positive expression, CDH16 is negative and declined to a greater extent than E-cadherin [166]. RGD motifs in CDH17 and CDH5 promote tumor migration and invasion by activating α2β1 integrin signal and CDH6 activating αIIbβ3 integrin signal [167, 168]. Placental adhesin (CDH3) promote the growth, migration and invasion of TC, which may play a role by up-regulating E-cadherin and down-regulating N-cadherin [169]. E-cadherin is not expressed in TPC1 and bCPAP, but N-cadherin is down-regulated by silencing CDH6 to inhibit EMT, which may be regulated by the inhibition of forming autophagosome by the combination of LC3 and Flag-GABARAP after CDH6 silencing [170].
Conclusion
The article reviews the expression of E-cadherin in TC and the related pathways regulating its expression. To sum up, it can be concluded that the low expression of E-cadherin in thyroid carcinoma promotes the invasion and migration of tumor cells, thus inducing the occurrence of EMT. The signaling pathways regulating this process include Wnt/β-catenin, PI3K/AKT, MAPK and TGF-β signaling pathways, as well as tumor microenvironment regulated by immune system, miRNA and lncRNA regulating oncogenes or tumor suppressor genes and extracellular matrix. By inhibiting E-cadherin, it reduces the contact between cells, induces the occurrence of EMT, and leads to the movement of thyroid tumor. From in vitro and in vivo studies, E-cadherin may be a promising biomarker for malignant phenotypes such as tumor invasiveness, distant metastasis and lymph node metastasis, which may make E-cadherin an indicator of early diagnosis and prognosis of TCs.
As the adhesion between protein-dimensional cells on the membrane, E-cadherin surface molecules show dysfunction when tumor cells metastasize and decompose into molecules soluble in extracellular matrix and blood, so the changes of E-cadherin can be detected in patient’s serum [171, 172]. Serum E-cadherin is higher in thyroid papillary carcinoma than in benign nodules and normal tissues, while the expression of E-cadherin in thyroid papillary carcinoma was lower than that in paracancerous tissues, and it was significantly correlated with malignant phenotypes such as lymphatic metastasis and distant metastasis [158, 172]. And the low expression of E-cadherin in cancer tissues may be related to the activation of its promoter methylation [13]. It has been found that thyroid cancers with lymphatic metastasis are more likely to have Ecadherin methylation and low expression of E-cadherin [13]. Thyroid tumors are divided into benign tumor, differentiated thyroid carcinoma, poorly differentiated thyroid carcinoma and undifferentiated thyroid carcinoma according to the pathological types [62]. With the increase of malignant degree, the positive expression rate of E-cadherin decreases gradually, which is negatively correlated with higher clinical stage, distant lymph node metastasis, extracapsular infiltration and lower disease-free survival rate [62, 173]. And in gastric cancer, breast cancer, ovarian cancer and other tumors also showed that the low expression of E-cadherin-induced EMT is related to the invasive characteristics of tumors, and become an independent prognostic factor of tumor patients [75]. Interestingly, in one study, there was no difference in the expression of E-cadherin among neoplastic, adenoma and non-neoplastic lesions [174]. We think this may be the result of the small sample size.
To sum up, it can be seen that the occurrence of malignant expression type of thyroid carcinoma is significantly related to the low expression of E-cadherin, whether in vivo or in vitro. This suggests that E-cadherin may provide a basis for the diagnosis of thyroid tumors and assist in malignant degree and pathological classification. By reviewing the mechanism of regulating E-cadherin, E-cadherin may provide a new idea for the treatment of thyroid cancer, which provides a wealth of options for treatment of TC, not limited to traditional surgery, radioiodine therapy and radiochemotherapy [175]. From E-cadherin to explore the treatment strategy of thyroid cancer include the following three aspects: (1) block the transduction of signal pathways related to the regulation of E-cadherin in thyroid carcinoma; (2) inhibition of expression of transcription factors upstream of E-cadherin; (3) immune activation regulates the tumor microenvironment of thyroid cancer.
The expression of E-cadherin is regulated by pathway inhibitors in thyroid carcinoma, which is mainly concentrated in Wnt/β-catenin, PI3K/AKT and MAPK signal pathways. The Wnt pathway inhibitor (DKK-1) reverses the deletion of E-cadherin expression in PTC cell membrane, thereby inducing EMT [176, 177]. High expression of β-catenin and abnormal localization of nucleus and cytoplasm were found in thyroid carcinoma [178]. In one study, the non-steroidal anti-inflammatory drug, sulinda hypothesis, decreased the expression of β-catenin in thyroid cancer cells and showed nuclear transfer to the cell membrane [178]. In addition, it was also found that the process of reversing β-catenin was only found in 8505murine C and KTC-1 cells with BRAF (V600E) mutation [178]. β-Catenin inhibitors can obviously inhibit the resistance of thyroid cancer cells to BRAF (V600E) inhibitors, thus inhibiting the occurrence of EMT [179]. Inhibition of EMT by inhibiting RTK and Akt/mTOR signaling pathways and promoting the expression of E-cadherin in thyroid cancer cells [180]. In clinical studies, kinase inhibitors Sorafenib, Vandetanib, Cabozantinib and Lenvatinib have been approved by FDA and the European Medical Association (EMA) for the treatment of medullary thyroid carcinoma and advanced RAI-R (refractory to radioiodine therapy) and poorly differentiated thyroid cancer [181]. A phase I clinical study found that thisirolimus, a mTOR inhibitor, enhanced the limited rate of sorafenib in the treatment of recurrent or metastatic radioiodine refractory thyroid cancer (RAITC), compared with sorafenib alone [182]. A retrospective study of clinical mortality and causes of death in patients with thyroid cancer found that the mortality rate of patients using tyrosine kinase inhibitor (TKI) (15.8%) was lower than that of patients undergoing surgery (64.2%), radiotherapy (53.3%) and cytotoxic chemotherapy (24.2%) [183]. Surgery and radiotherapy combined with TKI was the most effective, and the concomitant median survival rate was 34.3 months [183]. Distant metastasis has become the main cause of thyroid surface death in patients without TKI treatment [183].
A large number of studies have shown that malignant phenotype can be improved by regulating the expression of upstream transcription factors of E-cadherin. As an oncogene of thyroid cancer, BRAFV600E mutation can induce the development of thyroid cancer [39]. The carcinogenicity of BRAFV600E is caused by the activation of MAPK signal pathway [184]. The study found that thyroid cancer patients’ with BRAFV600E mutation were treated with BRAFV600E inhibitor (Dabrafenib) combined with MEK inhibitor (Trametinib) symptoms were relieved and the median survival time was increased [185, 186]. In addition, the efficacy of Dabrafenib combined with Trametinib in metastatic non-small cell lung cancer (NSCLC) and advanced melanoma with BRAFV600E mutation has also been confirmed, which can prolong the median progression-free survival (PFS) and global survival (OS) of tumor patients with less physical toxicity [187, 188]. In addition, there is the regulation of tumor suppressor factor PTEN. Resveratrol can promote PTEN expression and nuclear metastasis in thyroid cancer cells, and at the same time induce E-cadherin transfer from cytoplasm to cell membrane to enhance intercellular adhesion and inhibit the occurrence of EMT [189]. Green tea extract (EGCG) and Combretastatin A4 can directly inhibit transcription factors Snail, Slug, Zeb1 and Twist1 to up-regulate the expression of E-cadherin and inhibit the occurrence of EMT [190, 191].
It can be seen from the above that immunosuppression and immune escape are the main causes of immune system EMT in thyroid cancer. Then the corresponding immunotherapy includes immune checkpoint inhibitors and immunization. Immunosuppressive molecule PD-1 is overexpressed on activated T cell membrane and inhibits T cell function by binding to its ligand [89]. Therefore, the specific recognition of T cells to tumor immune antigens can be improved by immune checkpoint inhibitors in patients with T cell inflammatory phenotype [192, 193]. The therapeutic value of anti-PD-1 and PD-L1 drugs, pembrolizumab, nivolumab and atezolizumab, in recurrent or metastatic head and neck squamous cell carcinoma, non-small cell lung cancer and melanoma has been confirmed, showing that the patients have higher survival rate and safety [194,195,196]. And a meta analysis confirmed that anti-PD-1 drugs (nivolumab, pembrolizumab, atezolizumab) did not cause serious organ-specific immune adverse events such as pneumonia, colitis and hypophysitis, on the contrary, tumor-targeted drugs and chemotherapeutic drugs had a higher risk of organ-specific immune adverse events, with a high security [197]. The concentration of macrophages in tumor microenvironment was positively correlated with extraglandular invasion and extracapsular invasion, and negatively correlated with survival rate [198]. Therefore, inhibiting the polarization of macrophages may be a therapeutic idea. It has been found that zoledronic acid (ZA) can inhibit M2-like polarization of macrophages and inhibit the characteristics of thyroid cancer stem cells and the occurrence of EMT, which is characterized by low expression of stem cell markers CD133 and Oct4 and high expression of E-cadherin [199].
It can be seen from the above that in thyroid carcinoma, the expression of E-cadherin is affected by regulating signal pathways, upstream genes and immune microenvironment, thus affecting the occurrence of EMT. There are a variety of methods, whether they have been clinically proven, or are still in the experimental stage, or are simply studied in vitro. These results suggest that it is a new idea to regulate E-cadherin to affect the occurrence of thyroid EMT and improve the malignant expression of tumors.
Overall, the malignant phenotype of thyroid cancer is negatively correlated with E-cadherin, and its complex regulatory mechanisms and widely involved cytokines may provide new ideas for the early diagnosis, prognosis and treatment of thyroid cancer.
Availability of data and materials
Yes.
References
Liu Y, Lin Z, Sheng C, Zhu Y, Huang Y, Zhong N, Jia Z, Qu S. The prevalence of thyroid nodules in northwest China and its correlation with metabolic parameters and uric acid. Oncotarget. 2017;8(25):41555–62.
Meixner M, et al. Disease-free survival in papillary and follicular thyroid carcinoma. Comparison between UICC 5th and 7th classifications of T stage, and the prognostic value of primary tumour size. Nuklearmedizin. 2013;52(3):71–80.
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110.
Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29(3):212–26.
Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–35.
Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci. 2013;116:317–36.
Guillot C, Lecuit T. Mechanics of epithelial tissue homeostasis and morphogenesis. Science. 2013;340(6137):1185–9.
van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer. 2014;14(2):121–34.
Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci. 2007;8(1):11–20.
Tepass U, Truong K, Godt D, Ikura M, Peifer M. Cadherins in embryonic and neural morphogenesis. Nat Rev Mol Cell Biol. 2000;1(2):91–100.
Shrestha N, Chand L, Han MK, Lee SO, Kim CY, Jeong YJ. Glutamine inhibits CCl4 induced liver fibrosis in mice and TGF-β1 mediated epithelial–mesenchymal transition in mouse hepatocytes. Food Chem Toxicol. 2016;93:129–37.
Lourenco AR, Ban Y, Crowley MJ, Lee SB, Ramchandani D, Du W, Elemento O, George JT, Jolly MK, Levine H. Sheng, differential contributions of pre- and post-EMT tumor cells in breast cancer metastasis. Cancer Res. 2020;80(2):163–9.
Jensen K, et al. Dynamic changes in E-cadherin gene promoter methylation during metastatic progression in papillary thyroid cancer. Exp Ther Med. 2010;1(3):457–62.
Lv N, et al. The Expression of transcription factors is different in papillary thyroid cancer cells during TNF-alpha induced EMT. J Cancer. 2021;12(9):2777–86.
Sastre-Perona A, Santisteban P. Role of the wnt pathway in thyroid cancer. Front Endocrinol. 2012;3:31.
Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16(12):3153–62.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26.
Zhao C, et al. FOXN3 suppresses the growth and invasion of papillary thyroid cancer through the inactivation of Wnt/β-catenin pathway. Mol Cell Endocrinol. 2020;515: 110925.
Wu L, et al. Expression of lncRNA-HOTAIR in the serum of patients with lymph node metastasis of papillary thyroid carcinoma and its impact. Oncol Lett. 2020;20(1):907–13.
Izaguirre MF, Casco VH. E-cadherin roles in animal biology: a perspective on thyroid hormone-influence. Cell Commun Signal. 2016;14(1):27.
van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65(23):3756–88.
Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial–mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35.
Cho SW, Kim YA, Sun HJ, Ahn HY, Lee EK, Yi KH, Oh BC, Park DJ, Cho BY, Park YJ. Therapeutic potential of Dickkopf-1 in wild-type BRAF papillary thyroid cancer via regulation of β-catenin/E-cadherin signaling. J Clin Endocrinol Metab. 2014;99(9):1641–9.
Cho SW, Lee EJ, Kim H, Kim SH, Ahn HY, Kim YA, Yi KH, Park DJ, Shin CS, Ahn SH, Cho BY. Dickkopf-1 inhibits thyroid cancer cell survival and migration through regulation of β-catenin/E-cadherin signaling. Mol Cell Endocrinol. 2013;366(1):90–8.
Ivanova K, et al. Expression of E-cadherin/Beta-catenin in epithelial carcinomas of the thyroid gland. Open Access Maced J Med Sci. 2017;5(2):155–9.
Jiang BH, Liu LZ. Chapter 2 PI3K/PTEN signaling in angiogenesis and tumorigenesis. 2009. p. 19–65.
Jiang N, et al. Role of PI3K/AKT pathway in cancer: the framework of malignant behavior. Mol Biol Rep. 2020;47(6):4587–629.
Yin Y, et al. Extracellular vesicles from mast cells induce mesenchymal transition in airway epithelial cells. Respir Res. 2020;21(1):101.
Wang SC, et al. HPIP promotes thyroid cancer cell growth, migration and EMT through activating PI3K/AKT signaling pathway. Biomed Pharmacother. 2015;75:33–9.
Sun Y, Ye D, Li Y, Chen E, Hao R, Cai Y, Wang Q, Wang O, Zhang X. CUX2 functions as an oncogene in papillary thyroid cancer. Onco Targets Ther. 2018;12:217–24.
Cao HL, et al. miR-144-3p contributes to the development of thyroid tumors through the PTEN/PI3K/AKT pathway. Cancer Manag Res. 2020;12:9845–55.
Xiong J, et al. Deregulated expression of miR-107 inhibits metastasis of PDAC through inhibition PI3K/Akt signaling via caveolin-1 and PTEN. Exp Cell Res. 2017;361(2):316–23.
Ramirez-Moya J, Wert-Lamas L, Santisteban P. MicroRNA-146b promotes PI3K/AKT pathway hyperactivation and thyroid cancer progression by targeting PTEN. Oncogene. 2018;37(25):3369–83.
Gao W, Han J. Overexpression of ING5 inhibits HGF-induced proliferation, invasion and EMT in thyroid cancer cells via regulation of the c-Met/PI3K/Akt signaling pathway. Biomed Pharmacother. 2018;98:265–70.
Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, Van Roy F, Lee-Kwon W, Donowitz M, Tsichlis PN, Larue L. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63(9):2172–8.
Guigon CJ, et al. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene. 2009;28(4):509–17.
Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, Bernet V, Burman KD, Kohn LD, Saji M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61(16):6105–11.
Zhao L, Jiang R, Xu M, Zhu P, Mo XM, Wang N, Chen GG, Liu ZM. Concomitant high expression of BRAFV 600E, P-cadherin and cadherin 6 is associated with high TNM stage and lymph node metastasis in conventional papillary thyroid carcinoma. Clin Endocrinol. 2016;84(5):748–55.
Shimamura M, et al. Acceleration of BRAF(V600E)-induced thyroid carcinogenesis by TGFβ signal deficiency in mice. Endocrine. 2020;69(3):571–7.
Fujishita MAT. Oncogenic Roles of the PI3K/AKT/mTOR axis. Curr Top Microbiol Immunol. 2017;407:153–89.
Xia P, Xu XY. PI3K Akt mTOR signaling pathway in cancer stem cells from basic research to clinical application. Am J Cancer Res. 2015;5(5):1602–9.
Vara JÁ, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30(2):193–204.
Noorolyai S, et al. The relation between PI3K/AKT signalling pathway and cancer. Gene. 2019;698:120–8.
Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22(4):686–707.
Ramírez-Moya J, Wert-Lamas L, Santisteban P. MicroRNA-146b promotes PI3K/AKT pathway hyperactivation and thyroid cancer progression by targeting PTEN. Oncogene. 2018;37(25):3369–83.
Sun C, et al. F-box protein 11 promotes the growth and metastasis of gastric cancer via PI3K/AKT pathway-mediated EMT. Biomed Pharmacother. 2018;98:416–23.
Wang L, et al. Deltex3 inhibits epithelial mesenchymal transition in papillary thyroid carcinoma via promoting ubiquitination of XRCC5 to regulate the AKT signal pathway. J Cancer. 2021;12(3):860–73.
Lv J, et al. M2like tumourassociated macrophagesecreted IGF promotes thyroid cancer stemness and metastasis by activating the PI3K/AKT/mTOR pathway. Mol Med Rep. 2021;24(2):1–10.
Dhillon AS, et al. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–90.
Brzezianska E, Pastuszak-Lewandoska D. A minireview: the role of MAPK/ERK and PI3K/Akt pathways in thyroid follicular cell-derived neoplasm. Front Biosci. 2021;16:422–39.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–83.
van Beijnum JR, et al. miRNAs: micro-managers of anticancer combination therapies. Angiogenesis. 2017;20(2):269–85.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–63.
Lin X, et al. TFF3 contributes to epithelial–mesenchymal transition (EMT) in papillary thyroid carcinoma cells via the MAPK/ERK signaling pathway. J Cancer. 2018;9(23):4430–9.
Xie F, et al. TGF-beta signaling in cancer metastasis. Acta Biochim Biophys Sin. 2018;50(1):121–32.
Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harbor Perspect Biol. 2016;8(5): a021873.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96.
Hao Y, Baker D, Ten Dijke P. TGF-beta-mediated epithelial–mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20(11):2767.
Baquero P, Jiménez-Mora E, Santos A, Lasa M, Chiloeches A. TGFβ induces epithelial–mesenchymal transition of thyroid cancer cells by both the BRAF/MEK/ERK and Src/FAK pathways. Mol Carcinog. 2016;55(11):1639–54.
Kim DW, et al. Complex temporal changes in TGFβ oncogenic signaling drive thyroid carcinogenesis in a mouse model. Carcinogenesis. 2013;34(10):2389–400.
Bhatti MZ, et al. REGγ potentiates TGF-β/Smad signal dependent epithelial–mesenchymal transition in thyroid cancer cells. Cell Signal. 2019;64: 109412.
Min WP, Wei XF. Silencing SIX1 inhibits epithelial mesenchymal transition through regulating TGF-β/Smad2/3 signaling pathway in papillary thyroid carcinoma. Auris Nasus Larynx. 2020;48(3):487–95.
Li T, et al. Epigallocatechin gallate (EGCG) suppresses epithelial–mesenchymal transition (EMT) and invasion in anaplastic thyroid carcinoma cells through blocking of TGF-β1/Smad signaling pathways. Bioengineered. 2019;10(1):282–91.
Baquero P, et al. TGFβ induces epithelial–mesenchymal transition of thyroid cancer cells by both the BRAF/MEK/ERK and Src/FAK pathways. Mol Carcinog. 2016;55(11):1639–54.
Wieczorek-Szukala K, et al. Snail-1 overexpression correlates with metastatic phenotype in BRAF(V600E) positive papillary thyroid carcinoma. J Clin Med. 2020;9(9):2701.
Lopez-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1(6–7):303–14.
Huang L, Mellor AL. Metabolic control of tumour progression and antitumour immunity. Curr Opin Oncol. 2014;26(1):92–9.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6.
Li H, et al. The tumor microenvironment: an irreplaceable element of tumor budding and epithelial–mesenchymal transition-mediated cancer metastasis. Cell Adhes Migr. 2016;10(4):434–46.
Suarez-Carmona M, et al. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. 2017;11(7):805–23.
Redente EF, Dwyer-Nield LD, Merrick DT, Raina K, Agarwal R, Pao W, Rice PL, Shroyer KR, Malkinson AM. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am J Pathol. 2010;176:2972–85.
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73.
Coussens LM, Zitvogel L, Palucka A. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;6117(339):286–91.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Yan Y, et al. High tumor-associated macrophages infiltration is associated with poor prognosis and may contribute to the phenomenon of epithelial–mesenchymal transition in gastric cancer. Onco Targets Ther. 2016;9:3975–83.
Bonde AK, et al. Intratumoral macrophages contribute to epithelial–mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35.
Hu Y, et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Exp Clin Cancer Res. 2016;35:12.
Lv J, et al. M2-like tumor-associated macrophages-secreted Wnt1 and Wnt3a promotes dedifferentiation and metastasis via activating beta-catenin pathway in thyroid cancer. Mol Carcinog. 2021;60(1):25–37.
Lin X, et al. miR-195-5p/NOTCH2-mediated EMT modulates IL-4 secretion in colorectal cancer to affect M2-like TAM polarization. J Hematol Oncol. 2019;12(1):20.
Chen Y, Wen H, Zhou C, Su Q, Lin Y, Xie Y, Huang Y, Qiu Q, Lin J, Huang X, Tan W. TNF-α derived from M2 tumor-associated macrophages promotes epithelial–mesenchymal transition and cancer stemness through the Wnt/β-catenin pathway in SMMC-7721 hepatocellular carcinoma cells. Exp Cell Res. 2019;378(1):41–50.
Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1977;77(4):1033–79.
Mekori YA. The mastocyte: the “other” inflammatory cell in immunopathogenesis. J Allergy Clin Immunol. 2004;114(1):52–7.
Okano M, et al. Triple-negative breast cancer with high levels of annexin A1 expression is associated with mast cell infiltration, inflammation, and angiogenesis. Int J Mol Sci. 2019;20(17):4197.
Melillo RM, et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene. 2010;29(47):6203–15.
Visciano C, Liotti F, Prevete N, Franco R, Collina F, De Paulis A, Marone G, Santoro M, Melillo RM. Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene. 2015;34(40):5175–86.
Jiang Y, et al. Mast cell chymase affects the proliferation and metastasis of lung carcinoma cells in vitro. Oncol Lett. 2017;14(3):3193–8.
Ferrari SM, et al. Immune and inflammatory cells in thyroid cancer microenvironment. Int J Mol Sci. 2019;20(18):4413.
Aghajani MJ, et al. Epithelial-to-mesenchymal transition and its association with PD-L1 and CD8 in thyroid cancer. Endocr Connect. 2020;9(10):1028–41.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–12.
Alsuliman A, et al. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: significance in claudin-low breast cancer cells. Mol Cancer. 2015;14:149.
Wang L, et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun. 2018;9(1):3503.
Sánchez-Tilló E, Siles L, De Barrios O, Cuatrecasas M, Vaquero EC, Castells A, Postigo A. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer Res. 2011;7(1):897.
Rosenbloom KR, et al. The UCSC genome browser database: 2015 update. Nucleic Acids Res. 2015;43(Database issue):D670–6781.
Chen L, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241.
Noda K, Miyoshi E, Uozumi N, Gao CX, Suzuki K, Hayashi N, Hori M, Taniguchi N. High expression of alpha-1-6 fucosyltransferase during rat hepatocarcinogenesis. Int J Cancer. 1998;3(75):444–50.
Miyoshi E, Uozumi N, Noda K, Hayashi N, Hori M, Taniguchi N. Expression of alpha1-6 fucosyltransferase in rat tissues and human cancer cell lines. Int J Cancer. 1997;6(72):1117–21.
Ito Y, Miyauchi A, Yoshida H, Uruno T, Nakano K, Takamura Y, Miya A, Kobayashi K, Yokozawa T, Matsuzuka F, et al. Expression of α1,6-fucosyltransferase (FUT8) in papillary carcinoma of the thyroid: its linkage to biological aggressiveness and anaplastic transformation. Cancer Lett. 2003;2(200):167–72.
Osumi D, Takahashi M, Miyoshi E, Yokoe S, Lee SH, Noda K, Nakamori S, Gu J, Ikeda Y, Kuroki Y, Sengoku K, Ishikawa M, Taniguchi N. Core fucosylation of E-cadherin enhances cell–cell adhesion in human colon carcinoma WiDr cells. Cancer Sci. 2009;5(100):888–95.
Ma M, et al. Fucosyltransferase 8 regulation and breast cancer suppression by transcription factor activator protein 2gamma. Cancer Sci. 2021;112(8):3190–204.
Fei GE, Shi BZ, Yuan YF, Wu XZ. The expression of core fucosylated E-cadherin in cancer cells and lung cancer patients: prognostic implications. Cell Res. 2004;5(14):423–33.
Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, Nakano M, Yoshimura A. Blockage of core fucosylation reduces cell-surface expression of PD-1 and promotes anti-tumor immune responses of T cells. Cell Rep. 2017;5(20):1017–28.
Fuxe J, Karlsson MC. TGF-β-induced epithelial–mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol. 2012;5–6(22):455–61.
DeNardo DG, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67.
Baghdadi M, et al. Chemotherapy-induced IL34 enhances immunosuppression by tumor-associated macrophages and mediates survival of chemoresistant lung cancer cells. Cancer Res. 2016;76(20):6030–42.
Zhou SL, et al. miR-28-5p-IL-34-macrophage feedback loop modulates hepatocellular carcinoma metastasis. Hepatology. 2016;63(5):1560–75.
Zhang P, et al. IL-34 is a potential biomarker for the treatment of papillary thyroid cancer. J Clin Lab Anal. 2020;34(8): e23335.
Lv N, et al. Inflammatory mediators, tumor necrosis factor-α and interferon-γ, induce EMT in human PTC cell lines. Oncol Lett. 2015;10(4):2591–7.
Nilsson M, Husmark J, Bjorkman U, Ericson LE. Cytokines and thyroid epithelial integrity: interleukin-1α induces dissociation of the junctional complex and paracellular leakage in filter-cultured human thyrocytes. J Clin Endocrinol Metab. 1998;3(83):945–52.
Leonardi FPA. NF-kappaB in solid tumors. Biochem Pharmacol. 2006;72(9):1142–52.
Mann DA, Oakley F. NF-kappaB: a signal for cancer. J Hepatol. 2005;42(4):610–1.
Shostak K, Chariot A. EGFR and NF-kappaB: partners in cancer. Trends Mol Med. 2015;21(6):385–93.
Bommarito A, Richiusa P, Carissimi E, Pizzolanti G, Rodolico V, Zito G, Criscimanna A, et al. BRAFV600E mutation, TIMP-1 upregulation, and NF-κB activation: closing the loop on the papillary thyroid cancer trilogy. Endocr Relat Cancer. 2011;6(18):669–85.
Lv N, Shan Z, Gao Y, et al. Twist1 regulates the epithelial–mesenchymal transition via the NF-κB pathway in papillary thyroid carcinoma. Endocrine. 2016;3(51):469–77.
Li H, Chen A, Yuan Q, Chen W, Zhong H, Teng M, Xu C, Qiu Y, Cao J. NF-κB/Twist axis is involved in chysin inhibition of ovarian cancer stem cell features induced by co-treatment of TNF-α and TGF-β. Int J Clin Exp Pathol. 2019;1(12):101–12.
Lv N, et al. Twist1 regulates the epithelial–mesenchymal transition via the NF-κB pathway in papillary thyroid carcinoma. Endocrine. 2016;51(3):469–77.
Wang W, Zhang E, Lin C. MicroRNAs in tumor angiogenesis. Life Sci. 2015;136:28–35.
You A, et al. MicroRNA-203 restrains epithelial–mesenchymal transition, invasion and migration of papillary thyroid cancer by downregulating AKT3. Cell Cycle. 2020;19(10):1105–21.
Wen HL, et al. miR-597-3p inhibits invasion and migration of thyroid carcinoma SW579 cell by targeting RAB23. Endokrynol Pol. 2020;72(1):22–8.
Tamagawa S, et al. MicroRNA 200b promotes mesenchymal-to-epithelial transition in anaplastic thyroid carcinoma. Oncol Lett. 2020;20(4):3.
Li Y, Zeng QG, Qiu JL, Pang T, Wang H, Zhang XX. Propofol suppresses migration, invasion, and epithelial–mesenchymal transition in papillary thyroid carcinoma cells by regulating miR-122 expression. Eur Rev Med Pharmacol Sci. 2020;24(9):5101–10.
Hardin H, et al. The roles of the epithelial–mesenchymal transition marker PRRX1 and miR-146b-5p in papillary thyroid carcinoma progression. Am J Pathol. 2014;184(8):2342–54.
Liu C, et al. MiR-144-3p promotes the tumor growth and metastasis of papillary thyroid carcinoma by targeting paired box gene 8. Cancer Cell Int. 2018;18:54.
Zhang X, et al. MicroRNA 483-3p targets Pard3 to potentiate TGF-beta1-induced cell migration, invasion, and epithelial–mesenchymal transition in anaplastic thyroid cancer cells. Oncogene. 2019;38(5):699–715.
Wang DP, et al. microRNA-599 promotes apoptosis and represses proliferation and epithelial–mesenchymal transition of papillary thyroid carcinoma cells via downregulation of Hey2-depentent Notch signaling pathway. J Cell Physiol. 2020;235(3):2492–505.
Wang Y, Liu BG, Zhou CX. MicroRNA-31 inhibits papillary thyroid carcinoma cell biological rogression by directly targeting SOX11 and egulating epithelial-to-mesenchymal transition, ERK and Akt signaling pathways. Eur Rev Med Pharmacol Sci. 2019;23(13):5863–73.
Hao F, et al. miR-199a-5p suppresses epithelial–mesenchymal-transition in anaplastic thyroid carcinoma cells via targeting Snail signals. Cancer Biomark. 2020;29(3):317–26.
Hong S, et al. MiR-20b displays tumor-suppressor functions in papillary thyroid carcinoma by regulating the MAPK/ERK signaling pathway. Thyroid. 2016;26(12):1733–43.
Ren L, et al. miR-199b-5p-Stonin 2 axis regulates metastases and epithelial-to-mesenchymal transition of papillary thyroid carcinoma. IUBMB Life. 2019;71(1):28–40.
Han J, et al. miR-215 suppresses papillary thyroid cancer proliferation, migration, and invasion through the AKT/GSK-3beta/Snail signaling by targeting ARFGEF1. Cell Death Dis. 2019;10(3):195.
Fan X, Zhao Y. miR-451a inhibits cancer growth, epithelial–mesenchymal transition and induces apoptosis in papillary thyroid cancer by targeting PSMB8. J Cell Mol Med. 2019;23(12):8067–75.
Huang Y, et al. MiR-613 inhibits the proliferation, migration, and invasion of papillary thyroid carcinoma cells by directly targeting TAGLN2. Cancer Cell Int. 2021;21(1):494.
Pan XM, He XY, Yang YL, Jia WJ, Yang ZQ, Yan D, Ma JX. MiR-630 inhibits papillary thyroid carcinoma cell growth, metastasis, and epithelial–mesenchymal transition by suppressing JAK2 STAT3 signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(6):2453–60.
Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–69.
Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73(13):2491–509.
He H, Yang H, Liu D, Pei R. LncRNA NORAD promotes thyroid carcinoma rogression through targeting miR-202-5p. Am J Transl Res. 2019;11(1):290–9.
Xia S, Ji R, Zhan W. Long noncoding RNA papillary thyroid carcinoma susceptibility candidate 3 (PTCSC3) inhibits proliferation and invasion of glioma cells by suppressing the Wnt/beta-catenin signaling pathway. BMC Neurol. 2017;17(1):30.
Wang DP, et al. Overexpression of long noncoding RNA SLC26A4-AS1 inhibits the epithelial–mesenchymal transition via the MAPK pathway in papillary thyroid carcinoma. J Cell Physiol. 2020;235(3):2403–13.
Zhang R, et al. Long non-coding RNA Linc-ROR is upregulated in papillary thyroid carcinoma. Endocr Pathol. 2018;29(1):1–8.
Feng J, et al. A novel lncRNA n384546 promotes thyroid papillary cancer progression and metastasis by acting as a competing endogenous RNA of miR-145-5p to regulate AKT3. Cell Death Dis. 2019;10(6):433.
Wang XJ, et al. LINC00106 prevents against metastasis of thyroid cancer by inhibiting epithelial–mesenchymal transition. Eur Rev Med Pharmacol Sci. 2020;24(19):10015–21.
Zhou T, et al. LncRNA CASC2 expression is down-regulated in papillary thyroid cancer and promotes cell invasion by affecting EMT pathway. Cancer Biomark. 2018;23(2):185–91.
Pellecchia S, et al. The long non-coding RNA Prader Willi/Angelman region RNA5 (PAR5) is downregulated in anaplastic thyroid carcinomas where it acts as a tumor suppressor by reducing EZH2 activity. Cancers. 2020;12(1):235.
Wang Y, et al. lncRNA BANCR promotes EMT in PTC via the Raf/MEK/ERK signaling pathway. Oncol Lett. 2018;15(4):5865–70.
Zhang R, et al. MALAT1 long non-coding RNA expression in thyroid tissues: analysis by in situ hybridization and real-time PCR. Endocr Pathol. 2017;28(1):7–12.
Li D, Hao S, Zhang J. Long non-coding RNA UCA1 exerts growth modulation by miR-15a in human thyroid cancer TPC-1 cells. Artif Cells Nanomed Biotechnol. 2019;47(1):1815–22.
Lei H, Gao Y, Xu X. LncRNA TUG1 influences papillary thyroid cancer cell proliferation, migration and EMT formation through targeting miR-145. Acta Biochim Biophys Sin. 2017;49(7):588–97.
Chen D, et al. Knockdown of LINC02471 inhibits papillary thyroid carcinoma cell invasion and metastasis by targeting miR-375. Cancer Manag Res. 2020;12:8757–71.
Xia E, et al. lncRNA LINC00673 induces proliferation, metastasis and epithelial–mesenchymal transition in thyroid carcinoma via Kruppel-like factor 2. Int J Oncol. 2018;53(5):1927–38.
Gupta A, Williams BR, Hanash SM, Rawwas J. Cellular retinoic acid-binding protein II is a direct transcriptional target of MycN in neuroblastoma. Cancer Res. 2006;66:8100–8.
Zhao H, et al. LINC01816 promotes the migration, invasion and epithelial–mesenchymal transition of thyroid carcinoma cells by sponging miR34c5p and regulating CRABP2 expression levels. Oncol Rep. 2021;45(5):1–12.
Wang X-J, Zheng H-T, Xu J, Guo Y-W, Zheng G-B, Ma C, Hao S-L, Liu X-C, Chen H-J, Wei S-J, Wu G-C. LINC00106 prevents against metastasis of thyroid cancer by inhibiting epithelial–mesenchymal transition. Eur Rev Med Pharmacol Sci. 2020;24(19):10015–21.
Zhao X, Hu X. Downregulated long noncoding RNA LINC00313 inhibits the epithelial–mesenchymal transition, invasion, and migration of thyroid cancer cells through inhibiting the methylation of ALX4. J Cell Physiol. 2019;234(11):20992–1004.
Wang G, Le Y, Wei L. Cheng L CREB3 transactivates lncRNA ZFAS1 to promote papillary thyroid carcinoma metastasis by modulating miR-373-3p/MMP3 regulatory axis. Int J Endocrinol. 2021;2021: 9981683.
Cui N, Hu M, Khalil RA. Biochemical and biological attributes of matrix metalloproteinases. Prog Mol Biol Transl Sci. 2017;147:1–73.
Farina AR, Mackay AR. Gelatinase B/MMP-9 in tumour pathogenesis and progression. Cancers. 2014;6(1):240–96.
Radisky ES, Radisky DC. Matrix metalloproteinase-induced epithelial–mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15(2):201–12.
Gao H, et al. Relationships of MMP-9, E-cadherin, and VEGF expression with clinicopathological features and response to chemosensitivity in gastric cancer. Tumour Biol. 2017;39(5):1010428317698368.
Jablonska-Trypuc A, Matejczyk M, Rosochacki S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J Enzyme Inhib Med Chem. 2016;31(sup1):177–83.
Li Y, et al. Role of MMP-9 in epithelial–mesenchymal transition of thyroid cancer. World J Surg Oncol. 2020;18(1):181.
Agraval H, Yadav UC. MMP-2 and MMP-9 mediate cigarette smoke extract-induced epithelial–mesenchymal transition in airway epithelial cells via EGFR/Akt/GSK3beta/beta-catenin pathway: amelioration by fisetin. Chem Biol Interact. 2019;314: 108846.
Eckert MA, et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19(3):372–86.
Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009;1(6): a003129.
Kourtidis A, et al. A central role for cadherin signaling in cancer. Exp Cell Res. 2017;358(1):78–85.
Da C, Wu K, Yue C, Bai P, Wang R, Wang G, Zhao M, Lv Y, Hou P. N-cadherin promotes thyroid tumorigenesis through modulating major signaling pathways. Oncotarget. 2017;8(5):8131–42.
Calì G, et al. CDH16/Ksp-cadherin is expressed in the developing thyroid gland and is strongly down-regulated in thyroid carcinomas. Endocrinology. 2012;153(1):522–34.
Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol. 2005;21:381–410.
Casal JI, Bartolome RA. Beyond N-cadherin, relevance of cadherins 5, 6 and 17 in cancer progression and metastasis. Int J Mol Sci. 2019;20(13):3373.
Zhou Y, et al. Downregulated CDH3 decreases proliferation, migration, and invasion in thyroid cancer. Am J Transl Res. 2020;12(6):3057–67.
Gugnoni M, et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene. 2017;36(5):667–77.
Guixia W, Liwei Z. Advances of relationship between E-cadherin and gastric cancer. BME Clin Med. 2019;23(2):230–4.
Shuping Z, Wenjie T, Peng W. Serum E-cad and clinic pathological factors in patients with papillary thyroid carcinoma. China Health Stand Manage. 2020;11(20):122–4.
Na HY, et al. Expression of class III beta-tubulin is associated with invasive potential and poor prognosis in thyroid carcinoma. J Clin Med. 2020;9(12):3830.
El Demellawy D, Nasr A, Alowami S. Application of CD56, P63 and CK19 immunohistochemistry in the diagnosis of papillary carcinoma of the thyroid. Diagn Pathol. 2008;3:5.
Haugen BR, et al. 2015 American thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid. 2016;26(1):1–133.
Cho SW, et al. Therapeutic potential of Dickkopf-1 in wild-type BRAF papillary thyroid cancer via regulation of β-catenin/E-cadherin signaling. J Clin Endocrinol Metab. 2014;99(9):E1641–9.
Cho SW, et al. Dickkopf-1 inhibits thyroid cancer cell survival and migration through regulation of β-catenin/E-cadherin signaling. Mol Cell Endocrinol. 2013;366(1):90–8.
Cho NL, Lin CI, Whang EE, Carothers AM, Moore FD Jr, Ruan DT. Sulindac reverses aberrant expression and localization of beta-catenin in papillary thyroid cancer cells with the BRAFV600E mutation. Thyroid. 2010;20(6):615–22.
Wang N, Wen J, Ren W, Wu Y, Deng C. Upregulation of TRIB2 by Wnt/β-catenin activation in BRAF V600E papillary thyroid carcinoma cells confers resistance to BRAF inhibitor vemurafenib. Cancer Chemother Pharmacol. 2021;88(1):155–64.
Park CH, et al. Combined effects of baicalein and docetaxel on apoptosis in 8505c anaplastic thyroid cancer cells via downregulation of the ERK and Akt/mTOR pathways. Endocrinol Metab. 2018;33(1):121–32.
Naoum GE, Morkos M, Kim B, Arafat W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol Cancer. 2018;17:51.
Sherman EJ, et al. Phase 2 study evaluating the combination of sorafenib and temsirolimus in the treatment of radioactive iodine-refractory thyroid cancer. Cancer. 2017;123(21):4114–21.
Park J, Jung HA, Shim JH, Park WY, Kim TH, Lee SH, Kim SW, Ahn MJ, Park K, Chung JH. Multimodal treatments and outcomes for anaplastic thyroid cancer before and after tyrosine kinase inhibitor therapy: a real-world experience. Eur J Endocrinol. 2021;184(6):837–45.
Corcoran RB, André T, Atreya CE, Schellens JH, Yoshino T, Bendell JC, Hollebecque A, McRee AJ, Siena S, Middleton G, Muro K, Gordon MS, Tabernero J, Yaeger R, O’Dwyer PJ, Humblet Y, De Vosi F, ScottJung A, Brase JC, Jaeger S, Bettinger S, Mookerjee B, Rangwala F, Van Cutsem E. Combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600E-mutant colorectal cancer. Cancer Discov. 2018;8(4):428–43.
Iyer PC, et al. Real-world experience with targeted therapy for the treatment of anaplastic thyroid carcinoma. Thyroid. 2018;28(1):79–87.
Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JH, Soria JC, Wen PY, Zielinski C, Cabanillas ME, Urbanowitz G, Mookerjee B, Wang D, Rangwala F, Keam B. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36(1):7–13.
Planchard D, Smit EF, Groen HJ, Mazieres J, Besse B, Helland Å, Giannone V, D’Amelio AM Jr, Zhang P, Mookerjee B, Johnson BE. Dabrafenib plus trametinib in patients with previously untreated BRAF V600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16.
Menzies AM, Long GV. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin Cancer Res. 2014;20(8):2035–43.
Xiong L, et al. Biological implications of PTEN upregulation and altered sodium/iodide symporter intracellular distribution in resveratrol-suppressed anaplastic thyroid cancer cells. J Cancer. 2020;11(23):6883–91.
De Amicis F, et al. Epigallocatechin gallate inhibits growth and epithelial-to-mesenchymal transition in human thyroid carcinoma cell lines. J Cell Physiol. 2013;228(10):2054–62.
Liang W, et al. Combretastatin A4 regulates proliferation, migration, invasion, and apoptosis of thyroid cancer cells via PI3K/Akt signaling pathway. Med Sci Monit. 2016;22:4911–7.
Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17(12):e542–51.
Desrichard A, Snyder A, Chan TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res. 2016;22(4):807–12.
Larkin J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34.
Hellmann MD, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381(21):2020–31.
Ferris RL, et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018;81:45–51.
Baxi S, Yang A, Gennarelli RL, Khan N, Wang Z, Boyce L, Korenstein D. Immune-related adverse events for anti-PD-1 and anti-PD-L1 drugs: systematic review and meta-analysis. BMJ. 2018;360: k793.
Ryder M, et al. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer. 2008;15(4):1069–74.
Lv J, et al. Zoledronic acid inhibits thyroid cancer stemness and metastasis by repressing M2-like tumor-associated macrophages induced Wnt/β-catenin pathway. Life Sci. 2020;256: 117925.
Acknowledgements
The authors gratefully acknowledge the financial supports, editor and anonymous reviewers.
Funding
Scientific research projects in state administration of Traditional Chinese Medicine [Grant Number 2018ZY03005]. Shanghai Science and Technology Commission [17401933400].
Author information
Authors and Affiliations
Contributions
XZ is responsible for writing; XW leads the way of thinking; YG and JD are responsible for retrieving literature. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes.
Competing interests
The author declares that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, X., Wang, X., Gong, Y. et al. E-cadherin on epithelial–mesenchymal transition in thyroid cancer. Cancer Cell Int 21, 695 (2021). https://doi.org/10.1186/s12935-021-02344-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02344-6