
Abstract
Cancers are serious life-threatening diseases which annually are responsible for millions of deaths across the world. Despite many developments in therapeutic approaches for affected individuals, the rate of morbidity and mortality is high. The survival rate and life quality of cancer patients is still low. In addition, the poor prognosis of patients and side effects of the present treatments underscores that finding novel and effective complementary and alternative therapies is a critical issue. Melatonin is a powerful anticancer agent and its efficiency has been widely documented up to now. Melatonin applies its anticancer abilities through affecting various mechanisms including angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress and oxidative stress. Regarding the implication of mentioned cellular processes in cancer pathogenesis, we aimed to further evaluate the anticancer effects of melatonin via these mechanisms.
Similar content being viewed by others

Introduction
As the second cause of mortality worldwide, new cases of cancer have recently been reported to increase by 2025 (approximately 19.3 million annually) [1]. Cancer growth control, complete eradication and preventing its incidence are main purposes for cancer-associated investigations. Chemotherapy, radiotherapy and surgery are the major conventional anticancer treatments. The restricted efficiency of these treatments as well as their dangerous side effects have forced researchers to find novel effective anticancer therapies based on herbal extracts and natural compounds as single or combined therapies [2,3,4].
Melatonin, a multifunctional pleiotropic neurohormone secreted by the pineal gland and other organs including bone marrow, retina, and skin. It is an immune regulatory agent and powerful antioxidant with a capability of preventing cell death in oxidative stress situations. [5, 6]. Moreover, melatonin interrupts cell death mechanisms, inflammation, and redox activity probably resulting in cancer cells sensitization to chemotherapy and radiation [7].
Furthermore, in addition to diverse therapeutic potentials for several diseases [8, 9], melatonin has been shown to possess anticancer abilities against skin cancer [10], glioma [11], lung cancer [12], gastrointestinal cancers [13], gynecological cancers [14, 15], and hematological cancers [16, 17]. Although mechanistic impacts of melatonin on various cancers have been widely demonstrated, in the present review we discuss anticancer effects of melatonin with focusing on molecular pathways including angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress, and oxidative stress.
Melatonin, a neurohormone with a broad spectrum functions
Monitoring of circadian rhythm is one of the several properties of melatonin, which also possesses oncostatic, vasoregulation, antioxidant, anti-inflammatory, and immunomodulatory abilities [18, 19]. It has been demonstrated that the normally enhanced melatonin levels at night help in the organization of homeostatic metabolic rhythms of targeted organs and systems [20]. Of note, disruption of circadian rhythm has been shown as one of the contributing factors in cancer progression and development [21].
Melatonin, as an antioxidant agent, scavenges free radicals. Melatonin has protective effects on neurodegenerative disorders, epilepsy, and cancer through inhibiting oxidative stress in vitro and in vivo [22, 23]. Melatonin increases the activity and expression of enzymes, including catalase, superoxide dismutase and glutathione peroxidase, implicated in antioxidant abilities [24, 25]. Melatonin also has anti-inflammatory impacts and attenuates pathogenic inflammation through modulating different pathways, including reducing the secretion of tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-2) and interferon-gamma (IFN-γ), and enhancing the amounts of IL-4, IL-10 and IL-27. Melatonin alleviates pro-inflammatory cytokines secretion via suppressing nuclear factor kappa B (NF-κB) [26,27,28]. In addition, in neurodegenerative disorders, melatonin blocks cyclooxygenase-2 (COX-2) expression, a pro-inflammatory mediator [29]. Melatonin inhibits apoptosis through regulating Bax/Bcl2 and decreasing caspase-3 activity and expression, proposing that melatonin modulates apoptotic functions in the protection against malignancy and neurodegenerative disorders [30,31,32].
Melatonin regulates multiple physiological and neural functions (Fig. 1). Among of them, effects on blood lipid profile, glycemic control, gestation, reproduction, and fetal development, neural protection, immune system, and cardiovascular system have been widely documented [33, 34]. Melatonin prevents the growth and promotion of spontaneous and chemically mediated breast tumors [35, 36]. Moreover, at physiological concentrations, melatonin suppresses cell invasiveness and proliferation in breast cancer cells [37].

Melatonin with a broad spectrum functions
Melatonin and cancer: effect on different molecular mechanisms and cellular pathways
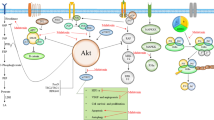
In this section we describe the effect of melatonin on oxidative stress and endoplasmic reticulum stress, and various signaling pathways including angiogenesis, apoptosis, autophagy affected by melatonin in different cancer cells (Fig. 2).

Effects of melatonin on various molecular mechanisms and cellular pathways
Melatonin and angiogenesis
Angiogenesis is a crucial event implicated in the progression of tumor as well as its metastasis [38]. Hypoxia in the central areas of solid tumor is a leading cause of angiogenesis via activation of angiogenic mediators [38, 39]. Vascular endothelial growth factor (VEGF), the specific mitogen of endothelial cells and the most active pro-angiogenic agent, is a powerful angiogenesis enhancer which increases vascular permeability. Numerous data suggest that, in tumor development, anti-VEGF therapy has important roles in the suppression of tumor cell growth, leading to a considerable amelioration in progression-free survival [40]. Hypoxia-inducible factor-1 (HIF-1) is another key factor in angiogenesis, which modulates hypoxia-activated genes transcription and consists of HIF-1α and HIF-1β heterodimer. The α subunit of HIF-1 is stabilized under hypoxia and degraded under normoxic situations, however, HIF-1β is expressed constitutively [41].
Melatonin has been shown to have regulatory role in angiogenesis process [42]. In other words, melatonin possesses various impacts on neovascularization under diverse pathological and physiological situations. In skin lesions, gastric ulcers, and some physiologic events, melatonin promotes angiogenesis, while in a hypoxic environment, in age-related ocular diseases, and in tumors melatonin suppresses neovascularization in tissues [43].
Melatonin exerts its antitumor potentials via inhibiting HIF-1-induced angiogenesis [44]. Furthermore, melatonin inhibits the accumulation of HIF-1α through suppressing the formation of ROS and the sphingosine kinase 1 (SPHK1) pathway in prostate cancer cells under hypoxic conditions [45]. Melatonin plays an important role in the paracrine interaction between proximal endothelial cells and malignant epithelial cells by a downmodulatory effect on the expression of VEGF in breast tumor cells, which reduces VEGF levels around endothelial cells [46].
Of note, anti-angiogenic potential of melatonin is a key factor resulting in the inhibition of proliferation of cancer cells, as demonstrated in various investigations. For instance, melatonin attenuates proliferation of prostate cancer cells triggered by epidermal growth factor [47]. Melatonin also hampers vasculogenic mimicry of oral cancer cells via inhibition of ROS-activated Akt and ERKs signaling pathway implicating the HIF-α pathway [48]. Melatonin up-regulates TGF-β1 expression in tumor tissues during the inhibition of gastric cancer tumor growth process [49]. Furthermore, apoptotic and anti-proliferative effects of melatonin on breast cancer cells are mediated by the simultaneous activation of the Apaf-1/caspase-dependent apoptotic pathway and the inhibition of PI3K/Akt, p300/NF-κB, and COX-2/PGE2 signaling pathways [32].
Endothelin-1 is a peptide acting as a survival factor in colon cancer, promoting angiogenesis and mediating cell proliferation. Melatonin suppresses endothelin-1 mRNA expression. Also, melatonin blocks the activity of endothelin-1 promoter modulated by NF-κβ and FoxO1 [50]. Melatonin represses ROCK-1, VEGF and HIF-1α genes expressions in oral cancer [51]. Melatonin alters the expression of inflammatory and angiogenic proteins in both co-culture and monoculture of cancer cells and cancer-associated fibroblasts [52]. Melatonin suppresses tumor angiogenesis and the growth of gastric cancer cells in tumor-bearing nude mice. Moreover, melatonin decreases the expression of VEGF and HIF-1α at translational and transcriptional levels within gastric cancer cells during tumorigenesis [53]. Reduced serum levels of VEGF have been reported in cancer subjects treated with melatonin [54]. Vimalraj et al. [55] showed that melatonin upregulates miR-424-5p expression in osteosarcoma cells suppressing VEGFA. Furthermore, it inhibits tumor angiogenesis, regulating surrounding endothelial cells migration and proliferation, and angiogenic growth factors and the morphology of blood vessels E (Table 1).
Melatonin and oxidative stress
In normal cellular condition, there is a balance between the production of oxidants, so called reactive oxygen species (ROS), and their neutralizing compounds, named antioxidants. The state of excess ROS, in which the oxidant content of the cells dominates the neutralizing capacity of antioxidants, is defined as oxidative stress [56, 57]. Sustained oxidative stress increases the risk of cancer development either through inducing mutagenesis or by promoting the expression of proto-oncogenes such as cyclin D1. It also plays a signaling role in the activation of several genes involved in the cancer progression including the mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK) and JUN N-terminal kinase (JNK) [58, 59].
Melatonin role as a natural ally against oxidative stress has been revealed in many in vitro and in vivo studies. Detoxification of oxidants by melatonin is triggered by several direct or indirect mechanisms. In direct scenario, melatonin neutralizes the oxidants by its nonreceptor‐mediated free radical scavenging capacity. As indirect scenario, melatonin reduces the oxidative content through several mechanisms such as activating anti-oxidative enzymes and suppressing pro‐oxidative enzymes. It also stabilizes the mitochondrial inner membrane, thereby maintaining mitochondrial integrity leading to a reduced electron leakage and ROS generation [60, 61].
The inducing role of oxidative stress in cancer progression and preventive role of melatonin in the production and function of oxidants indicated a possible oncostatic property for melatonin [62]. Subsequently, it was revealed that melatonin reduces the oxidative damage to cellular components under conditions where toxic oxygen derivatives are acknowledged to be released [63, 64]. Moreover, in vitro studies demonstrated that melatonin treatment reduces the amount of oxidative contents in a variety of cancer cells, which was further supported by in vivo studies (Table 2).
Melatonin and endoplasmic reticulum stress
Endoplasmic reticulum (ER) is an entry site for secretory proteins and most integral membrane proteins for proper folding and covalent modifications to assemble into a functional complex. In addition to the processing of proteins, ER is involved in various cellular functions including lipid synthesis, fatty acid turnover, detoxification, Ca2+ homeostasis. The ER network extends into all cell compartments to sense intrinsic and extrinsic perturbations and integrate the stress signals for maintenance of cellular homeostasis to preserve proper cellular and organismal function [65, 66]. However, the ER function can be impacted and disturbed by a multitude of exogenous and endogenous factors, leading to the accumulation of mis/unfolded proteins in the ER. This causes the imbalance between the client proteins load in the lumen of ER and the folding capacity of this organelle leading to the failure of the ER to cope with unusual high protein folding load, which is termed 'ER stress' [67]. To restore protein homeostasis, an adaptive signal transduction pathway called the unfolded protein response (UPR) is activated to induce compensatory responses to stressors for recovering normal ER function [68]. Signaling proteins sensing UPR include inositol-requiring protein-1α (IRE1α), activating transcription factor 6α (ATF6α) and protein kinase RNA (PKR)-like ER kinase (PERK). In nonstressed cells, UPR stress sensors are maintained in an inactive state through direct binding to the ER chaperone proteins, Bip (78-kDa glucose regulated protein, GRP78). Upon ER stress, aggregation of misfolded proteins leads to the dissociation of UPR sensors from Bip, which causes activation of UPR signals [69]. Although activation of UPR signaling pathways is a cellular strategy to increase survival, this pathway will instead activate cell death signaling pathways when the intensity or duration of cellular stress increases. Therefore, certain anti-cancer patterns may activate ER stress/UPR pathway to induce apoptosis in cancer cells [70].
Melatonin induces mitochondria-mediated apoptosis in colorectal cancer cells through reducing the expression of PrPC and PINK1 resulting in the enhancement of superoxide production and induction of ER stress [71]. Melatonin also ameliorates ER-stress mediated insulin resistance. ER stress induces autophagy in pancreatic β cells, which this pathway plays an important role in insulin production and secretion. In the glucose analog 2-DG-treated rat insulinoma INS-1E cells, melatonin reduces insulin production via ER stress-induced autophagy [72]. Combination of melatonin with ER stress inducer tunicamycin increases the sensitivity of cancer cells to apoptosis through inhibiting the expression of COX-2 and increasing the Bax/Bcl-2 ratio and CHOP levels [73]. Selective inhibition of ATF-6 by melatonin results in the suppression of COX-2 production and enhancement of cancer cells to ER-stress induced apoptosis [74].
Melatonin increases apoptosis through enhancing caspase-3, -8 and -9 activities, Bax/Bcl-2 ratio, PARP cleavage and cytochrome c, p53 and Fas-L proteins concentrations in hepatocellular carcinoma, which this effect is mediated by the elevation of ER stress characterized by up-regulation of ATF6, CHOP and Bip [75]. Furthermore, melatonin increases the sensitivity of hepatocellular carcinoma cells to sorafenib through targeting the PERK-ATF4-Beclin1 pathway [76]. The same results have been reported in gastric cancer; melatonin inhibits cell proliferation through inducing activation of the IRE/JNK/Beclin1 signaling [77].
Melatonin in combination with the ER stressor thapsigargin increases the expression level of nuclear mammalian RNA-binding protein (HuD) resulting in the reduction of intracellular biosynthesis of insulin. Suppression of AKT/PI3K pathway and induction of nuclear mTOR (Ser2481, Ser2448) expressions by melatonin sensitizes rat insulinoma INS-1E cells to insulin through increasing the expression of insulin receptor substrate [78]. In contrast with these reports, melatonin has been reported to inhibit tunicamycin-induced ER stress in human hepatocellular carcinoma cells and increase the response of these cells to cytotoxic effects of doxorubicin; this is accompanied by inhibition of the PI3K/AKT pathway, elevation of CHOP and reduction of Survivin [79]. These evidences suggest that melatonin could improve the toxic effect of anti-cancer agents on cancer cells through regulating ER stress in cells (Table 3).
Melatonin and autophagy
Autophagy is a complicated process maintaining intracellular homeostasis by eliminating degraded proteins and organelles during cellular stress. Autophagy is principally considered as a pro-survival process, but, excessive or inappropriate autophagy contributes to the cell death, a process known as autophagic cell death or type II programmed cell death [80].
Autophagy is a complicated process, which consists of five sequential steps, including: (a) initiation complex formation and double-membrane phagophore (nucleation) maturation; (b) membrane elongation and autophagosome formation sequestering cargo; (c) fusion with lysosome; (d) inner membrane disruption leading to degradation of cargo by hydrolases; and (e) macromolecular component utilization [81]. These steps of the autophagy pathway are regulated by more than 35 autophagy related genes (ATGs) and proteins most of which function in complexes. The initiation phase is regulated by Unc-51-like kinase1 (mammalian homologues of Atg1, ULK1)–Atg13–Atg101–FIP200 (mammalian homologues of Atg17) protein complex. Unc-51-like kinase1 phosphorylates and activates Beclin-1 (mammalian homologue of Atg6). Beclin-1 is a part of multiprotein-complex, class III PI3-kinase Vps34–p150 (mammalian homolog of Vps15)–Atg14-like protein (Atg14L)–Beclin-1, promoting nucleation [81,82,83]. The elongation phase is regulated by two ubiquitin-like conjugation systems, Atg12 and LC3 (mammalian homologue of Atg8). In the first conjugation system, Atg12 is activated by Atg7 (E1-like enzyme), transferred to Atg10 (E2-like enzyme) and conjugated to Atg5. The Atg12–Atg5 conjugates further couples with Atg16 (Atg16L in mammals) to form the E3-like complex. In the LC3 conjugation system, LC3 is cleaved by a cysteine protease, Atg4, forming LC3-I. Thereafter, LC3-I is activated by Atg7 (E1-like enzyme), transferred to Atg3 (E1-like enzyme) and conjugated to phosphatidylethanolamine (PE) to form LC3-II; this process is facilitated by the E3-like complex. This lipidated form of LC3, LC3-II, is recruited to the autophagosome membrane. Finally, the Atg9 dependent pathway promotes autophagosome membrane expansion [81,82,83].
Cargo sequestration can be selective or non-selective; the selectivity is based on autophagy receptors such as P62/SQSTM1, NBR1, NDP52, NIX/BNIP3L, BNIP3 and FUNDC1 [82]. Fusion of autophagosome with lysosome is the next step. The inner vesicle is degraded by lysosomal hydrolases, including cathepsin B, D (a homolog of proteinase A), and L. The degradation products are released to the cytosol and used in different anabolic pathways [84]. ER stress-induced activation of UPR pathways promotes induction of autophagy [85]. Activated PERK/ATF4 pathway up-regulates the expression of ATG genes including ATG5, ATG7, and ATG10 [86]. The conversion of LC3-I conversion to LC3-II is also induced by PERK pathway [87]. Activation of IRE1α pathway induces the expression of Beclin1 and the phosphorylation of Bcl-2 by JNK, which subsequently results in the Bcl-2–Beclin 1 dissociation [88,89,90]. The release of Ca2+ from ER to cytosol triggers autophagy pathway through activating several mechanisms including (I) inhibition of mTOR by Ca2+/calmodulin dependent kinase kinase-β-mediated activation of AMP-activated protein kinase (AMPK) [91], and (II) dissociation of Bcl-2–Beclin 1 by inducing death-associated protein kinase (DAPK) 3-mediated Beclin 1 phosphorylation [92].
Melatonin has a modulatory effect on autophagy in various cell types and different conditions. Melatonin indirectly modulates autophagy through affecting oxidative stress, ER stress and inflammation [69]. Melatonin enhances the effectiveness of cisplatin and radiotherapy in head and neck squamous cell carcinoma, which this effect is mediated by the excessive activation of mitochondria leading to the over-production of ROS and subsequent induction of autophagy and apoptosis [93]. Melatonin also increases cytotoxic effects of rapamycin in cancer cells. Combination of rapamycin and melatonin suppresses AKT/mTOR pathway activation, which this effect leads to the enhancement of mitochondrial function and ROS production resulting in the induction of apoptosis and mitophagy [94]. Melatonin induces autophagy in clear cell renal cell carcinoma through activating transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1A (PGC1A) and uncoupling protein 1 (UCP1); this is associated with the elimination of lipid deposits without generating ATP, which subsequently leads to the tumor size reduction [95].
Melatonin reduces the viability liver cancer cells through transient induction autophagy by up-regulating JNK phosphorylation. However, ATG5 silencing sensitizes cancer cells to melatonin-induced apoptosis. This suggests that modulation of autophagy by melatonin has dual effect on cell death [96]. Similarly, disruption of autophagy sensitizes glioblastoma cells and tongue squamous cell carcinoma to melatonin-induced apoptosis [97]. Melatonin-induced autophagy is suggested to be mediated by activation of melatonin membrane receptor in tongue squamous cell carcinoma and suppression of melatonin membrane receptor-dependent autophagy may be strategy for treatment of tongue squamous cell carcinoma [98].
Several studies indicate that melatonin may induce apoptosis in cancer cells through inhibiting autophagy pathway. Melatonin down-regulates Beclin-1 and p62 expressions and LC3B-II/LC3B-I ratio in colitis-associated colon carcinogenesis in mice; this effect is associated with the increased level of Nrf2 and its downstream antioxidant enzymes including NAD(P)H:quinone oxidoreductase (NQO-1) and heme oxygenase-1 (HO-1). These suggest that the ameliorative effect of melatonin on inflammation and oxidative stress results in the reduction of autophagy [99]. Induction of ER stress is associated with the activation of autophagy in sorafenib-treated hepatocellular carcinoma cells, which this contributes to the resistance of cancer cells to apoptosis. Combination of melatonin with sorafenib inhibits ER stress-related autophagy through suppressing the PERK-ATF4-Beclin1 pathway leading to the sensitivity of hepatocellular carcinoma cells to sorafenib [76]. Co-stimulation of cancer cells with cisplatin and melatonin induce apoptosis in HeLa cells, which this effect is accompanied by inactivating mitophagy via blockade of JNK/Parkin pathway [100]. In contrast with this report, melatonin has been found to reversed the effects of cisplatin in HepG2 cells through suppression of mTOR and DNA excision repair cross complementary 1 (ERCC1) proteins expressions and up-regulation of Beclin-1 and LC3II expressions [101]. Taken together, different effects of melatonin on autophagy may be related to type of cancer cells, the stage of cancer and dose of melatonin (Table 4).
Melatonin and apoptosis
The balance between cell proliferation and death in tissues is maintained by apoptosis, a classical form of programmed cell death. Apoptosis is associated with the disassembly of apoptotic cells into membrane-enclosed vesicles, which are removed by macrophages without inducing inflammatory responses. Apoptosis is mediated by two principle signaling pathways, including extrinsic and intrinsic pathways [102]. The extrinsic apoptotic signaling pathways, defined as death receptor pathways, are initiated by the interaction of transmembrane death receptors (Fas, TNFR1, DR4 and DR5) with extracellular ligands (FasL, TNFα, TRAIL, and TNFSF10) resulting in the activation of adaptor proteins such as Fas-associated death domain (FADD). Activated FADD recruits initiator caspases (caspase 8 and caspase 10) to form the death-inducing signal complex (DISC). Formation of DISC leads to the proteolytic activation of caspase 8, which is the main initiator caspase of the extrinsic apoptotic signaling pathway. Caspase 8 activates executioner caspases (caspase 3, caspase 6, and caspase 7) and cleaves Bid, a BH3-only domain member of the B cell lymphoma-2 (Bcl-2) family. Truncated Bid (tBid) translocates to mitochondria and activates other proapoptotic Bcl-2 family members including Bak or Bax [102, 103].
The intrinsic apoptosis pathway, defined as mitochondrial-mediated apoptotic pathway, is activated by exogenous and endogenous stimuli such as DNA damage, oxidative stress, chemotherapy and radiotherapy. This apoptosis pathway is mediated by insertion of pro-apoptotic Bcl-2 family members (Bax/Bak) into mitochondrial membrane leading to the mitochondrial outer membrane permeabilization and release of pro-apoptotic factors such as cytochrome c, Smac/DIABLO, the nuclease EndoG, the oxidoreductase AIF, and the protease HtrA2/Omi [104]. Therefore, activation of pro-apoptotic Bcl-2 family members (Bax/Bak) is essential for cancer therapy. In contrast, elevation of anti-apoptotic Bcl-2 family proteins inhibits apoptosis in cancer cells through heterodimerization with Bax/Bak preventing the release of pro-apoptotic factors from mitochondria; this could result in the resistance of cancer cells to immune-surveillance [105, 106]. Once in the cytosol, cytochrome c combines with Apaf–1 and procaspase-9 to drive the assembly of the apoptosome; this molecular platform activates caspase 9, which this is followed by the activation of caspase-3 cascade of apoptosis [107]. Smac/DIABLO and HtrA2/Omi induce apoptosis through degrading inhibitor of apoptosis protein (IAP) family, neutralizing the inhibitory effect of IAPs on caspases [108]. The nuclease EndoG and the oxidoreductase AIF translocate to the nucleus, where they trigger internucleosomal DNA fragmentation independently of caspases [109].
As mentioned earlier, UPR signaling may promote the apoptotic pathways. Upon ER stress, apoptosis signal-regulating kinase 1 (ASK1) is recruited by IRE1α-TNF receptor-associated factor 2 (TRAF2) complex, causing activation of ASK1 and the downstream JNK pathway. Activation of JNK results in the phosphorylation of Bcl-2 and Bax; phosphorylation of Bcl-2 family suppresses antiapoptotic activity of Bcl-2, while induces mitochondrial translocation of Bax and activation of apoptosis pathway. Activated JNK also activates C/EBP homologous protein (CHOP), a stress-induced transcription factor inducing the expression of pro-apoptotic Bcl-2 family members. Furthermore, IRE1α-TRAF2 complex triggers the activation of caspase-12, which this caspase translocates from the ER to the cytosol, where it activates caspase-9, independent from the apoptosome pathway [110]. Furthermore, Activated PERK phosphorylates eIF2α, promoting the expression of activating transcriptional factor 4 (ATF4); ATF4 translocates to the nucleus where it induces CHOP expression [111].
Melatonin is reported to restrict tumor growth and cancer cell proliferation through inducing apoptosis in cancer cells (Table 5). As a powerful antioxidant melatonin inhibits ROS-induced activation of extracellular-regulated protein kinases (ERKs) and Akt pathways which are involved in the cancer cell survivor; inactivation of ROS-dependent Akt signaling contributes to the down-regulation of cyclin D1, PCNA, and Bcl-2 and up-regulation of Bax in cancer cells [48]. Inhibition of MDM2 expression is a mechanism by which melatonin induces apoptosis through upregulating the activity of caspase-3 and -9; MDM2 is an E3 ubiquitin ligase, which negatively regulates the p53 tumor suppressor [112, 113]. Under hypoxic conditions, tumor cells become resistant to TRAIL-induced cell apoptosis; this contributes to the up-regulation of anti-apoptotic protein expression and reduction of pro-apoptotic protein expression. Treatment with melatonin blocks hypoxic responses leading to the induction of apoptosis in TRAIL resistance tumor cells by the regulation of mitochondrial transmembrane potential and induction of Bax translocation [114]. Melatonin inhibits cancer cell growth by increasing cell cycle arrest in the G2/M phase, which this effect is coincident with the induction of apoptosis through up-regulating the expression of p53, p21, caspase-3/8/9, PARP, cytochrome c, Bax, JNK 1,-2 and -3 and p38 MAPKs in cancer cells [115]. Melatonin triggers two distinct apoptotic processes including TGFβ1 and caspase-independent early apoptosis and TGFβ1 and caspases-dependent late apoptosis. Early apoptosis is associated with the elevation level of p53/MDM2 ratio and up-regulation of AIF release; this process is independent to caspase activity or cleavage of PARP. Late apoptosis is associated with elevation of caspases-9 and -7 activity and cleaved-PARP level as well as reduction of Bcl-2/Bax ratio [116]. Melatonin also induces apoptosis through simultaneous suppression of COX-2/PGE2, p300/NF-κB, and PI3K/Akt signaling pathway. Inhibition of these pathways leads to the induction of Apaf-1 expression triggering cytochrome c release, and caspase-3 and -9 activation and cleavage [32].
Melatonin induces dephosphorylation and nuclear import of histone deacetylase 4 (HDAC4) in cancer cells; melatonin exerts this effect through inactivation of Ca2+/calmodulin-dependent protein kinase II alpha (CaMKIIα), leading to the H3 acetylation on Bcl-2 promoter and subsequent reduction of Bcl-2 expression [117]. Furthermore, inhibition of HDAC9 expression is a mechanism of melatonin to promote apoptosis in non-small cell lung cancer; the increased level of HDAC9 in patients with non-small cell lung cancer is correlated with worse overall survival and poor prognosis [118]. Melatonin promotes TNF-α-mediated apoptosis via inhibiting mitophagy in tumor cells. Since activation of mitophagy suppresses mitochondrial apoptosis, inhibition of mitophagy by melatonin results in the repression of mitochondrial potential, elevation of ROS generation, augmentation of mPTP opening rate and up-regulation of cytochrome c expression and caspases activity. Melatonin inhibits autophagy in tumor cells through inhibiting CaMKII activity leading to the suppression of Parkin expression [119]. In diethylnitrosamine (DEN)-induced hepatocellular carcinoma (HCC), melatonin increases therapeutic potential of mesenchymal stem cells (MSCs) through reduction of oxidative stress and inflammation, and induction of apoptosis [120].
Melatonin has been reported to increase therapeutic potential of anti-cancer agents, which this effect may result from its stimulatory effect on apoptosis. Co-treatment of melatonin and pterostilbene in colorectal cancer cells synergically enhances ROS production and apoptosis. Combination of these two agents upregulates the mRNA level of miR-25-5p, which this results in the activation of PARP and sex-determining region Y-Box10 (SOX10), and attenuation of Bcl-xL, neural precursor cell expressed developmentally downregulated protein 9 (NEDD9), and SOX9 expressions [121]. Melatonin synergically enhances anticancer potential of cisplatin through inducing apoptosis; melatonin increases the effect of cisplatin to the inhibition of ERK phosphorylation and induction of 90-kDa ribosomal S6 kinase (p90RSK) and heat shock protein 27 (HSP27) dephosphorylation [122]. Treatment with melatonin enhances ER stress–mediated apoptosis in tunicamycin-treated cancer cells; this effect is associated with the down-regulation of COX-2 and Bcl-2 expressions and up-regulation of Bim, CHOP and Bax expressions [73]. Melatonin inhibits tunicamycin-induced COX-2 activation in tumor cells through inhibiting NF-ĸB and p38 MAPK activation and p65 nuclear translocation [123]. Combination of melatonin with phenylarsine oxide also induces endoplasmic reticulum stress-induced cell death, accompanied by JNK activation, PARP cleavage, ROS generation and caspase-3 activation [124].
Conclusions
This review summarizes the anti-carcinogenic potentials of melatonin by evaluating various signaling pathways. Melatonin inhibits proliferation of cancer cells through triggering cell cycle arrest and causes cell death by induction of apoptosis. Melatonin suppresses metastasis angiogenesis, and proliferation of cancer cells through affecting various signaling pathways in tumor cells. Melatonin also regulates autophagy pathway in cancer cell by affecting oxidative stress condition in tumor cells. These findings suggest that melatonin may increase the sensitivity of cancer cells to anti-cancer agents and may be a potential treatment for cancers either alone or in combination with other anti-cancer drugs. However, further clinical studies are needed to clarify the effect of this molecule in different cancers and obtain affective dose of melatonin for patients with cancer.
Availability of data and materials
Not applicable.
Abbreviations
- JNK:
-
C-Jun N-terminal kinase
- ROS:
-
Reactive oxygen species
- VEGF:
-
Vascular endothelial growth factor
- TNF-α:
-
Tumor necrosis factor-α
- IL-2:
-
Interleukin-2
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- Apaf-1:
-
Apoptotic protease activating factor-1
- COX-2:
-
Cyclooxygenase-2
- Sirt1:
-
Sirtuin
- HIF:
-
Hypoxia-inducible factor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- PARP-1:
-
Poly [ADP-ribose] polymerase 1
- ATG:
-
Autophagy related genes
- IRE1:
-
Inositol-requiring enzyme 1
- ATF6:
-
Activating transcription factor 6
- ERK:
-
Extracellular signal-regulated kinase
- MEK:
-
Mitogen-activated protein kinase kinase
- MAPK:
-
Mitogen-activated protein kinase
- CHOP:
-
CCAAT-enhancer-binding proteins homologous protein
- PUMA:
-
P53-upregulated modulator of apoptosis
- Bcl-2:
-
B cell lymphoma-2
- Bim:
-
Bcl-2-interacting mediator of cell death
- PI3K:
-
Phosphatidylinositol-3-kinase
- Akt:
-
Protein kinase B
- UPR:
-
Unfolded protein response
- ATF6α:
-
Activating transcription factor 6α
- ER:
-
Endoplasmic reticulum
- PERK:
-
Protein kinase RNA-like ER kinase
- ERK:
-
Extracellular signal-regulated kinase
- SPHK1:
-
Sphingosine kinase 1
- IFN-γ:
-
Interferon-gamma
- PGC1A:
-
Peroxisome proliferator-activated receptor gamma coactivator 1A
- NQO-1:
-
NAD(P)H:quinone oxidoreductase
- HO-1:
-
Heme oxygenase-1
- FADD:
-
Fas-associated death domain
- tBid:
-
Truncated Bid
- ASK1:
-
Apoptosis signal-regulating kinase 1
- TRAF2:
-
IRE1α-TNF receptor-associated factor 2
- ERKs:
-
Extracellular-regulated protein kinases
- CaMKIIα:
-
Ca2+/calmodulin-dependent protein kinase II alpha
- DEN:
-
Diethylnitrosamine
- HCC:
-
Hepatocellular carcinoma
- MSCs:
-
Mesenchymal stem cells
- NEDD9:
-
Neural precursor cell expressed developmentally downregulated protein 9
- p90RSK:
-
90-KDa ribosomal S6 kinase
- HSP27:
-
Heat shock protein 27
References
Block KI, Gyllenhaal C, Lowe L, Amedei A, Amin A, Amin A, Aquilano K, Arbiser J, Arreola A, Arzumanyan A, et al. Designing a broad-spectrum integrative approach for cancer prevention and treatment. Seminars Cancer Biol. 2015;35 Suppl(Suppl):S276–304.
Alobaedi OH, Talib WH, Basheti IA. Antitumor effect of thymoquinone combined with resveratrol on mice transplanted with breast cancer. Asian Pac J Trop Med. 2017;10(4):400–8.
Talib WH. Consumption of garlic and lemon aqueous extracts combination reduces tumor burden by angiogenesis inhibition, apoptosis induction, and immune system modulation. Nutrition (Burbank, Los Angeles County, Calif). 2017;43–44:89–97.
Falah RR, Talib WH, Shbailat SJ. Combination of metformin and curcumin targets breast cancer in mice by angiogenesis inhibition, immune system modulation and induction of p53 independent apoptosis. Therapeutic Adv Med Oncol. 2017;9(4):235–52.
Slominski A, Fischer TW, Zmijewski MA, Wortsman J, Semak I, Zbytek B, Slominski RM, Tobin DJ. On the role of melatonin in skin physiology and pathology. Endocrine. 2005;27(2):137–48.
Pourhanifeh MH, Mehrzadi S, Hosseinzadeh A. Melatonin and regulation of miRNAs: novel targeted therapy for cancerous and noncancerous disease. Epigenomics. 2020;13:65–81.
Pourhanifeh MH, Hosseinzadeh A, Juybari KB, Mehrzadi S. Melatonin and urological cancers: a new therapeutic approach. Cancer Cell Int. 2020;20(1):444.
Bahrampour Juybari K, Pourhanifeh MH, Hosseinzadeh A, Hemati K, Mehrzadi S. Melatonin potentials against viral infections including COVID-19: Current evidence and new findings. Virus Res. 2020;287:198108.
Mehrzadi S, Karimi MY, Fatemi A, Reiter RJ, Hosseinzadeh A. SARS-CoV-2 and other coronaviruses negatively influence mitochondrial quality control: beneficial effects of melatonin. Pharmacol Ther. 2021;224:107825–107825.
Pourhanifeh MH, Mahdavinia M, Reiter RJ, Asemi Z. Potential use of melatonin in skin cancer treatment: a review of current biological evidence. J Cell Physiol. 2019;234(8):12142–8.
Anderson G. The effects of melatonin on signaling pathways and molecules involved in glioma: melatonin and glioblastoma: pathophysiology and treatment. Fundam Clin Pharmacol. 2020;34(2):189–91.
Pourhanifeh MH, Sharifi M, Reiter RJ, Davoodabadi A, Asemi Z. Melatonin and non-small cell lung cancer: new insights into signaling pathways. Cancer Cell Int. 2019;19:131.
Pourhanifeh MH, Mehrzadi S, Kamali M, Hosseinzadeh A. Melatonin and gastrointestinal cancers: Current evidence based on underlying signaling pathways. Eur J Pharmacol. 2020. https://doi.org/10.1016/j.ejphar.2020.173471.
Zhao Q, Wang W, Cui J. Melatonin enhances TNF-α-mediated cervical cancer HeLa cells death via suppressing CaMKII/Parkin/mitophagy axis. Cancer Cell Int. 2019;19:58.
Bu S, Wang Q, Sun J, Li X, Gu T, Lai D. Melatonin suppresses chronic restraint stress-mediated metastasis of epithelial ovarian cancer via NE/AKT/β-catenin/SLUG axis. Cell Death Dis. 2020;11(8):644.
Yan G, Lei H, He M, Gong R, Wang Y, He X, Li G, Pang P, Li X, Yu S, et al. Melatonin triggers autophagic cell death by regulating RORC in Hodgkin lymphoma. Biomed Pharmacother. 2020;123:109811.
Tian T, Li J, Li Y, Lu YX, Tang YL, Wang H, Zheng F, Shi D, Long Q, Chen M, et al. Melatonin enhances sorafenib-induced cytotoxicity in FLT3-ITD acute myeloid leukemia cells by redox modification. Theranostics. 2019;9(13):3768–79.
Reiter RJ, Tan DX, Fuentes-Broto L. Melatonin: a multitasking molecule. Prog Brain Res. 2010;181:127–51.
Bonnefont-Rousselot D, Collin F. Melatonin: action as antioxidant and potential applications in human disease and aging. Toxicology. 2010;278(1):55–67.
Slominski RM, Reiter RJ, Schlabritz-Loutsevitch N, Ostrom RS, Slominski AT. Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol. 2012;351(2):152–66.
Stevens RG, Brainard GC, Blask DE, Lockley SW, Motta ME. Breast cancer and circadian disruption from electric lighting in the modern world. CA Cancer J Clin. 2014;64(3):207–18.
He R, Cui M, Lin H, Zhao L, Wang J, Chen S, Shao Z. Melatonin resists oxidative stress-induced apoptosis in nucleus pulposus cells. Life Sci. 2018;199:122–30.
Reiter RJ, Tan DX, Erren TC, Fuentes-Broto L, Paredes SD. Light-mediated perturbations of circadian timing and cancer risk: a mechanistic analysis. Integr Cancer Ther. 2009;8(4):354–60.
Pourhanifeh MH, Hosseinzadeh A, Dehdashtian E, Hemati K, Mehrzadi S. Melatonin: new insights on its therapeutic properties in diabetic complications. Diabetol Metab Syndr. 2020;12:1–20.
Dehdashtian E, Pourhanifeh MH, Hemati K, Mehrzadi S, Hosseinzadeh A. Therapeutic application of nutraceuticals in diabetic nephropathy: current evidence and future implications. Diabetes Metab Res Rev. 2020;36:e3336.
Rosales-Corral S, Tan DX, Reiter RJ, Valdivia-Velázquez M, Martínez-Barboza G, Acosta-Martínez JP, Ortiz GG. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: a comparative, in vivo study versus vitamin C and E. J Pineal Res. 2003;35(2):80–4.
Hosseinzadeh A, Javad-Moosavi SA, Reiter RJ, Hemati K, Ghaznavi H, Mehrzadi S. Idiopathic pulmonary fibrosis (IPF) signaling pathways and protective roles of melatonin. Life Sci. 2018;201:17–29.
Daryani A, Montazeri M, Pagheh AS, Sharif M, Sarvi S, Hosseinzadeh A, Reiter RJ, Hadighi R, Joghataei MT, Ghaznavi H. The potential use of melatonin to treat protozoan parasitic infections: a review. Biomed Pharmacother. 2018;97:948–57.
Yokota O, Terada S, Ishizu H, Ishihara T, Ujike H, Nakashima H, Nakashima Y, Kugo A, Checler F, Kuroda S. Cyclooxygenase-2 in the hippocampus is up-regulated in Alzheimer’s disease but not in variant Alzheimer’s disease with cotton wool plaques in humans. Neurosci Lett. 2003;343(3):175–9.
Juybari KB, Hosseinzadeh A, Ghaznavi H, Kamali M, Sedaghat A, Mehrzadi S, Naseripour M. Melatonin as a modulator of degenerative and regenerative signaling pathways in injured Retinal Ganglion Cells. Curr Pharm Des. 2019;25(28):3057–73.
Menéndez-Menéndez J, Hermida-Prado F, Granda-Díaz R, González A, García-Pedrero JM, Del-Río-Ibisate N, González-González A, Cos S, Alonso-González C, Martínez-Campa C. Deciphering the molecular basis of melatonin protective effects on breast cells treated with doxorubicin: TWIST1 a transcription factor involved in EMT and metastasis, a novel target of melatonin. Cancers. 2019;11(7):1011.
Wang J, Xiao X, Zhang Y, Shi D, Chen W, Fu L, Liu L, Xie F, Kang T, Huang W, et al. Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J Pineal Res. 2012;53(1):77–90.
Cipolla-Neto J, Amaral FGD. Melatonin as a hormone: new physiological and clinical insights. Endocr Rev. 2018;39(6):990–1028.
Pourhanifeh MH, Dehdashtian E, Hosseinzadeh A, Sezavar SH, Mehrzadi S. Clinical application of melatonin in the treatment of cardiovascular diseases: current evidence and new insights into the cardioprotective and cardiotherapeutic properties. Cardiovasc Drugs Ther. 2020;14:1–25.
Sánchez-Barceló EJ, Cos S, Fernández R, Mediavilla MD. Melatonin and mammary cancer: a short review. Endocr Relat Cancer. 2003;10(2):153–9.
Cos S, Sánchez-Barceló EJ. Melatonin and mammary pathological growth. Front Neuroendocrinol. 2000;21(2):133–70.
Cos S, Fernández R, Güézmes A, Sánchez-Barceló EJ. Influence of melatonin on invasive and metastatic properties of MCF-7 human breast cancer cells. Can Res. 1998;58(19):4383–90.
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–10.
Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9(Suppl 5):10–7.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42.
Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111(5):709–20.
Mirza-Aghazadeh-Attari M, Reiter RJ, Rikhtegar R, Jalili J, Hajalioghli P, Mihanfar A, Majidinia M, Yousefi B. Melatonin: an atypical hormone with major functions in the regulation of angiogenesis. IUBMB Life. 2020;72(8):1560–84.
Ma Q, Reiter RJ, Chen Y. Role of melatonin in controlling angiogenesis under physiological and pathological conditions. Angiogenesis. 2020;23(2):91–104.
Park SY, Jang WJ, Yi EY, Jang JY, Jung Y, Jeong JW, Kim YJ. Melatonin suppresses tumor angiogenesis by inhibiting HIF-1alpha stabilization under hypoxia. J Pineal Res. 2010;48(2):178–84.
Cho SY, Lee HJ, Jeong SJ, Lee HJ, Kim HS, Chen CY, Lee EO, Kim SH. Sphingosine kinase 1 pathway is involved in melatonin-induced HIF-1α inactivation in hypoxic PC-3 prostate cancer cells. J Pineal Res. 2011;51(1):87–93.
Alvarez-García V, González A, Alonso-González C, Martínez-Campa C, Cos S. Regulation of vascular endothelial growth factor by melatonin in human breast cancer cells. J Pineal Res. 2013;54(4):373–80.
Siu SW, Lau KW, Tam PC, Shiu SY. Melatonin and prostate cancer cell proliferation: interplay with castration, epidermal growth factor, and androgen sensitivity. Prostate. 2002;52(2):106–22.
Liu R, Wang HL, Deng MJ, Wen XJ, Mo YY, Chen FM, Zou CL, Duan WF, Li L, Nie X. Melatonin inhibits reactive oxygen species-driven proliferation, epithelial-mesenchymal transition, and vasculogenic mimicry in oral cancer. Oxid Med Cell Longev. 2018;2018:3510970.
Liu H, Zhu Y, Zhu H, Cai R, Wang KF, Song J, Wang RX, Zhou RX. Role of transforming growth factor β1 in the inhibition of gastric cancer cell proliferation by melatonin in vitro and in vivo. Oncol Rep. 2019;42(2):753–62.
León J, Casado J, Jiménez Ruiz SM, Zurita MS, González-Puga C, Rejón JD, Gila A, Muñoz de Rueda P, Pavón EJ, Reiter RJ, et al. Melatonin reduces endothelin-1 expression and secretion in colon cancer cells through the inactivation of FoxO-1 and NF-κβ. J Pineal Res. 2014;56(4):415–26.
Goncalves Ndo N, Rodrigues RV, Jardim-Perassi BV, Moschetta MG, Lopes JR, Colombo J, Zuccari DA. Molecular markers of angiogenesis and metastasis in lines of oral carcinoma after treatment with melatonin. Anticancer Agents Med Chem. 2014;14(9):1302–11.
Maschio-Signorini LB, Gelaleti GB, Moschetta MG, Borin TF, Jardim-Perassi BV, Lopes JR, Lacerda JZ, Roela RA, Bordin NA, Corrêa LA, et al. Melatonin regulates angiogenic and inflammatory proteins in MDA-MB-231 cell line and in co-culture with cancer-associated fibroblasts. Anticancer Agents Med Chem. 2016;16(11):1474–84.
Wang RX, Liu H, Xu L, Zhang H, Zhou RX. Melatonin downregulates nuclear receptor RZR/RORγ expression causing growth-inhibitory and anti-angiogenesis activity in human gastric cancer cells in vitro and in vivo. Oncol Lett. 2016;12(2):897–903.
Lissoni P, Rovelli F, Malugani F, Bucovec R, Conti A, Maestroni GJ. Anti-angiogenic activity of melatonin in advanced cancer patients. Neuro Endocrinol Lett. 2001;22(1):45–7.
Vimalraj S, Saravanan S, Raghunandhakumar S, Anuradha D. Melatonin regulates tumor angiogenesis via miR-424-5p/VEGFA signaling pathway in osteosarcoma. Life Sci. 2020;256:118011.
Duracková Z. Some current insights into oxidative stress. Physiol Res. 2010;59(4):459–69.
Bagherifard A, Kadijani AA, Yahyazadeh H, Rezazadeh J, Azizi M, Akbari A, Mirzaei A. The value of serum total oxidant to the antioxidant ratio as a biomarker of knee osteoarthritis. Clin Nutr ESPEN. 2020;38:118–23.
Saha SK, Lee SB, Won J, Choi HY, Kim K, Yang G-M, Dayem AA, Cho S-G. Correlation between oxidative stress, nutrition, and cancer initiation. Int J Mol Sci. 2017;18(7):1544.
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49(11):1603–16.
Zhang HM, Zhang Y. Melatonin: a well-documented antioxidant with conditional pro-oxidant actions. J Pineal Res. 2014;57(2):131–46.
Galano A, Tan DX, Reiter RJ. Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res. 2011;51(1):1–16.
Li Y, Li S, Zhou Y, Meng X, Zhang J-J, Xu D-P, Li H-B. Melatonin for the prevention and treatment of cancer. Oncotarget. 2017;8(24):39896–921.
Sun TC, Liu XC, Yang SH, Song LL, Zhou SJ, Deng SL, Tian L, Cheng LY. Melatonin inhibits oxidative stress and apoptosis in cryopreserved ovarian tissues via Nrf2/HO-1 signaling pathway. Front Mol Biosci. 2020;7:163.
He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44(2):532–53.
Yoo YS, Han HG, Jeon YJ. Unfolded protein response of the endoplasmic reticulum in tumor progression and immunogenicity. Oxid Med Cell Longev. 2017;2017:2969271.
Wu H, Carvalho P, Voeltz GK. Here, there, and everywhere: the importance of ER membrane contact sites. Science. 2018;361(6401):eaan5835.
Schönthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica. 2012;2012:857516.
Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–17.
Mehrzadi S, Hemati K, Reiter RJ, Hosseinzadeh A. Mitochondrial dysfunction in age-related macular degeneration: melatonin as a potential treatment. Expert Opin Ther Targets. 2020;24(4):359–78.
Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249.
Yun CW, Kim S, Lee JH, Lee SH. Melatonin promotes apoptosis of colorectal cancer cells via superoxide-mediated ER stress by inhibiting cellular prion protein expression. Anticancer Res. 2018;38(7):3951–60.
Kim HS, Han TY, Yoo YM. Melatonin-mediated intracellular insulin during 2-deoxy-d-glucose treatment is reduced through autophagy and EDC3 protein in insulinoma INS-1E cells. Oxid Med Cell Longev. 2016;2016:2594703.
Zha L, Fan L, Sun G, Wang H, Ma T, Zhong F, Wei W. Melatonin sensitizes human hepatoma cells to endoplasmic reticulum stress-induced apoptosis. J Pineal Res. 2012;52(3):322–31.
Bu LJ, Yu HQ, Fan LL, Li XQ, Wang F, Liu JT, Zhong F, Zhang CJ, Wei W, Wang H, et al. Melatonin, a novel selective ATF-6 inhibitor, induces human hepatoma cell apoptosis through COX-2 downregulation. World J Gastroenterol. 2017;23(6):986–98.
Moreira AJ, Ordoñez R, Cerski CT, Picada JN, García-Palomo A, Marroni NP, Mauriz JL, González-Gallego J. Melatonin activates endoplasmic reticulum stress and apoptosis in rats with diethylnitrosamine-induced hepatocarcinogenesis. PLoS ONE. 2015;10(12):e0144517.
Zhou B, Lu Q, Liu J, Fan L, Wang Y, Wei W, Wang H, Sun G. Melatonin increases the sensitivity of hepatocellular carcinoma to sorafenib through the PERK-ATF4-beclin1 pathway. Int J Biol Sci. 2019;15(9):1905–20.
Zheng Y, Tu J, Wang X, Yu Y, Li J, Jin Y, Wu J. The therapeutic effect of melatonin on GC by inducing cell apoptosis and autophagy induced by endoplasmic reticulum stress. Onco Targets Ther. 2019;12:10187–98.
Yoo YM. Melatonin-mediated insulin synthesis during endoplasmic reticulum stress involves HuD expression in rat insulinoma INS-1E cells. J Pineal Res. 2013;55(2):207–20.
Fan L, Sun G, Ma T, Zhong F, Lei Y, Li X, Wei W. Melatonin reverses tunicamycin-induced endoplasmic reticulum stress in human hepatocellular carcinoma cells and improves cytotoxic response to doxorubicin by increasing CHOP and decreasing survivin. J Pineal Res. 2013;55(2):184–94.
Zhu B, Ma AQ, Yang L, Dang XM. Atorvastatin attenuates bleomycin-induced pulmonary fibrosis via suppressing iNOS expression and the CTGF (CCN2)/ERK signaling pathway. Int J Mol Sci. 2013;14(12):24476–91.
Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85:685–713.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12.
Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101–22.
Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501.
Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol. 2017;232:2977–84.
B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683–99.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14(2):230–9.
Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288(2):859–72.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39.
Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19(12):8469–78.
Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25(2):193–205.
Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy. 2009;5(5):720–2.
Fernandez-Gil BI, Guerra-Librero A, Shen YQ, Florido J, Martínez-Ruiz L, García-López S, Adan C, Rodríguez-Santana C, Acuña-Castroviejo D, Quiñones-Hinojosa A, et al. Melatonin enhances cisplatin and radiation cytotoxicity in head and neck squamous cell carcinoma by stimulating mitochondrial ROS generation, apoptosis, and autophagy. Oxid Med Cell Longev. 2019;2019:7187128.
Shen YQ, Guerra-Librero A, Fernandez-Gil BI, Florido J, García-López S, Martinez-Ruiz L, Mendivil-Perez M, Soto-Mercado V, Acuña-Castroviejo D, Ortega-Arellano H, et al. Combination of melatonin and rapamycin for head and neck cancer therapy: Suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. J Pineal Res. 2018;64(3):e12461.
Xiao W, Xiong Z, Xiong W, Yuan C, Xiao H, Ruan H, Song Z, Wang C, Bao L, Cao Q, et al. Melatonin/PGC1A/UCP1 promotes tumor slimming and represses tumor progression by initiating autophagy and lipid browning. J Pineal Res. 2019;67(4):e12607.
Ordonez R, Fernandez A, Prieto-Dominguez N, Martinez L, Garcia-Ruiz C, Fernandez-Checa JC, Mauriz JL, Gonzalez-Gallego J. Ceramide metabolism regulates autophagy and apoptotic cell death induced by melatonin in liver cancer cells. J Pineal Res. 2015;59(2):178–89.
Zhou N, Wei ZX, Qi ZX. Inhibition of autophagy triggers melatonin-induced apoptosis in glioblastoma cells. BMC Neurosci. 2019;20(1):63.
Fan T, Pi H, Li M, Ren Z, He Z, Zhu F, Tian L, Tu M, Xie J, Liu M, et al. Inhibiting MT2-TFE3-dependent autophagy enhances melatonin-induced apoptosis in tongue squamous cell carcinoma. J Pineal Res. 2018;64(2):e12457.
Trivedi PP, Jena GB, Tikoo KB, Kumar V. Melatonin modulated autophagy and Nrf2 signaling pathways in mice with colitis-associated colon carcinogenesis. Mol Carcinog. 2016;55(3):255–67.
Chen L, Liu L, Li Y, Gao J. Melatonin increases human cervical cancer HeLa cells apoptosis induced by cisplatin via inhibition of JNK/Parkin/mitophagy axis. In Vitro Cell Dev Biol Anim. 2018;54(1):1–10.
Bennukul K, Numkliang S, Leardkamolkarn V. Melatonin attenuates cisplatin-induced HepG2 cell death via the regulation of mTOR and ERCC1 expressions. World J Hepatol. 2014;6(4):230–42.
Fattman CL. Apoptosis in pulmonary fibrosis: too much or not enough? Antioxid Redox Signal. 2008;10(2):379–86.
Huang K, Zhang J, O’Neill KL, Gurumurthy CB, Quadros RM, Tu Y, Luo X. Cleavage by Caspase 8 and mitochondrial membrane association activate the BH3-only protein bid during TRAIL-induced apoptosis. J Biol Chem. 2016;291(22):11843–51.
Hollville E, Romero SE, Deshmukh M. Apoptotic cell death regulation in neurons. FEBS J. 2019;286(17):3276–98.
Hattori T, Ookawa N, Fujita R, Fukuchi K. Heterodimerization of Bcl-2 and Bcl-X(L) with Bax and Bad in colorectal cancer. Acta oncologica (Stockholm, Sweden). 2000;39(4):495–500.
Thomas S, Quinn BA, Das SK, Dash R, Emdad L, Dasgupta S, Wang X-Y, Dent P, Reed JC, Pellecchia M. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets. 2013;17(1):61–75.
Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3(4):350–6.
Suzuki Y, Takahashi-Niki K, Akagi T, Hashikawa T, Takahashi R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004;11(2):208–16.
Sevrioukova IF. Apoptosis-inducing factor: structure, function, and redox regulation. Antioxid Redox Signal. 2011;14(12):2545–79.
Kim C, Kim B. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: a review. Nutrients. 2018;10(8):1021.
Liao Y, Fung TS, Huang M, Fang SG, Zhong Y, Liu DX. Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J Virol. 2013;87(14):8124–34.
Song J, Ma SJ, Luo JH, Liu H, Li L, Zhang ZG, Chen LS, Zhou RX. Downregulation of AKT and MDM2, melatonin induces apoptosis in AGS and MGC803 Cells. Anatomical Rec. 2019;302(9):1544–51.
Proietti S, Cucina A, Dobrowolny G, D’Anselmi F, Dinicola S, Masiello MG, Pasqualato A, Palombo A, Morini V, Reiter RJ, et al. Melatonin down-regulates MDM2 gene expression and enhances p53 acetylation in MCF-7 cells. J Pineal Res. 2014;57(1):120–9.
Lee YJ, Lee JH, Moon JH, Park SY. Overcoming hypoxic-resistance of tumor cells to TRAIL-induced apoptosis through melatonin. Int J Mol Sci. 2014;15(7):11941–56.
Martín-Renedo J, Mauriz JL, Jorquera F, Ruiz-Andrés O, González P, González-Gallego J. Melatonin induces cell cycle arrest and apoptosis in hepatocarcinoma HepG2 cell line. J Pineal Res. 2008;45(4):532–40.
Cucina A, Proietti S, D’Anselmi F, Coluccia P, Dinicola S, Frati L, Bizzarri M. Evidence for a biphasic apoptotic pathway induced by melatonin in MCF-7 breast cancer cells. J Pineal Res. 2009;46(2):172–80.
Wei JY, Li WM, Zhou LL, Lu QN, He W. Melatonin induces apoptosis of colorectal cancer cells through HDAC4 nuclear import mediated by CaMKII inactivation. J Pineal Res. 2015;58(4):429–38.
Ma Z, Liu D, Di S, Zhang Z, Li W, Zhang J, Xu L, Guo K, Zhu Y, Li X, et al. Histone deacetylase 9 downregulation decreases tumor growth and promotes apoptosis in non-small cell lung cancer after melatonin treatment. J Pineal Res. 2019;67(2):e12587.
Zhao Q, Wang W, Cui J. Melatonin enhances TNF-alpha-mediated cervical cancer HeLa cells death via suppressing CaMKII/Parkin/mitophagy axis. Cancer Cell Int. 2019;19:58.
El-Magd MA, Mohamed Y, El-Shetry ES, Elsayed SA, Abo Gazia M, Abdel-Aleem GA, Shafik NM, Abdo WS, El-Desouki NI, Basyony MA. Melatonin maximizes the therapeutic potential of non-preconditioned MSCs in a DEN-induced rat model of HCC. Biomed Pharmacother. 2019;114:108732.
Jung JH, Shin EA, Kim JH, Sim DY, Lee H, Park JE, Lee HJ, Kim SH. NEDD9 inhibition by miR-25–5p activation is critically involved in co-treatment of melatonin- and pterostilbene-induced apoptosis in colorectal cancer cells. Cancers. 2019;11(11):1684.
Kim JH, Jeong SJ, Kim B, Yun SM, Choi DY, Kim SH. Melatonin synergistically enhances cisplatin-induced apoptosis via the dephosphorylation of ERK/p90 ribosomal S6 kinase/heat shock protein 27 in SK-OV-3 cells. J Pineal Res. 2012;52(2):244–52.
Woo SM, Min KJ, Kwon TK. Melatonin-mediated Bim up-regulation and cyclooxygenase-2 (COX-2) down-regulation enhances tunicamycin-induced apoptosis in MDA-MB-231 cells. J Pineal Res. 2015;58(3):310–20.
Zakki SA, Muhammad JS, Li JL, Sun L, Li ML, Feng QW, Li YL, Cui ZG, Inadera H. Melatonin triggers the anticancer potential of phenylarsine oxide via induction of apoptosis through ROS generation and JNK activation. Metallomics Integr Biometal Sci. 2020;12:396–407.
Gonzalez-Gonzalez A, Gonzalez A, Alonso-Gonzalez C, Menendez-Menendez J, Martinez-Campa C, Cos S. Complementary actions of melatonin on angiogenic factors, the angiopoietin/Tie2 axis and VEGF, in cocultures of human endothelial and breast cancer cells. Oncol Rep. 2018;39(1):433–41.
Kumari R, Rawat K, Kumari A, Shrivastava A. Amelioration of Dalton’s lymphoma-induced angiogenesis by melatonin. Tumour Biol. 2017;39(6):1010428317705758.
Zonta YR, Martinez M, Camargo IC, Domeniconi RF, Lupi Junior LA, Pinheiro PF, Reiter RJ, Martinez FE, Chuffa LG. Melatonin reduces angiogenesis in serous papillary ovarian carcinoma of ethanol-preferring rats. Int J Mol Sci. 2017;18(4):763.
Gelaleti GB, Borin TF, Maschio-Signorini LB, Moschetta MG, Hellmen E, Viloria-Petit AM, Zuccari D. Melatonin and IL-25 modulate apoptosis and angiogenesis mediators in metastatic (CF-41) and non-metastatic (CMT-U229) canine mammary tumour cells. Vet Comp Oncol. 2017;15(4):1572–84.
González A, González-González A, Alonso-González C, Menéndez-Menéndez J, Martínez-Campa C, Cos S. Melatonin inhibits angiogenesis in SH-SY5Y human neuroblastoma cells by downregulation of VEGF. Oncol Rep. 2017;37(4):2433–40.
Wang RX, Liu H, Xu L, Zhang H, Zhou RX. Melatonin downregulates nuclear receptor RZR/RORgamma expression causing growth-inhibitory and anti-angiogenesis activity in human gastric cancer cells in vitro and in vivo. Oncol Lett. 2016;12(2):897–903.
Jardim-Perassi BV, Lourenco MR, Doho GM, Grigolo IH, Gelaleti GB, Ferreira LC, Borin TF, Moschetta MG, Pires de Campos Zuccari DA. Melatonin regulates angiogenic factors under hypoxia in breast cancer cell lines. Anticancer Agents Med Chem. 2016;16(3):347–58.
Talib WH, Saleh S. Propionibacterium acnes augments antitumor, anti-angiogenesis and immunomodulatory effects of melatonin on breast cancer implanted in mice. PLoS ONE. 2015;10(4):e0124384.
Sohn EJ, Won G, Lee J, Lee S, Kim SH. Upregulation of miRNA3195 and miRNA374b mediates the anti-angiogenic properties of melatonin in hypoxic PC-3 prostate cancer cells. J Cancer. 2015;6(1):19–28.
Carbajo-Pescador S, Ordonez R, Benet M, Jover R, Garcia-Palomo A, Mauriz JL, Gonzalez-Gallego J. Inhibition of VEGF expression through blockade of Hif1alpha and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br J Cancer. 2013;109(1):83–91.
Kim KJ, Choi JS, Kang I, Kim KW, Jeong CH, Jeong JW. Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J Pineal Res. 2013;54(3):264–70.
Jardim-Perassi BV, Arbab AS, Ferreira LC, Borin TF, Varma NR, Iskander AS, Shankar A, Ali MM, de Campos Zuccari DA. Effect of melatonin on tumor growth and angiogenesis in xenograft model of breast cancer. PLoS ONE. 2014;9(1):e85311.
Marques JHM, Mota AL, Oliveira JG, Lacerda JZ, Stefani JP, Ferreira LC, Castro TB, Aristizabal-Pachon AF, Zuccari D. Melatonin restrains angiogenic factors in triple-negative breast cancer by targeting miR-152-3p: In vivo and in vitro studies. Life Sci. 2018;208:131–8.
Lacerda JZ, Ferreira LC, Lopes BC, Aristizabal-Pachon AF, Bajgelman MC, Borin TF, Zuccari D. Therapeutic potential of melatonin in the regulation of MiR-148a-3p and angiogenic factors in breast cancer. MicroRNA (Shariqah, United Arab Emirates). 2019;8(3):237–47.
Hsieh MJ, Lin CW, Su SC, Reiter RJ, Chen AW, Chen MK, Yang SF. Effects of miR-34b/miR-892a upregulation and inhibition of ABCB1/ABCB4 on melatonin-induced apoptosis in VCR-resistant oral cancer cells. Mol Ther Nucleic Acids. 2020;19:877–89.
Talib WH. A ketogenic diet combined with melatonin overcomes cisplatin and vincristine drug resistance in breast carcinoma syngraft. Nutrition (Burbank, Los Angeles County, Calif). 2019;72:110659.
Amin AH, El-Missiry MA, Othman AI, Ali DA, Gouida MS, Ismail AH. Ameliorative effects of melatonin against solid Ehrlich carcinoma progression in female mice. J Pineal Res. 2019;67(2):e12585.
Hao J, Fan W, Li Y, Tang R, Tian C, Yang Q, Zhu T, Diao C, Hu S, Chen M, et al. Melatonin synergizes BRAF-targeting agent vemurafenib in melanoma treatment by inhibiting iNOS/hTERT signaling and cancer-stem cell traits. J Exp Clin Cancer Res. 2019;38(1):48.
Sonehara NM, Lacerda JZ, Jardim-Perassi BV, de Paula R, Moschetta-Pinheiro MG, Souza YST, de Andrade JCJ, De Campos Zuccari DAP. Melatonin regulates tumor aggressiveness under acidosis condition in breast cancer cell lines. Oncol Lett. 2019;17(2):1635–45.
Leja-Szpak A, Nawrot-Porabka K, Goralska M, Jastrzebska M, Link-Lenczowski P, Bonior J, Pierzchalski P, Jaworek J. Melatonin and its metabolite N1-acetyl-N2-formyl-5-methoxykynuramine (afmk) enhance chemosensitivity to gemcitabine in pancreatic carcinoma cells (PANC-1). Pharmacol Rep. 2018;70(6):1079–88.
Kocak N, Donmez H, Yildirim IH. Effects of melatonin on apoptosis and cell differentiation in MCF-7 derived cancer stem cells. Cell Mol Biol. 2018;64(12):56–61.
Krestinina O, Fadeev R, Lomovsky A, Baburina Y, Kobyakova M, Akatov V. Melatonin can strengthen the effect of retinoic acid in HL-60 cells. Int J Mol Sci. 2018;19(10):2873.
Odeh LH, Talib WH, Basheti IA. Synergistic effect of thymoquinone and melatonin against breast cancer implanted in mice. J Cancer Res Ther. 2018;14(Supplement):S324-s330.
Safaroghli-Azar A, Pourbagheri-Sigaroodi A, Bashash D, Nooshinfar E, Anjam-Najmedini A, Sadeghi S, Rezaie-Tavirani M, Akbari ME. Stimulatory effect of indolic hormone on As2O3 cytotoxicity in breast cancer cells: NF-kappaB-dependent mechanism of action of melatonin. Int J Mol Cell Med. 2018;7(3):158–68.
Fang Z, Jung KH, Yan HH, Kim SJ, Rumman M, Park JH, Han B, Lee JE, Kang YW, Lim JH, et al. Melatonin synergizes with sorafenib to suppress pancreatic cancer via melatonin receptor and PDGFR-beta/STAT3 Pathway. Cell Physiol Biochem. 2018;47(5):1751–68.
Franco DG, Moretti IF, Marie SKN. Mitochondria transcription factor A: a putative target for the effect of melatonin on u87mg malignant glioma cell line. Molecules. 2018;23(5):1129.
Wei X, Qi Y, Jia N, Zhou Q, Zhang S, Wang Y. Hyperbaric oxygen treatment sensitizes gastric cancer cells to melatonin-induced apoptosis through multiple pathways. J Cell Biochem. 2018;119(8):6723–31.
Zou ZW, Liu T, Li Y, Chen P, Peng X, Ma C, Zhang WJ, Li PD. Melatonin suppresses thyroid cancer growth and overcomes radioresistance via inhibition of p65 phosphorylation and induction of ROS. Redox Biol. 2018;16:226–36.
Song J, Ma SJ, Luo JH, Zhang H, Wang RX, Liu H, Li L, Zhang ZG, Zhou RX. Melatonin induces the apoptosis and inhibits the proliferation of human gastric cancer cells via blockade of the AKT/MDM2 pathway. Oncol Rep. 2018;39(4):1975–83.
Yu Z, Tian X, Peng Y, Sun Z, Wang C, Tang N, Li B, Jian Y, Wang W, Huo X, et al. Mitochondrial cytochrome P450 (CYP) 1B1 is responsible for melatonin-induced apoptosis in neural cancer cells. J Pineal Res. 2018;65(1):e12478.
Zhu C, Huang Q, Zhu H. Melatonin inhibits the proliferation of gastric cancer cells through regulating the miR-16-5p-Smad3 pathway. DNA Cell Biol. 2018;37(3):244–52.
Chovancova B, Hudecova S, Lencesova L, Babula P, Rezuchova I, Penesova A, Grman M, Moravcik R, Zeman M, Krizanova O. Melatonin-induced changes in cytosolic calcium might be responsible for apoptosis induction in tumour cells. Cell Physiol Biochem. 2017;44(2):763–77.
Gatti G, Lucini V, Dugnani S, Calastretti A, Spadoni G, Bedini A, Rivara S, Mor M, Canti G, Scaglione F, et al. Antiproliferative and pro-apoptotic activity of melatonin analogues on melanoma and breast cancer cells. Oncotarget. 2017;8(40):68338–53.
Shrestha S, Zhu J, Wang Q, Du X, Liu F, Jiang J, Song J, Xing J, Sun D, Hou Q, et al. Melatonin potentiates the antitumor effect of curcumin by inhibiting IKKbeta/NF-kappaB/COX-2 signaling pathway. Int J Oncol. 2017;51(4):1249–60.
Pariente R, Bejarano I, Rodriguez AB, Pariente JA, Espino J. Melatonin increases the effect of 5-fluorouracil-based chemotherapy in human colorectal adenocarcinoma cells in vitro. Mol Cell Biochem. 2018;440(1–2):43–51.
Lu JJ, Fu L, Tang Z, Zhang C, Qin L, Wang J, Yu Z, Shi D, Xiao X, Xie F, et al. Melatonin inhibits AP-2beta/hTERT, NF-kappaB/COX-2 and Akt/ERK and activates caspase/Cyto C signaling to enhance the antitumor activity of berberine in lung cancer cells. Oncotarget. 2016;7(3):2985–3001.
Li W, Fan M, Chen Y, Zhao Q, Song C, Yan Y, Jin Y, Huang Z, Lin C, Wu J. Melatonin induces cell apoptosis in AGS Cells through the activation of JNK and P38 MAPK and the suppression of nuclear Factor-Kappa B: a novel therapeutic implication for gastric cancer. Cell Physiol Biochem. 2015;37(6):2323–38.
Chuffa LG, Alves MS, Martinez M, Camargo IC, Pinheiro PF, Domeniconi RF, Junior LA, Martinez FE. Apoptosis is triggered by melatonin in an in vivo model of ovarian carcinoma. Endocr Relat Cancer. 2016;23(2):65–76.
Pariente R, Pariente JA, Rodríguez AB, Espino J. Melatonin sensitizes human cervical cancer HeLa cells to cisplatin-induced cytotoxicity and apoptosis: effects on oxidative stress and DNA fragmentation. J Pineal Res. 2016;60(1):55–64.
Codenotti S, Battistelli M, Burattini S, Salucci S, Falcieri E, Rezzani R, Faggi F, Colombi M, Monti E, Fanzani A. Melatonin decreases cell proliferation, impairs myogenic differentiation and triggers apoptotic cell death in rhabdomyosarcoma cell lines. Oncol Rep. 2015;34(1):279–87.
Tosun M, Soysal Y, Mas NG, Karabekir HS. Comparison of the effects of 13-cis retinoic acid and melatonin on the viabilities of SH-SY5Y neuroblastoma cell line. J Korean Neurosurg Soc. 2015;57(3):147–51.
Laothong U, Hiraku Y, Oikawa S, Intuyod K, Murata M, Pinlaor S. Melatonin induces apoptosis in cholangiocarcinoma cell lines by activating the reactive oxygen species-mediated mitochondrial pathway. Oncol Rep. 2015;33(3):1443–9.
Plaimee P, Weerapreeyakul N, Barusrux S, Johns NP. Melatonin potentiates cisplatin-induced apoptosis and cell cycle arrest in human lung adenocarcinoma cells. Cell Prolif. 2015;48(1):67–77.
Xu L, Jin QD, Gong X, Liu H, Zhou RX. Anti-gastric cancer effect of melatonin and Bcl-2, Bax, p21 and p53 expression changes. Sheng li xue bao : [Acta physiologica Sinica]. 2014;66(6):723–9.
Plaimee P, Weerapreeyakul N, Thumanu K, Tanthanuch W, Barusrux S. Melatonin induces apoptosis through biomolecular changes, in SK-LU-1 human lung adenocarcinoma cells. Cell Prolif. 2014;47(6):564–77.
Kim W, Jeong JW, Kim JE. CCAR2 deficiency augments genotoxic stress-induced apoptosis in the presence of melatonin in non-small cell lung cancer cells. Tumour Biol. 2014;35(11):10919–29.
Hong Y, Won J, Lee Y, Lee S, Park K, Chang KT, Hong Y. Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J Pineal Res. 2014;56(3):264–74.
Park EJ, Woo SM, Min KJ, Kwon TK. Transcriptional and post-translational regulation of Bim controls apoptosis in melatonin-treated human renal cancer Caki cells. J Pineal Res. 2014;56(1):97–106.
Perdomo J, Cabrera J, Estevez F, Loro J, Reiter RJ, Quintana J. Melatonin induces apoptosis through a caspase-dependent but reactive oxygen species-independent mechanism in human leukemia Molt-3 cells. J Pineal Res. 2013;55(2):195–206.
Zhang S, Qi Y, Zhang H, He W, Zhou Q, Gui S, Wang Y. Melatonin inhibits cell growth and migration, but promotes apoptosis in gastric cancer cell line, SGC7901. Biotechn Histochem. 2013;88(6):281–9.
Wang J, Guo W, Chen W, Yu W, Tian Y, Fu L, Shi D, Tong B, Xiao X, Huang W, et al. Melatonin potentiates the antiproliferative and pro-apoptotic effects of ursolic acid in colon cancer cells by modulating multiple signaling pathways. J Pineal Res. 2013;54(4):406–16.
Xu C, Wu A, Zhu H, Fang H, Xu L, Ye J, Shen J. Melatonin is involved in the apoptosis and necrosis of pancreatic cancer cell line SW-1990 via modulating of Bcl-2/Bax balance. Biomed Pharmacother. 2013;67(2):133–9.
Li W, Wang Z, Chen Y, Wang K, Lu T, Ying F, Fan M, Li Z, Wu J. Melatonin treatment induces apoptosis through regulating the nuclear factor-kappaB and mitogen-activated protein kinase signaling pathways in human gastric cancer SGC7901 cells. Oncol Lett. 2017;13(4):2737–44.
Gu J, Lu Z, Ji C, Chen Y, Liu Y, Lei Z, Wang L, Zhang HT, Li X. Melatonin inhibits proliferation and invasion via repression of miRNA-155 in glioma cells. Biomed Pharmacother. 2017;93:969–75.
Gelaleti GB, Borin TF, Maschio-Signorini LB, Moschetta MG, Jardim-Perassi BV, Calvinho GB, Facchini MC, Viloria-Petit AM, de Campos Zuccari DAP. Efficacy of melatonin, IL-25 and siIL-17B in tumorigenesis-associated properties of breast cancer cell lines. Life Sci. 2017;183:98–109.
Bejarano I, Espino J, Marchena AM, Barriga C, Paredes SD, Rodríguez AB, Pariente JA. Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukaemia HL-60 cells. Mol Cell Biochem. 2011;353(1–2):167–76.
Um HJ, Park JW, Kwon TK. Melatonin sensitizes Caki renal cancer cells to kahweol-induced apoptosis through CHOP-mediated up-regulation of PUMA. J Pineal Res. 2011;50(4):359–66.
Leja-Szpak A, Jaworek J, Pierzchalski P, Reiter RJ. Melatonin induces pro-apoptotic signaling pathway in human pancreatic carcinoma cells (PANC-1). J Pineal Res. 2010;49(3):248–55.
Casado-Zapico S, Rodriguez-Blanco J, García-Santos G, Martín V, Sánchez-Sánchez AM, Antolín I, Rodriguez C. Synergistic antitumor effect of melatonin with several chemotherapeutic drugs on human Ewing sarcoma cancer cells: potentiation of the extrinsic apoptotic pathway. J Pineal Res. 2010;48(1):72–80.
Martín V, García-Santos G, Rodriguez-Blanco J, Casado-Zapico S, Sanchez-Sanchez A, Antolín I, Medina M, Rodriguez C. Melatonin sensitizes human malignant glioma cells against TRAIL-induced cell death. Cancer Lett. 2010;287(2):216–23.
Jang SS, Kim WD, Park WY. Melatonin exerts differential actions on X-ray radiation-induced apoptosis in normal mice splenocytes and Jurkat leukemia cells. J Pineal Res. 2009;47(2):147–55.
Raza H, John A, Brown EM, Benedict S, Kambal A. Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol Appl Pharmacol. 2008;226(2):161–8.
García-Santos G, Antolín I, Herrera F, Martín V, Rodriguez-Blanco J, Carrera MdP, Rodriguez C. Melatonin induces apoptosis in human neuroblastoma cancer cells. J Pineal Res. 2006;41(2):130–5.
Majsterek I, Gloc E, Blasiak J, Reiter RJ. A comparison of the action of amifostine and melatonin on DNA-damaging effects and apoptosis induced by idarubicin in normal and cancer cells. J Pineal Res. 2005;38(4):254–63.
Wenzel U, Nickel A, Daniel H. Melatonin potentiates flavone-induced apoptosis in human colon cancer cells by increasing the level of glycolytic end products. Int J Cancer. 2005;116(2):236–42.
Cos S, Mediavilla MD, Fernández R, González-Lamuño D, Sánchez-Barceló EJ. Does melatonin induce apoptosis in MCF-7 human breast cancer cells in vitro? J Pineal Res. 2002;32(2):90–6.
Lu Y-X, Chen D-L, Wang D-S, Chen L-Z, Mo H-Y, Sheng H, Bai L, Wu Q-N, Yu H-E, Xie D. Melatonin enhances sensitivity to fluorouracil in oesophageal squamous cell carcinoma through inhibition of Erk and Akt pathway. Cell Death Dis. 2016;7(10):e2432–e2432.
Lin PH, Tung YT, Chen HY, Chiang YF, Hong HC, Huang KC, Hsu SP, Huang TC, Hsia SM. Melatonin activates cell death programs for the suppression of uterine leiomyoma cell proliferation. J Pineal Res. 2020;68(1):e12620.
Lee WJ, Chen LC, Lin JH, Cheng TC, Kuo CC, Wu CH, Chang HW, Tu SH, Ho YS. Melatonin promotes neuroblastoma cell differentiation by activating hyaluronan synthase 3-induced mitophagy. Cancer Med. 2019;8(10):4821–35.
Nopparat C, Sinjanakhom P, Govitrapong P. Melatonin reverses H(2) O(2) -induced senescence in SH-SY5Y cells by enhancing autophagy via sirtuin 1 deacetylation of the RelA/p65 subunit of NF-κB. Journal of pineal research 2017;63(1).
Martín V, Sanchez-Sanchez AM, Puente-Moncada N, Gomez-Lobo M, Alvarez-Vega MA, Antolín I, Rodriguez C. Involvement of autophagy in melatonin-induced cytotoxicity in glioma-initiating cells. J Pineal Res. 2014;57(3):308–16.
Liu C, Jia Z, Zhang X, Hou J, Wang L, Hao S, Ruan X, Yu Z, Zheng Y. Involvement of melatonin in autophagy-mediated mouse hepatoma H22 cell survival. Int Immunopharmacol. 2012;12(2):394–401.
Sagrillo-Fagundes L, Bienvenue-Pariseault J, Vaillancourt C. Melatonin: The smart molecule that differentially modulates autophagy in tumor and normal placental cells. PLoS ONE. 2019;14(1):e0202458.
Galley HF, McCormick B, Wilson KL, Lowes DA, Colvin L, Torsney C. Melatonin limits paclitaxel-induced mitochondrial dysfunction in vitro and protects against paclitaxel-induced neuropathic pain in the rat. J Pineal Res. 2017;63(4):e12444.
Waseem M, Sahu U, Salman M, Choudhury A, Kar S, Tabassum H, Parvez S. Melatonin pre-treatment mitigates SHSY-5Y cells against oxaliplatin induced mitochondrial stress and apoptotic cell death. PLoS ONE. 2017;12(7):e0180953.
Koşar PA, Nazıroğlu M, Övey İS, Çiğ B. Synergic effects of doxorubicin and melatonin on apoptosis and mitochondrial oxidative stress in MCF-7 breast cancer cells: involvement of TRPV1 channels. J Membr Biol. 2016;249(1–2):129–40.
Plaimee P, Khamphio M, Weerapreeyakul N, Barusrux S, Johns NP. Immunomodulatory effect of melatonin in SK-LU-1 human lung adenocarcinoma cells co-cultured with peripheral blood mononuclear cells. Cell Prolif. 2014;47(5):406–15.
Ruiz-Rabelo J, Vázquez R, Arjona A, Perea D, Montilla P, Túnez I, Muntané J, Padillo J. Improvement of capecitabine antitumoral activity by melatonin in pancreatic cancer. Pancreas. 2011;40(3):410–4.
Lin ZY, Chuang WL. Pharmacologic concentrations of melatonin have diverse influence on differential expressions of angiogenic chemokine genes in different hepatocellular carcinoma cell lines. Biomed Pharmacother. 2010;64(10):659–62.
Padillo FJ, Ruiz-Rabelo JF, Cruz A, Perea MD, Tasset I, Montilla P, Túnez I, Muntané J. Melatonin and celecoxib improve the outcomes in hamsters with experimental pancreatic cancer. J Pineal Res. 2010;49(3):264–70.
Moselhy SS, Al mslmani MA. Chemopreventive effect of lycopene alone or with melatonin against the genesis of oxidative stress and mammary tumors induced by 7,12 dimethyl(a)benzanthracene in sprague dawely female rats. Mol Cell Biochem. 2008;319(1–2):175–80.
Ruiz-Rabelo JF, Vázquez R, Perea MD, Cruz A, González R, Romero A, Muñoz-Villanueva MC, Túnez I, Montilla P, Muntané J, et al. Beneficial properties of melatonin in an experimental model of pancreatic cancer. J Pineal Res. 2007;43(3):270–5.
Zhu H, Chen Y, Bai LC, Cao XR, Xu R. Different effects of melatonin on X-rays-irradiated cancer cells in a dose-dependent manner. Dose-Response. 2019;17(3):1559325819877271.
Chen YT, Yang CC, Shao PL, Huang CR, Yip HK. Melatonin-mediated downregulation of ZNF746 suppresses bladder tumorigenesis mainly through inhibiting the AKT-MMP-9 signaling pathway. J Pineal Res. 2019;66(1):e12536.
Gurunathan S, Jeyaraj M, Kang MH, Kim JH. Melatonin Enhances Palladium-Nanoparticle-Induced Cytotoxicity and Apoptosis in Human Lung Epithelial Adenocarcinoma Cells A549 and H1229. Antioxidants (Basel, Switzerland). 2020;9(4):357.
Li M, Wu C, Muhammad JS, Yan D, Tsuneyama K, Hatta H, Cui ZG, Inadera H. Melatonin sensitises shikonin-induced cancer cell death mediated by oxidative stress via inhibition of the SIRT3/SOD2-AKT pathway. Redox Biol. 2020;36:101632.
Zakki SA, Muhammad JS, Li JL, Sun L, Li ML, Feng QW, Li YL, Cui ZG, Inadera H. Melatonin triggers the anticancer potential of phenylarsine oxide via induction of apoptosis through ROS generation and JNK activation. Metallomics Integr Biometal Sci. 2020;12(3):396–407.
Acknowledgements
Not applicable.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conception and design: AH and SM. Performing the literature search: MHP, AM and FM. Drafting the manuscript: all authors. Approving the final version: all authors. AH is responsible for the integrity of the work as a whole. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mehrzadi, S., Pourhanifeh, M.H., Mirzaei, A. et al. An updated review of mechanistic potentials of melatonin against cancer: pivotal roles in angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress and oxidative stress. Cancer Cell Int 21, 188 (2021). https://doi.org/10.1186/s12935-021-01892-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-01892-1