
Abstract
Background
Because its metastasis to the lymph nodes are closely related to poor prognosis, miRNAs and mRNAs can serve as biomarkers for the diagnosis, prognosis, and therapy of colorectal cancer (CRC). This study aimed to identify novel gene signatures in the lymph node metastasis of CRC.
Methods
GSE56350, GSE70574, and GSE95109 datasets were downloaded from the Gene Expression Omnibus (GEO) database, while data from 569 colorectal cancer cases were also downloaded from The Cancer Genome Atlas (TCGA) database. Differentially expressed miRNAs (DE-miRNAs) were calculated using R programming language (Version 3.6.3), while gene ontology and enrichment analysis of target mRNAs were performed using FunRich (http://www.funrich.org). Furthermore, the mRNA–miRNA network was constructed using Cytoscape software (Version 3.8.0). Gene expression levels were verified using the GEO datasets. Similarly, quantitative real-time PCR (qPCR) was used to examine expression profiles from 20 paired non-metastatic and metastatic lymph node tissue samples obtained from patients with CRC.
Results
In total, five DE-miRNAs were selected, and 34 mRNAs were identified after filtering the results. Moreover, two key miRNAs (hsa-miR-99a, hsa-miR-100) and one gene (heparan sulfate-glucosamine 3-sulfotransferase 2 [HS3ST2]) were identified. The GEO datasets analysis and qPCR results showed that the expression of key miRNA and genes were consistent with that obtained from the bioinformatic analysis. A novel miRNA–mRNA network capable of predicting the prognosis and confirmed experimentally, hsa-miR-99a-HS3ST2-hsa-miR-100, was found after expression analysis in metastasized lymph node tissue from CRC samples.
Conclusion
In summary, miRNAs and genes with potential as biomarkers were found and a novel miRNA–mRNA network was established for CRC lymph node metastasis by systematic bioinformatic analysis and experimental validation. This network may be used as a potential biomarker in the development of lymph node metastatic CRC.
Similar content being viewed by others

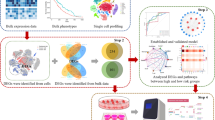

Background
Colorectal cancer (CRC) is a serious health threat worldwide. Compared to the early stage of disease, the treatment response and overall survival of patients with advanced CRC is still very poor. The 5-year survival rate of patients with advanced CRC is reduced from 50 to 10% [1]. Surgical tumor resectioning is still the cornerstone of the treatment of localized, advanced CRC. There is no cure for metastatic tumors that cannot be surgically removed, or that respond poorly to the effects of chemotherapy and radiotherapy [2]. At present, the AJCC’s TNM staging system has limited value in predicting recurrence [3, 4]. Moreover, while lymph node metastasis is not the only form of metastasis in patients with advanced CRC, it is one of the most essential prognostic risk factors [5]. So as to promote the prognosis and individualized treatment, it is therefore urgent to determine the key factors influencing lymph node metastasis in CRC.
MicroRNAs (miRNAs) are small noncoding RNAs, associated with post-transcriptional gene regulation [6]. According to previous studies, miRNAs can regulate many target genes, or many genes can regulate one type of miRNA [7]. Notably, Sin et al. found that some miRNAs can improve the therapeutic effect by improving the drug sensitivity of cancer cells [8]. Ma et al. found that miR-374a, miR-92a, and miR-106a increased drug resistance and promoted growth and metastasis of lung cancer [9]. Similarly, Kania et al. reported that miR-9-3p and miR-9-5p decreased DNA topoisomerase IIα expression levels in acquired resistance to etoposide and may act as a biomarker of responsiveness to TOP2-targeted therapy [10]. However, the mechanisms of miRNAs in the transformation of adenomas to adenocarcinoma remain unknown.
In recent years, an accumulating number of studies have documented that bioinformatics analyses have provided a deeper understanding of the aberrant genetic pathways in the development, progression, and metastasis of various human cancers. Amongst others, these included investigations into breast [11], lung [12], liver [13], and colorectal cancer [14]. There are many reports describing the identification of key genes or pathways in CRC or for predicting CRC prognosis by integrated bioinformatics analysis [15,16,17,18]. Although Zhang et al. identified key candidate genes and constructed novel miRNA–mRNA regulatory axes in CRC liver metastasis, and found that miR-885 promoted CRC cell migration by decreasing the expression of von Willebrand factor (vWF) and insulin-like growth factor binding protein 5 (IGFBP5) by using integrated bioinformatics analysis and in vitro experiments [19], studies identifying biomarkers associated with CRC metastasis by bioinformatics analysis are scare. In particular, the systematic analyses of mRNAs and miRNAs in CRC lymph node metastasis is still not adequate enough to definitively determine aberrant genetic pathways. In this study, a systematic bioinformatics analysis identified two miRNAs (hsa-miR-99a and hsa-miR-100) and one gene (heparan sulfate-glucosamine 3-sulfotransferase 2 [HS3ST2]), through which a novel mRNA–miRNA regulatory network in lymph node metastasis of CRC was established. This model may be used in the early diagnosis and therapy of metastatic CRC.
Materials and methods
Microarray data
The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo) is a public, functional genomics data repository that allows users to download and import gene expression data. In this study, gene expression data were obtained from GEO for GSE56350, GSE70574, and GSE95109.
GSE56350 includes data from eight primary CRC tissue types derived from stage II–III CRC patients with (n = 20) or without (n = 15) lymph node metastasis. MicroRNA expression profiling analysis of these samples was performed using an Agilent-021827 Human miRNA Microarray assay [miRNA_107_Sep09_2_105]. GSE70574included data from 16 T1-stage CRCs comprised of seven lymph node-positive and nine lymph node-negative tumors that were processed using an Agilent-031181 Unrestricted_Human_miRNA_V16.0_Microarray 030840 assay (Feature Number version). GSE95109 contained data from 13 lymph node-negative patients and nine lymph node-positive patients. The mRNA profiles of all 22 patients were analyzed using Agilent microarray technology to explore the differential expression between lymph node-negative and lymph node-positive subgroups.
Differentially expressed miRNAs and mRNAs analysis
R programming language (Version 3.6.3) was used to compare the two groups of tissue. Cut-off criteria were established by |log2FC|≥ 1, while P < 0.05 indicated significant statistical differences [20].
Functional and pathway enrichment analyses
In this study, transcription factor enrichment analysis was conducted and transcription factors that may regulate differentially expressed miRNAs (DE-miRNAs) were identified by FunRich (http://www.funrich.org) which is a publicly accessible software with the ability to identify enriched transcription factors. While, the target genes of the DE-miRNAs were also analyzed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis through the DIANA-miRPath v3.0 database (http://www.microrna.gr/miRPathv3/).
Construction of PPI network and clustered subnetworks
The exploration of protein interactions helps to reveal the underlying pathological mechanism of CRC. In this study, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://string-db.org/) was used to construct a protein–protein interaction network.
Prediction of miRNA target genes and the miRNA–mRNA regulatory network
A previous study suggested that the function of miRNAs lies in the regulation of target genes. Therefore, the prediction of target genes is particularly important as it can indirectly elucidate the biological function and enrichment pathway of the associated miRNAs. The miRNA enrichment function in FunRich was used for miRNA targeting predictions. By combining the FunRich and differential analysis results from the GSE95109 data, screened genes were identified, and the miRNA–mRNA regulatory network built using Cytoscape software (Version 3.8.0).
Construction of a prognostic signature model
To determine the influence of differential expression of miRNAs on the prognosis of CRC patients, univariate and multivariate Cox proportional risk regression analysis was performed for different miRNA expression levels. Those miRNAs found to be related to CRC prognosis were selected, and a linear risk model established based on The Cancer Genome Atlas (TCGA) dataset. Collectively, data from 569 CRC patients downloaded from TCGA were randomly divided into training and test groups. A model capable of predicting the genetic features of input data was built into the training group, with validity testing being performed using the test group data. First, the training group was analyzed by univariate Cox regression analysis to select the prognoses-related differentially expressed genes (DEGs). Through further functional analysis and development of potential risk characteristics, the least absolute shrinkage and selection operator (LASSO) method was used to regress the high-dimensional prediction factors as reported previously [21, 22]. The R “glmnet” package was used to calculate the coefficient and partial likelihood deviation [23]. Through multivariate Cox regression analysis, the identified miRNAs were further studied to determine significant targets and build a linear risk model. To better understand the relationship between the selected miRNAs and the prognosis of CRC patients, a risk prediction model was constructed. By using the “survival ROC” package in R programming language (Version 3.6.3), the AUCs of the ROC curves associated with 3 and 5 year survival were constructed to assess the predictive power of the identified miRNAs, respectively.
Hematoxylin and eosin (H&E) staining and analysis
Fresh colorectal carcinoma and lymph node tissue samples were fixed in 10% formalin and embedded in paraffin before sectioning and staining. Tissue sections that were 4 μm thick were respectively deparaffinized and rehydrated in a series of xylene and ethanol steps. H&E staining was performed according to standard protocols.
Verification of miRNA and mRNA expression
Twenty non-metastatic, and 20 metastatic lymph node CRC tissue samples were obtained from patients with CRC, respectively. Ethical approval was obtained from the Second Affiliated Hospital of Soochow University ethics committee. Total RNA was extracted from the tissue samples using TRIzol reagent (Mesgen Biotech Co., Shanghai, China), and treated with RNase-free DNase I (Takara Biotech Co., Dalian, China) to eliminate traces of mixed DNA. Then, PCR was employed to confirm that there was no genomic DNA contamination using the specific primers of target genes. The reverse transcription performed according to the manufacturer’s protocol. Real-time PCR was performed on the QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific, Shanghai, China) using qPCR SYBR Green master mix (Vazyme Biotech Co., Nanjing, China). The primers are shown in Additional file 2: Table S1. The expression levels of the miRNAs were normalized against U6 (internal standard control) and calculated using the 2−ΔΔCt method. All experiments were performed in triplicate.
Statistical analysis
Negative and positive lymph node tests were performed to evaluate the statistical significance between the two groups. All data analysis was performed using R programming language (version 3.6.3) and GraphPadPrism6 (GraphPad Software, Inc., La Jolla, California, USA). The correlation between the miRNAs and their possible targeted mRNA among individual samples were also assessed. P < 0.05 was viewed as indicating statistical significance. Using the CORR function, the correlation analysis between the RT-qPCR and RNA-seq results was calculated in Excel 2013 (Microsoft Corporation, Redmond, WA, USA).
Results
Identification of the miRNAs between non-metastatic and metastatic lymph node tissues
We performed a comprehensive bioinformatics analysis to elucidate the key miRNA–mRNA axis in the metastatic lymph node of CRC (flow chart was shown in Additional file 1: Figure S1). Using R programming language (Version 3.6.3) to research the gene expression profiles, a total of three GEO datasets (GSE56350, GSE70574 and GSE95109) were selected and downloaded. In total, 47 DE-miRNAs (13 upregulated and 34 downregulated), and 30 DE-miRNAs (13 upregulated and 17 downregulated) were obtained from the GSE56350 and GSE70574 datasets, respectively. Similarly, 34 DEGs (29 upregulated and 5 downregulated, Additional file 3: Table S2) were identified from the GSE95109 dataset according to the cut-off criteria for this study (P < 0.05 and |log2FC|≥ 1). The identified DE-miRNAs and DEGs are shown in heat maps as well as a volcano plots (Fig. 1A–F). Furthermore, a total of five common DE-miRNAs were screened from the GSE56350 and GSE70574 datasets. This included five downregulated miRNAs (Fig. 2), the details of which are listed in Additional file 4: Table S3.

Volcano and heat maps of GSE56350, GSE70754 and GSE95109. A Unsupervised clustering analysis of differentially-expressed miRNAs (DE-miRNAs) in GSE56350. B Volcano plots of miRNAs in GSE56350. C Unsupervised clustering analysis of DE-miRNAs in GSE70754. D Volcano plots of miRNAs in GSE70754. E Unsupervised clustering analysis of the DE-mRNAs in GSE95109. F Volcano plots of mRNAs in GSE95109. A, C, E Red dots indicate significantly up-regulated miRNAs or mRNAs, green dots indicate significantly down-regulated miRNAs or mRNAs. B, D, F Red dots indicate up-regulated DE-miRNAs or DE-mRNAs, green dots indicate down-regulated DE-miRNAs or DE-mRNAs, black dots indicate non-differentially expressed miRNAs or mRNAs

Venn diagram of GSE56350 and GSE70574
Screening of potential transcription factors and enrichment analysis
To identify the shared transcriptional factor signatures associated with the DE-miRNAs, FunRich was used. As shown in Fig. 3A, the top 10 transcription factors that were found to be enriched for included SP1, TEAD1, TCF3, SOX1, HNF4A, TFAP4, KLF7, NHLH1, HENMT1, and RREB1. The results of GO analysis for the target genes of DE-miRNAs showed that DE-miRNAs were most enriched in the cellular nitrogen compound metabolic process, protein complex, ion binding, etc. (Fig. 3B). In addition, these target genes were mainly enriched in the pathways including fatty acid metabolism, fatty acid biosynthesis, hippo signaling pathway, proteoglycans in cancer, lysine degradation and so on by KEGG pathway analysis (Fig. 3C). The construction of the protein–protein interaction (PPI) network is shown in Fig. 4. Under the conditions that the comprehensive Gt score > 0.7 and unconnected points were removed, this network included 598 nodes and 1004 edges.

Screening of potential transcription factors and target genes of DE-miRNAs. A Identification of the potential transcription factors of DE-miRNAs by FunRich software. B The top 10 of biological process, cellular component, and molecular function of the target genes of DE-miRNAs. C KEGG pathway enriched by potential target mRNAs of DE-miRNAs

The PPI network of the target genes of the identified DE-miRNAs
Construction of miRNA–mRNA regulatory network
The 598 potential target genes of screened DE-miRNAs were predicted using FunRich. The subsequent Venn diagram analysis of the target mRNA and GSE 95109-derived DEGs identified one gene of interest (Fig. 5A). To show the composition and relationship between target genes more intuitively, a complete network of target genes was constructed using Cytoscape (Version 3.8.0). Finally, two essential miRNA–mRNA regulatory networks that demonstrated the crucial effects of lymph node metastasis in CRC were identified (Fig. 5B).

The miRNA–mRNA network of CRC lymph node metastasis. A Venn diagram of target mRNAs of DE-miRNAsand DE-mRNAs of GSE95109. B The miRNA–mRNA regulatory network in the lymph node metastasis of CRC
Construction of a prognostic risk model and predictability assessment
To identify the best prognostic miRNAs, the LASSO Cox regression algorithm was applied to 20 prognosis-related miRNAs. Nine miRNAs were selected to build the risk signature based on the minimum standard (Fig. 6A). Multivariate Cox proportional risk regression analysis was subsequently conducted using the nine candidate prognostic miRNAs to assess their independent prognostic values. According to the Cox model, seven candidate miRNAs (hsa-miR-125a-5p, hsa-miR-377, hsa-miR-100, hsa-miR-455-3p, hsa-miR-126, hsa-miR-199a, and hsa-miR-99a) showed significance and were selected as independent prognostic factors. These seven prognostic miRNAs were then combined to build a model to predict patient outcomes. The AUC for 3 year survival using the 7-miRNA signature achieved a value of 0.809, while the 5 year survival AUC value was calculated as 0.981. This demonstrated that the model performed well in predicting the survival risk of CRC patients (Fig. 6B). According to this risk model, patients were divided into high and low risk groups. The results showed that this model accurately predicted the clinical outcomes of patients. The risk scores, survival status and distribution of the expression levels of the seven miRNAs in each patient were also analyzed (Fig. 6C, D).

Identification of the signature significantly associated with the survival of patients with CRC in the training group. A LASSO Cox regression algorithm was used to reduce the scope. B Time-dependent ROC curves analysis. C, D Risk score distribution and survival status for patients in high- and low-risk groups by the signature. LASSO least absolute shrinkage and selection operator, ROC receiver operating characteristic
Validation of the expression of DE-miRNAs and DEGs
In order to further identify the key genes in CRC lymph node metastasis, the expression of hsa-miR-99a, hsa-miR-100, and HS3ST2 were assessed using the GEO database. Two different GEO datasets (GSE108153 and GSE126093) showed that hsa-miR-99a and hsa-miR-100 were down-regulated in CRC tissue compared to normal tissue (Fig. 7A–D, P < 0.001), while data from GSE146587 and GSE110224 demonstrated that HS3ST2 was up-regulated in CRC tissue compared to the normal tissue(Fig. 7E, P < 0.001; F, P < 0.01). Moreover, the expression of hsa-miR-99a, hsa-miR-100, and HS3ST2 was validated by qPCR using 20 CRC tissue samples with lymph node metastasis and 20 CRC tissue samples without lymph node metastasis (Fig. 8A–D). As shown in Fig. 8E–G, the expression of hsa-miR-99a and hsa-miR-100 decreased significantly in CRC tissue with lymph node metastasis compared to CRC tissue without lymph node metastasis (P < 0.01, P < 0.05), while the expression of HS3ST2 greatly increased in CRC tissue with lymph node metastasis (P < 0.05). Generally, the data indicated that hsa-miR-100, hsa-miR-99a, and HS3ST2 could be the candidate biomarkers for CRC lymph node metastasis.

The differential expression of hsa-miR-99a and hsa-miR-100 in CRC tissues and their corresponding normal-appearing tissues. A–D Validation of hsa-miR-99a and hsa-miR-100 in GEO datasets GSE108153 and GSE126093, respectively. E, F Validation of HS3ST2 in GEO datasets GSE146587 and GSE110224, respectively. **P < 0.01, ***P < 0.001

The expression levels of hsa-miR-100, hsa-miR-99a and HS3ST2 in CRC tissues and lymph node metastatic CRC tissues. A Hematoxylin and eosin (H&E) staining of CRC tissues without lymph node metastasis. B H&E staining of CRC tissues in patients with lymph node metastatic CRC. C H&E staining of lymph nodes in patients with CRC. D H&E staining of metastatic lymph nodes in patients with CRC. E–G Validation of hsa-miR-99a, hsa-miR-100 and HS3ST2 in GEO datasets GSE146587 and GSE110224, respectively. *P < 0.05, **P < 0.01
Discussion
CRC can occur through the progression of adenomas, which is the result of genetic and epigenetic events in epithelial cells. Some microarray-based studies have identified gene expression profiles in adenomas and cancers [24,25,26]. In this study, five DE-miRNAs were identified, and it suggested that these five DE-miRNAs played a role in promoting the development of CRC. Functional annotation indicated that these five DE-miRNAs were mostly related to the cellular nitrogen compound metabolic process, protein complex and RNA binding. This is consistent with the recognition that the cellular nitrogen compound metabolic process and protein complex play important roles in tumor development [27, 28]. KEGG pathway analysis indicated that the target genes of five DE-miRNAs were mainly enriched in ten pathways, including fatty acid metabolism, fatty acid biosynthesis, hippo signaling pathway, proteoglycans in cancer, lysine degradation and so on. It was reported that the dysregulation of fatty acids synthesis/catabolism played a regulatory role in the metabolic regulation that supports cancer cell growth [29, 30]. Hippo signaling pathway is a key regulator of organ size, tissue hemostasis and regeneration. Dysregulation of the Hippo pathway has been recognized in a variety of human cancers, including pancreatic cancer [31]. Proteoglycans are dominant components of the extracellular matrix, which are not only a highly heterogeneous proteome, but also an attractive pharmacological target in cancer [32]. However, these pathways have not been reported in the pathogenesis of CRC or in the lymph node metastasis of CRC yet.
A miRNA–mRNA regulatory network was built following FunRich and Cytoscape software analysis. Five DE-miRNAs (hsa-miR-100, hsa-miR-375, hsa-miR-125b, hsa-miR-143 and hsa-miR-99a) and one potential DEG were identified by combining two screening results. It has previously been reported that down-regulation of hsa-miR-100 is involved in tumorigenesis and progression of multiple cancer types [33] In addition, low expression of miR-99a significantly predicts poor prognosis in head and neck squamous cell carcinoma and regulates cancer cell migration and invasion [34]. Xu et al. also demonstrated that miR-99a inhibited the migratory and invasive abilities of cancer cells by regulating the expression of the insulin-like growth factor 1 receptor. It has therefore been concluded that the miR-99a/IGF1R axis may provide novel insight into the pathogenesis of gastric cancer [35]. Hsa-miR-125b, as a tumor suppressor, can contribute to prostate tumorigenesis by modulations in the PI3K/AKT and MAPK/ERK signaling pathways. These key pathways simultaneously influence prostate cancer progression [36]. Hsa-miR-125b can also inhibit the development of bladder cancer by inhibiting SIRT7 and MALAT1 [37], and has additionally been found to play an essential role in the progression of oral squamous cell cancer (OSCC), as well as the target genes and transcription factors associated with hsa-miR-125b [38]. In this study, survival analysis indicated that overexpression of hsa-miR-125b was related to worse overall survival in patients with CRC by using KM-plot software.
Through the combination of the GEO and TCGA analysis, two miRNAs (hsa-miR-100 and hsa-miR-99a) were especially notable. We found that hsa-miR-100 and hsa-miR-99a had different expression levels in non-metastasized and metastasized lymph node tissue through qPCR analysis. Interestingly, hsa-miR-100 and hsa-miR-99a both targeted HS3ST2. HS3ST2, an enzyme mediating 3-O-sulfation of heparan sulfate, is present in all cell types and tissues. It interacts with growth factors, tyrosine kinase receptors, matrix metalloproteinases and extracellular matrix proteins to modulate cell adhesion, proliferation and motility [39, 40]. In breast, colorectal, lung, cervical, pancreatic and recurrent prostate cancers, HS3ST2 is silenced due to hypermethylation, suggesting that it may play an important role in multiple cancers [41]. A previous study has confirmed that abnormal HS3ST2 methylation levels were important in endometrial hyperplasia and carcinogenesis [42]. The prognostic significance of HS3ST2 mRNA expression in several cancer types has been evaluated [43, 44]. HS3ST2 protein expression could be used as a favorable prognostic tissue biomarker in patients with primary advanced-stage lung cancer [44]. For gastric cancer, statistical analyses using a Chi-squared test showed that there is a significant difference in HS3ST2 methylation levels between gastric cancer and non-cancerous patients. It has therefore been concluded that HS3ST2 methylation may act as a novel cancer-related molecular mechanism for the detection of new treatment strategies [45]. In this study, we found that HS3ST2 was upregulated in the metastatic lymph node CRC tissue compared to CRC samples. Moreover, a novel miRNA–mRNA network, hsa-miR-99a-HS3ST2-hsa-miR-100, has been described for the first time in the metastasized lymph node samples derived from CRC.
Our findings proved that many differentially expressed mRNAs and miRNAs were involved in the lymph node metastasis of CRC by select signaling pathways, and that they have prognostic significance. Because most of our data were generated by applying bioinformatics tools to data from the GEO database and TCGA platform, and given that a limited number of relevant samples were available for analysis, more data analysis and clinical experiments should be performed to further develop these potential biomarkers for predicting the recurrence of CRC.
Taken together, this study revealed potential mechanisms associated with the development of CRC. Several differentially expressed mRNAs and miRNAs were identified across non-metastasized and metastasized lymph node tissues by using bioinformatics methods. Hsa-miR-100, hsa-miR-99a and HS3ST2 were determined to be potential biomarkers for predicting the recurrence of CRC.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- GEO:
-
Gene Expression Omnibus
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- miRNA:
-
MicroRNAs
- TCGA:
-
The Cancer Genome Atlas
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- qPCR:
-
Quantitative real-time PCR
- DE-miRNAs:
-
Differentially expressed miRNAs
- DEGs:
-
Differentially expressed genes
- CRC:
-
Colorectal cancer
- HS3ST2:
-
Heparan sulfate-glucosamine 3-sulfotransferase 2
References
Steele SR, et al. Practice guideline for the surveillance of patients after curative treatment of colon and rectal cancer. Dis Colon Rectum. 2015;58(8):713–25.
Vogel JD, Eskicioglu C, Weiser MR, Feingold DL, Steele SR. The American Society of Colon and Rectal Surgeons clinical practice guidelines for the treatment of colon cancer. Dis Colon Rectum. 2017;60(10):999–1017.
Balch C, Ramapuram JB, Tiwari AK. The epigenomics of embryonic pathway signaling in colorectal cancer. Front Pharmacol. 2017;8:267.
Dienstmann R, et al. Prediction of overall survival in stage II and III colon cancer beyond TNM system: a retrospective, pooled biomarker study. Ann Oncol. 2017;28(5):1023–31.
Bosch SL, et al. Predicting lymph node metastasis in pT1 colorectal cancer: a systematic review of risk factors providing rationale for therapy decisions. Endoscopy. 2013;45(10):827–41.
Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29(5):903–6.
Ren P, et al. MicroRNA-92a promotes growth, metastasis, and chemoresistance in non-small cell lung cancer cells by targeting PTEN. Tumor Biol. 2016;37(3):3215–25.
Sin TK, et al. Implications of microRNAs in the treatment of gefitinib-resistant non-small cell lung cancer. Int J Mol Sci. 2016;17(2):237.
Ma X, Liang A-L, Liu Y-J. Research progress on the relationship between lung cancer drug-resistance and microRNAs. J Cancer. 2019;10(27):6865–75.
Kania EE, et al. Hsa-miR-9-3p and hsa-miR-9-5p as post-transcriptional modulators of DNA topoisomerase IIα in human leukemia K562 cells with acquired resistance to etoposide. Mol Pharmacol. 2020;97(3):159–70.
Fridrichova I, Zmetakova I. MicroRNAs contribute to breast cancer invasiveness. Cells. 2019;8(11):1361.
Ma Q, et al. Identification and validation of key genes associated with non-small-cell lung cancer. J Cell Physiol. 2019;234(12):22742–52.
Zeng L, et al. Bioinformatics analysis based on multiple databases identifies hub genes associated with hepatocellular carcinoma. Curr Genomics. 2019;20(5):349–61.
Liu X, et al. Expression patterns and prognostic value of m6A-related genes in colorectal cancer. Am J Transl Res. 2019;11(7):3972–91.
Ding X, Duan H, Luo H. Identification of core gene expression signature and key pathways in colorectal cancer. Front Genet. 2020;11:45.
Hozhabri H, Lashkari A, Razavi SM, Mohammadian A. Integration of gene expression data identifies key genes and pathways in colorectal cancer. Med Oncol. 2021;38(1):7.
Dai GP, Wang LP, Wen YQ, Ren XQ, Zuo SG. Identification of key genes for predicting colorectal cancer prognosis by integrated bioinformatics analysis. Oncol Lett. 2020;19(1):388–98.
Pirim D. Integrative analyses of molecular pathways and key candidate biomarkers associated with colorectal cancer. Cancer Biomark. 2020;27(4):555–68.
Zhang T, et al. Identifying the key genes and microRNAs in colorectal cancer liver metastasis by bioinformatics analysis and in vitro experiments. Oncol Rep. 2019;41(1):279–91.
Li L, et al. Identification of key genes and pathways associated with obesity in children. Exp Ther Med. 2017;14(2):1065–73.
Ikeda N, et al. Clinical significance of aminopeptidase N/CD13 expression in human pancreatic carcinoma. Clin Cancer Res. 2003;9(4):1503–8.
Veenman CJ, Tax DM. LESS: a model-based classifier for sparse subspaces. IEEE Trans Pattern Anal Mach Intell. 2005;27(9):1496–500.
Pollock BE, et al. Comparative analysis of arteriovenous malformation grading scales in predicting outcomes after stereotactic radiosurgery. J Neurosurg. 2017;126(3):852–8.
Kitahara O, et al. Alterations of gene expression during colorectal carcinogenesis revealed by cDNA microarrays after laser-capture microdissection of tumor tissues and normal epithelia. Cancer Res. 2001;61(9):3544–9.
Lechner S, et al. Gene expression pattern of laser microdissected colonic crypts of adenomas with low grade dysplasia. Gut. 2003;52(8):1148–53.
Notterman DA, Alon U, Sierk AJ, Levine AJ. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 2001;61(7):3124–30.
Chen X, et al. Screening and functional prediction of key candidate genes in Hepatitis B Virus-associated hepatocellular carcinoma. BioMed Res Int. 2020;2020:7653506.
Hill VK, Kim JS, Waldman T. Cohesin mutations in human cancer. Biochim Biophys Acta. 2016;1866(1):1–11.
Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153–61.
Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. 2016;73(2):377–92.
Ansari D, et al. The Hippo signaling pathway in pancreatic cancer. Anticancer Res. 2019;39(7):3317–21.
Espinoza-Sánchez NA, Götte M. Role of cell surface proteoglycans in cancer immunotherapy. Semin Cancer Biol. 2020;62:48–67.
Qin C, Huang RY, Wang ZX. Potential role of miR-100 in cancer diagnosis, prognosis, and therapy. Tumour Biol. 2015;36(3):1403–9.
Okada R, et al. Regulation of oncogenic targets by miR-99a-3p (passenger strand of miR-99a-duplex) in head and neck squamous cell carcinoma. Cells. 2019;8(12):1535.
Xu X, Guo A, Pan Q, Chang AM, Zhao C. MiR-99a suppresses cell migration and invasion by regulating IGF1R in gastric cancer. Eur Rev Med Pharmacol Sci. 2019;23(17):7375–82.
Budd WT, et al. Dual action of miR-125b as a tumor suppressor and OncomiR-22 promotes prostate cancer tumorigenesis. PLoS ONE. 2015;10(11):e0142373.
Han Y, et al. Hsa-miR-125b suppresses bladder cancer development by down-regulating oncogene SIRT7 and oncogenic long non-coding RNA MALAT1. FEBS Lett. 2013;587(23):3875–82.
Yan Z, Luo Z, Zhang L, Li J, Liu J. Integrated analysis and MicroRNA expression profiling identified seven miRNAs associated with progression of oral squamous cell carcinoma. J Cell Physiol. 2017;232(8):2178–85.
Afratis N, et al. Glycosaminoglycans: key players in cancer cell biology and treatment. FEBS J. 2012;279(7):1177–97.
Kumar AV, et al. HS3ST2 modulates breast cancer cell invasiveness via MAP kinase-and Tcf4 (Tcf7l2)-dependent regulation of protease and cadherin expression. Int J Cancer. 2014;135(11):2579–92.
Danková Z, et al. Methylation status of KLF4 and HS3ST2 genes as predictors of endometrial cancer and hyperplastic endometrial lesions. Int J Mol Med. 2018;42(6):3318–28.
Zuo Q, et al. Methylation in the promoters of HS3ST2 and CCNA1 genes is associated with cervical cancer in Uygur women in Xinjiang. Int J Biol Markers. 2014;29(4):354–62.
Hellec C, Delos M, Carpentier M, Denys A, Allain F. The heparan sulfate 3-O-sulfotransferases (HS3ST) 2, 3B and 4 enhance proliferation and survival in breast cancer MDA-MB-231 cells. PLoS ONE. 2018;13(3):e0194676.
Hwang J-A, et al. Epigenetic inactivation of heparan sulfate (glucosamine) 3-O-sulfotransferase 2 in lung cancer and its role in tumorigenesis. PLoS ONE. 2013;8(11):e79634.
Eyvazi S, et al. CpG islands methylation analysis of CDH11, EphA5, and HS3ST2 genes in gastric adenocarcinoma patients. J Gastrointest Cancer. 2019;51(2):579–83.
Acknowledgements
This study was supported by the Natural Science Foundation of Jiangsu province, China (BK20181173), Gusu health youth talents program of Suzhou (GSWS2019039, GSWS2020030), Jiangsu youth medical talents program (QNRC-866,867), Discipline Construction Program of The Second Affiliated Hospital of Soochow University (XKTJ-TD202001), Innovation and Entrepreneurship Training Program for College Students in Jiangsu Province(202010285125Y), the Medical Research Programs of Health Commission Foundation of Jiangsu Province (H2019071), and the Science and Technology Program of Suzhou (SLT201934, SYS2020023, SS202056).
Funding
This study is funded by "Basic Research Program of Jiangsu Province (Grant No. BK20181173)".
Author information
Authors and Affiliations
Contributions
Conception and design: HFZ, ZFC, XW. Development of methodology: XW, GYG, ZRC. Acquisition of data: XW, GYG, ZRC. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): XW, GYG, ZHC, MXH, HD. Writing, review and/or revision of the manuscript: XW, HFZ, ZFC. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): MXH, XLX, QYJ. Study supervision: HFZ, ZFC. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
We obtained the approval from the ethics committee of Second Affiliated Hospital of Soochow University.
Consent for publication
This study has not been published previously, and the authors agree the consent for publication.
Competing interests
The authors have declared that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
A whole flow chart of this study.
Additional file 2: Table S1.
The primers for verification of miRNA expression and mRNA expression.
Additional file 3: Table S2.
Identification of DEGs associated with lymph node metastasis of CRC
Additional file 4: Table S3.
Identification of DE-miRNAs associated with lymph node metastasis of CRC
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Wang, X., Gao, G., Chen, Z. et al. Identification of the miRNA signature and key genes in colorectal cancer lymph node metastasis. Cancer Cell Int 21, 358 (2021). https://doi.org/10.1186/s12935-021-02058-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02058-9