
Abstract
Background
We used bioinformatic tools to dichotomize 157 non-M3 AML patients from the TCGA dataset based on the presence or absence of TP53 mutations, and screened out a key gene related to TP53 mutation for future analysis.
Methods
DEGs were analyzed by R package “DESeq2” and then run GSEA, GO enrichment, KEGG pathway and PPI network. Hub genes were selected out according to MCC. Log-rank (Mantel–Cox) test was used for survival analysis. Mann–Whitney U’s nonparametric t test and Fisher’s exact test was used for continuous and categorical variables respectively. p value< 0.05 was considered to be statistical significance.
Results
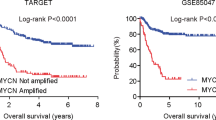
TNFRSF4 was final screened out as a key gene. Besides TP53 mutation (p = 0.0118), high TNFRSF4 was also associated with FLT3 mutation (p = 0.0102) and NPM1 mutation (p = 0.0024). Elevated TNFRSF4 was significantly related with intermediate (p = 0.0004) and poor (p = 0.0011) risk stratification as well as relapse statute (p = 0.0099). Patients with elevated TNFRSF4 expression had significantly shorter overall survival (median survival: 2.35 months vs. 21 months, p < 0.0001). Based on our clinical center data, TNFRSF4 expression was significantly higher in non-M3 AML patients than HDs (p = 0.0377) and MDS patients (EB-1, 2; p = 0.0017).
Conclusions
Elevated TNFRSF4 expression was associated with TP53, FLT3 and NPM1 mutation as well as poor clinical outcome. TNFRSF4 expression was significantly higher in non-M3 AML patients than HDs and MDS (EB-1, 2) patients. TNFRSF4 is need for future functional and mechanistic studies to investigate the role in non-M3 AML.
Similar content being viewed by others

Background
Acute myeloid leukemia (AML) is a heterogenous and hematologic malignant disease which is characterized by infiltration of the bone marrow, blood, and other tissues by proliferative, clonal, abnormally differentiated, and occasionally poorly differentiated cells of the hematopoietic system [1]. Abnormal accumulated blasts replace the normal hematopoietic tissue and trigger out cytopenia [2]. With the advance of microarray and next-generation sequencing, recognition of the molecular heterogeneity of AML has enormously increased [3, 4]. However, current understanding of the molecular mechanisms underlying the development and progression of AML is limited, and early diagnosis remains difficult, which may lead to treatment delays. Therefore, the identification of key mechanisms regulating AML management and patient survival may contribute to the development of AML specific targeted therapies.
Since 1989, TP53 has been identified as a tumor suppressor gene [5], which encodes tumor suppressor p53 protein regarded as “guardian of the genome” that plays an important role in maintaining genome stability under cellular stress, and participating in various processes of development, differentiation, aging, and disease [6, 7]. TP53 mutations account for ~ 10% of de novo AML patients [8], 20–37% of secondary AML, therapy-relate AML patients [9] and 60% of complex karyotype patients. TP53 mutations are also increasingly common appearance in relapsed or refractory AML cases which predicts poor clinical outcome [10, 11].
Tumor necrosis factor receptor superfamily member 4 (TNFRSF4), as known as OX40 or CD134 is expressed primarily on activated T cells [12]. TNFRSF4 can activate the NF-kappa-B pathway by mediating TRAF2 and TRAF5 [13]. The PI3K/PKB and NFAT pathway also have been identified as the downstream of TNFRSF4 [12, 14]. The most remarkable function of TNFRSF4 is to enhance division, proliferation, survival and cytokine production of T cells by activating the pathways described above. Series researches have investigated that TNFRSF4 as a therapeutic agent plays a significant role in immunotherapy of preclinical tumor models [15,16,17].
It has been found that TP53 mutations promote the immunogenicity of breast cancer, and elevated TNFRSF4 expression is also associated with TP53 mutations [18]. On the other hand, TNFRSF4 expression in CD8-positive (CD8+) T cells and Tregs is significantly increased in relapsed AML patients compared with healthy donors (HDs) [19]. We analyzed the differentially expressed genes (DEGs) function or pathways between TP53-mutated and TP53-wildtype non-M3 AML based on the Cancer Genome Atlas (TCGA) transcriptome data [3]. TNFRSF4 was finally screened out as a key gene associated with poor clinical outcome. In addition, based on our clinical center data, we validated TNFRSF4 expression level of non-M3 AML patients was significantly higher than that of HDs and Myelodysplastic syndrome (MDS) Excess blasts (EB)-1,2 patients. Furthermore, the expression level was positive related with the percentage of bone marrow blasts after combined with MDS patient data.
Methods
Patient datasets
Complete clinical data and RNA sequence data of 157 newly diagnosis adult non-M3 AML patients obtained from TCGA dataset [3] downloaded from cBioPortal (https://www.cbioportal.org) including normalized Z-score data and median expression data [20]. Z-score indicates the number of standard deviations away from the mean of expression in the reference population. The subtypes which were classified according to the French–American–British (FAB) classification systems in which M3 subtype was removed from present research considering the unique attributes [21]. The risk group stratification was according to National Comprehensive Cancer Network (NCCN) guidelines. Patients included in the study were assessed for the most frequently found somatic mutations in AML, such as FLT3, NPM1, IDH1/2, and TET.
Approval of the code of ethics and consent to participation are not necessary because all data is public to identify and all datasets analyzed in this study were available from cBioportal.
DEGs analysis
Patients were dichotomized based on the presence and absence of TP53 mutation. DEGs were filtrated by R package “DESeq2” [22]. The screening condition was to satisfy both log2FoldChange(log2FC) > 1 or < − 1, and adjusted p value < 0.05. All genes were visualized using volcanic map plotted by R package “ggplot2” [23].
KEGG, GO and GSEA analysis
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis and Gene Ontology (GO) enrichment analysis were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID https://david.ncifcrf.gov) online tool [24]. False discovery rate (FDR) < 0.05 was considered to indicate a statistically significant difference.
All genes were performed Gene set enrichment analysis (GSEA) with a cut off nominal p value< 0.05 and FDR < 0.10. The reference gene set from the Molecular Signatures Database (MSigDB) of c6, the oncogenic signatures which were generated directly from microarray data from National Center for Biotechnology Information Gene Expression Omnibus or from internal unpublished profiling experiments involving perturbation of known cancer genes [25].
Protein–protein interaction (PPI) network analysis
Proteins and their functional interactions networks of selected enrichment genes were acquired from the STRING database (https://string-db.org) [26]. Genes with minimum interaction score more than 0.4 were selected to visualize in Cytoscape which is an open source software project to integrate biomolecular interaction networks with high throughput expression data and other molecular states into a unified conceptual framework [27]. We utilized CytoHubba plug-ins for ranking nodes in a network by their network features with the Maximal Clique Centrality (MCC) methods. Wayne diagram was produced by webtool Bioinformatics & Evolutionary Genomics (http://bioinformatics.psb.ugent.be/webtools/Venn/) to overlap genes.
Clinical patients and reverse transcribed quantitative PCR (RT-qPCR)
To analyze the mRNA expression of TNFRSF4 in human bone marrow cells (BMCs), We collected non-M3 de novo AML patients, MDS (EB-1, 2) and HDs from our clinical center. The written informed consents were provided by all the patients in accordance with the Declaration of Helsinki before enrollment in the study.
The total RNA was isolated by Trizol (Invitrogen, USA), and was reverse transcribed into cDNA using the PrimeScript™ RT Master Mix (Perfect Real Time) (TaKaRa, Dalian, China). RT-qPCR was performed using TaKaRa SYBR Supermix (TaKaRa, Dalian, China) on a StepOne Plus analysis system (Applied Biosystems, Foster City, CA, USA). The amplification conditions were as follows: pre-denaturation (95 °C for 30 s), 40 cycles of denaturation (95 °C for 30 s), and annealing and extension (60 °C for 34 s). The primers were designed and synthesized with the following sequences:
sense, 5′-ACAACGACGTGGTCAGCTCCAA-3′,
antisense, 5′-CAGCGGCAGACTGTGTCCTGT-3′(TNFRSF4);
sense, 5′-GTAACCCGTTGAACCCCATT-3′,
antisense, 5′-CCATCCAATCGGTAGTAGCG-3′ (18 s RNA).
The relative expression levels of the target genes were calculated by the comparative Ct method presented as 2−ΔCt. The experiments were conducted in triplicate.
Statistical analysis
All data were analyzed with the IBM SPSS statistics 26 and GraphPad Prism 8 software. Log-rank (Mantel–Cox) test was used to compare Overall survival (OS) and disease free survival (DFS) between patients with Z ≥ 0 (high) and Z < 0 (low) TNFRSF4 expression. Additionally, Kaplan–Meier survival curves were generated for patients with Z ≥ 0 and Z < 0 TNFRSF4 expression after stratification by FAB classification, risk stratification, age, cytogenetic status, transplant status, TP53, FLT3, NPM1 and RUNX1 mutation status. Mann–Whitney U’s nonparametric t test and Fisher’s exact test was used for continuous and categorical variables respectively. Spearman rank correlation was used to analyze the correlation between TNFRSF4 and bone marrow blasts. p value< 0.05 was considered to be statistical significance, and all statistical methods were list in Additional file 1: Table S1.
Results
DEGs analysis
The present study was conducted as a multiple strategy to select hub genes correlated with TP53 mutation from TCGA non-M3 AML patient dataset for further analysis (Fig. 1a). Patients were divided into TP53 mutation and TP53 wild type groups. 1449 DEGs were screened out by R package “DESeq2” and displayed in a volcanic map (Fig. 1b).

a The schematic view of the procedure of present study. b Volcanic maps of all genes. Red spot, the expression is up-regulated; Blue spot, the expression is down-regulated; Gray spot, no significantly dysregulated. c, d The bubble chart showed the top 10 pathways with significant difference. c The KEGG enrichment analysis. d The GO biological process enrichment analysis. e IL15 signaling pathway screened out from GSEA with the reference gene sets from the MSigDB of the oncogenic signatures
KEGG and GO enrichment analysis
DEGs were conducted the KEGG as well as GO biological process enrichment analysis and the cytokine–cytokine receptor interaction pathway (p = 8.26E−10, FDR = 1.08E−06) and the immune response pathway (p = 2.53E−13, FDR = 4.68E−10) were screened out respectively (Fig. 1c, d).
GSEA analysis
GSEA with the advantage of analyzing the genes obtained in the TCGA dataset instead of the DEGs. The IL15 signaling pathway was final screened out (NES = 1.81, p < 0.0001, FDR = 0.08, Fig. 1e). We next utilized the Wayne diagram and found 5 same genes, TNFRSF4, TNFRSF9, CCL4, LIF and IL18RAP by overlapping DEGs from cytokine–cytokine receptor interaction pathway, immune response pathway and the IL15 signaling pathway (Additional file 2: Figure S1A).
PPI network
In order to find the hub genes, we performed the PPI analysis of the DEGs from 3 most significant pathways aforementioned utilized the “String” website tool (Fig. 2a–c). Then we imported PPI networks into Cytoscape plug-ins to rank nodes and found out the candidate genes (Fig. 2d–f). We listed the 15 candidate genes of each pathways (Additional file 1: Table S2) by using CytoHubba plug-ins from Cytoscape software, and then screened out TNFRSF4 by overlapping candidate genes from each pathway (Fig. 2g).

a–c The PPI analysis at STRING (https://string-db.org). d–f Cytoscape analysis candidate genes after PPI network analysis. a, d genes from IL15 signaling pathway; b, e genes from cytokine–cytokine receptor interaction pathway; c, f genes from immune response pathway. g Venn diagram showed the overlapping gene of candidate genes. h The TNFRSF4 mRNA relative expression was calculated by the comparative Ct method presented as 2−ΔCt. *p < 0.05, **p < 0.01. i Spearman correlation coefficient analysis between bone marrow blasts percentage and TNFRSF4 expression
TNFRSF4 expression in bone marrow simples
We checked the TCGA normal and The Genotype-Tissue Expression AML datasets at GEPIA2 (http://gepia2.cancer-pku.cn) and found the TNFRSF4 expression in AML is significantly higher than HDs (Additional file 2: Figure S1B). Subsequently, BM samples of 39 non-M3 AML, 29 MDS (EB-1, 2) patients and 18 HDs were collected from our clinical center, Southeast University affiliate Zhongda Hospital, from 1 February 2016 to 1 August 2019. RT-qPCR was performed to detect the TNFRSF4 mRNA expression. The expression of TNFRSF4 mRNA in AML patients was significantly higher compared with HDs (p = 0.0377) and MDS (EB-1, 2; p = 0.0017; Fig. 2h) patients respectively. There no statistically significant difference of TNFRSF4 mRNA expression between HDs and MDS (EB-1, 2; p = 0.1243) patients.
Additionally, combined with MDS (EB-1, 2) patients blasts results, we found TNFRSF4 expression level was positively related with bone marrow blasts percentage (Spearman’s Rho = 0.372, p = 0.002; Fig. 2i).
TNFRSF4 expression and clinical characters
TNFRSF4 expression data and clinical data of 157 non-M3 patients were download from TCGA dataset. Histograms representing the distribution of TNFRSF4 mRNA log2-transformed data and TNFRSF4 scores are provided in Additional file 2: Figure S1C, D. The scatterplot of TNFRSF4 log2-transformed mRNA expression against TNFRSF4 Z score is shown in Additional file 2: Figure S1E (Pearson’s r = 0.8, p < 0.001). TNFRSF4 mRNA levels were compared among patients classified according to the FAB and there was significant difference of TNFRSF4 expression among the subgroups (Additional file 2: Figure S1F; p = 0.0055). We also analyzed TNFRSF4 expression according to the NCCN AML classification based on their molecular and cytogenetic risk status into good, intermediate, and poor risk stratifications. TNFRSF4 expression of good stratification patients was significantly lower than intermediate (p = 0.0004) and poor (p = 0.0011, Additional file 2: Figure S1G) stratification patients. Additionally, we used the Vizome data analysis tool [28], which contains data from the BEAT AML cohort, and examined the level of TNFRSF4 expression of relapsed (N = 22) was significantly higher than de novo non-M3 AML (N = 214) samples (p = 0.0099, Additional file 2: Figure S1H).
We dichotomized the patients in the TCGA data set based on their TNFRSF4 mRNA expression Z score (RNA Seq V2 RSEM) into high (Z score ≥ 0, N = 20) and low (Z score < 0, N = 137). Patients with high TNFRSF4 expression had significantly higher age (median 66.5 vs. 57, p = 0.012) and were widely distributed in poor group (20% vs. 0%, p = 0.044) compared with the good group (Table 1).
TNFRSF4 and mutations
TNFRSF4 was screened out from non-M3 AML patients based on absence or presence of TP53 mutation. We verified the expression of TNFRSF4 was significantly higher in patients with TP53 mutation (N = 15) than TP53 wide type (N = 142, p = 0.0118, Fig. 3a). To understand other potential molecular genetic aberrations that may lead to or be associated with high TNFRSF4, we analyzed the expression with respect to the other mutational status of patients. TNFRSF4 was significantly higher in patients with NPM1 mutation (N = 49) than in patients with NPM1 wild type (N = 108, p = 0.0024, Fig. 3b). TNFRSF4 was also significantly higher in patients with FLT3 mutation (ITD and point mutations) (N = 45) than in patients carrying FLT3 wild type (N = 112, p = 0.0102, Fig. 3c). Additionally, TNFRSF4 was significantly lower in the patients with RUNX1 mutation (N = 28) than in patients with the wild-type RUNX1 (N = 129, p = 0.0311, Fig. 3d). No significant association was observed between TNFRSF4 expression and mutations in DNMT3A, IDH1, IDH2, TET2, CEBPA, WT1, and NRAS.

Association of TNFRSF4 expression with patient mutational status. Relative TNFRSF4 log2 mRNA expression in a patients with TP53 mutations versus wild type; b patients with NPM1 mutations versus wild type; c patients with FLT3 mutations versus wild type; and d patients with RUNX1 mutations versus wild type. *p < 0.05. **p < 0.01
When we dichotomized patients according to TNFRSF4 Z scores, we found a higher frequency of TP53 mutations in TNFRSF4 (Z ≥ 0) patients than in low TNFRSF4 (Z < 0) patients (25% vs. 7.3%, Fisher exact, p = 0.026). No other association was found between TNFRSF4 upregulation and mutations in DNMT3A, IDH1, IDH2, TET2, NPM1, CEBPA, WT1, and NRAS (Table 2).
Additionally, we examined the association between TNFRSF4 expression and clinical outcome in patients with TP53, NPM1, FLT3 and RUNX1 mutations. We stratified patients according to mutational status and performed survival analysis in each group. We found that in patients with wild type TP53, NPM1, FLT3 and RUNX1, high TNFRSF4 expression (Z ≥ 0) was associated with a significantly shorter OS (median survival: TP53, 7 months vs. 24.1 months, p = 0.024; NPM1, 3 months vs. 19.2 months, p < 0.0001; FLT3, 1.9 months vs. 24.1 months, p < 0.0001; RUNX1, 7 months vs. 21.5 months, p = 0.0305 Fig. 4a–d). There was a similar trend but not statistically significant association was observed with high TNFRSF4 expression and shorter DFS (median survival: TP53, 15 months vs. 16.1 months, p = 0.0507; NPM1, 12 months vs. 17 months, p = 0.0661; FLT3, 12 months vs. 17.3 months, p = 0.6943; RUNX1, 12 months vs. 14.2 months, p = 0.0650, Additional file 3: Figure S2A–D). We also found that only in patients with RUNX1 mutation, high TNFRSF4 expression was associated with a significantly shorter OS (median survival: 1 months vs. 17.4 months, p < 0.0001, Fig. 4e).

Survival analysis of patients with respect to TNFRSF4 expression after stratification based on TP53, NPM1, FLT3 and RUNX1 mutation status. Overall survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) among patients with aTP53, bNPM1, cFLT3 and dRUNX1 wild-type gene. e Overall survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) among patients with RUNX1 mutated gene
TNFRSF4 expression and clinical outcome
The OS of TNFRSF4 high group (Z ≥ 0) was significantly shorter than that of low expression patients (median survival: 2.35 months vs. 21 months, p < 0.0001, Fig. 5a), A similar trend was observed in DFS in patients with high TNFRSF4 expression (median survival: 12 months vs. 14.6 months, p = 0.0868; Additional file 3: Figure S2E). To further validate the association between high TNFRSF4 and poor clinical outcome, we stratified patients into Z ≥ 0.5 and Z < 0.5 for survival analysis. Patients with high TNFRSF4 (Z ≥ 0.5) expression had significantly shorter OS than patients with low TNFRSF4 (median survival: 0.8 months vs. 19 months, p < 0.0001; Additional file 3: Figure S2F). We also analyzed the TCGA data set using TNFRSF4 median expression to dichotomize patients into high and low expression groups. We found that patients with high TNFRSF4 expression had shorter OS (median survival: months 11.5 vs. 22 months, p = 0.0235; Additional file 3: Figure S2G).

Survival analysis of patients with respect to TNFRSF4 expression. a Overall survival of 157 non-M3 AML patients with TNFRSF4 Z score ≥ 0 and TNFRSF4 Z score < 0. Survival analysis of patients with respect to TNFRSF4 expression based on patient risk stratification. b Overall survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) in patients with intermediate risk stratification. c Overall survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) in patients with poor risk stratification. Survival analysis of patients with respect to TNFRSF4 expression after stratification based on patient transplant status. d Overall survival and e disease-free survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) in patients who received a transplant. Survival analysis of patients with respect to TNFRSF4 expression based on age. f Overall survival of patients < 60 years of age with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0)
Non-M3 patients were also grouped according to FAB classification system. Among M0, M1, M2 and M4 subtypes, patients with high TNFRSF4 (Z ≥ 0) had significantly shorter OS (median survival: M0, 2.4 months vs. 26 months, p < 0.0001; M1, 4 months vs. 27 months, p = 0.0163; M2, 0.8 months vs. 19 months, p = 0.004; M4, 4.85 months vs. 19 months, p = 0.0101; Additional file 4: Figure S3A–D). Furthermore, there was a significant decrease in the DFS of TNFRSF4 high patients (Z ≥ 0) compared with TNFRSF4 low patients (median survival: months 5.15 vs. 13 months, p = 0.004; Additional file 4: Figure S3E) in M1 subgroup but not in the rest subgroups.
When patients were stratified according to their risk stratification, we found that among intermediated and poor risk stratification patients, those TNFRSF4 high (Z ≥ 0) had shorter OS than those with TNFRSF4 low (median survival: intermediated, months 4.9 vs. 22 months, p = 0.0136; Poor, 1.9 vs. 11 months, p < 0.0001; Fig. 5b, c). A similar but no statistically significant trend of DFS was observed in poor risk stratification (median survival: 9 months vs. 17 months, p = 0.3376; Additional file 5: Figure S4A). However, no significant difference was observed in DFS between TNFRSF4 high and low group in intermediated risk stratification.
When patients were stratified according to whether they received a transplant or not, we found that only in patients who received a transplant with TNFRSF4 high expression (Z ≥ 0) associated with significantly shorter OS and DFS (median OS: 7 months vs. 32.3 months, p < 0.0001; median DFS: 9 months vs. 14.2 months, p = 0.0302; Fig. 5d, e). No significant difference but similar trend was observed in OS and DFS between the TNFRSF4 high and low in patients who did not receive a transplant (median OS: 3 months vs. 9.55 months, p = 0.1503; median DFS: 15 months vs. 17 months, p = 0.4539; Additional file 5: Figure S4B, C).
We performed survival analysis in patients with non-M3 AML stratified by their age into younger patients (< 60) and older patients (≥ 60). In patients < 60, high TNFRSF4 (Z ≥ 0) expression was associated with shorter OS compared with low TNFRSF4 (median survival: 6 months vs. 45.8 months, p < 0.0001; Fig. 5f) and a decrease in DFS though not significant (median survival: 6.5 months vs. 16.1 months, p = 0.2047, Additional file 5: Figure S4D). A similar trend but not statistically significant association of OS and DFS was observed in older patients (median OS: 6 months vs. 10.5 months; p = 0.6007; median DFS: 12 months vs. 13.4 months; p = 0.1207; Additional file 5: Figure S4E, F).
Discussion
In present study, we analyzed DEGs between TP53-mutated and wildtype non-M3 AML patients based on TCGA dataset. IL15 signaling pathway as well as cytokine–cytokine receptor interaction pathway and immune response pathway were screened out after enrichment analysis. We also utilized the “String” website to complete the PPI networks of DEGs from each pathway and imported PPI networks into Cytoscape plug-ins to found out the candidate genes. Subsequently, TNFRSF4 was screened out by overlapping candidate genes and was used for further study.
Here we reported that elevated TNFRSF4 mRNA expression is significantly associated with poorer OS of non-M3 AML patients. This is particularly relevant with patients < 60 years of age, patients received transplant and patients in intermediate and poor risk stratification. The significant shorter OS of high TNFRSF4 mRNA expression patients is also relevant with TP53, NPM1, FLT3 and RUNX1 wild type. Additionally, combined with our clinical BM simples, TNFRSF4 mRNA expression was higher in AML patients than HDs and MDS (EB-1, 2) patients and the expression is positively related with BM blasts percentage. Therefore, the identification of outcome predictors and possible viable targets in aforementioned subset of patients will greatly affect disease understanding and treatment outcome.
Cancer immunotherapy is emerging as a promising approach for cancer treatment and immune checkpoint inhibitors have advanced rapidly over the past decade. Anti-cytotoxic T-lymphocyte antigen 4 (CTLA4) and anti-programmed death 1 (PD1)/Programmed death-ligand 1 (PD-L1) monoclonal antibodies have produced long-lasting anti-tumor immune responses that translate into clinical benefits for many cancer types [29]. Previous research has found that p53 can transactivate a number of immunosuppressive genes including programmed death–ligand 1 (PD-L1), thus activating one of the major immunological checkpoints. p53 also transactivates the expression of forkhead box P3 (FOXP3), a transcription factor that is essential for the generation and function of regulatory T cells, a subpopulation that maintains tolerance to self-antigens [30]. Breast cancers with TP53 mutation also show significantly higher activities of a wide variety of immune cells, functions, and pathways than TP53 wildtype group [18]. In AML, TP53 mutation is associated with complex karyotype, poor standard therapy response and short overall survival [10, 11]. However, the role of TP53 in AML is enigmatic. In our research, TNFRSF4 was screened out by dividing patients based on absence or presence of TP53 mutation, thereby we speculated that the mechanism between TNFRSF4 and TP53 maybe associated with IL15 related immune response or cytokine–cytokine receptor interaction. We found that beside TP53, NPM1 and FLT3 mutation also associated with high TNFRSF4 expression. The mechanistic interplay between TNFRSF4 and the mutation genes is yet to be determined.
TNFRSF4/TNFSF4 signaling serves a key role in the development, differentiation and physiological functions of T cells and other immunological cells [31]. In various tumor models, anti-TNFRSF4 has been shown to enhance CD8+ T cells infiltration and reduce Treg cells infiltration into the tumor [17, 32, 33]. Another research also found a dependence on direct TNFRSF4 ligation on CD8+ T cells to increase tumor specific cytotoxicity in vivo [16]. Research has found that AML patients CD8+ T cell dysfunction was in part reversible upon PD-1 blockade or TNFRSF4 co-stimulation in vitro [34]. Besides T cells, NK cells are a second cytotoxic lymphocyte subset that contributes to antitumor immunity, particularly in leukemia [35]. Research reported that TNFRSF4 is expressed on AML blasts, depending on TNFRSF4/TNFSF4 signaling promoted NK-cell activation, cytokine production and cytotoxicity which can against primary AML cells [36]. In combination with our results, we suggested that immunotherapy may product more therapeutic effect in patients with high TNFRSF4 expression.
Conclusion
TNFRSF4 was screened out as a key gene related with TP53 mutation based on non-M3 AML TCGA data set. TNFRSF4 was higher in intermediate, poor risk stratification and related with relapse status. Additionally, high TNFRSF4 expression was also associated with NPM1, FLT3 mutation. Based our clinical data, we found TNFRSF4 expression was significant higher in non-M3 AML patients than HDs and MDS (EB-1, 2) patients. The expression level was positively related with blasts percentage. Our findings demonstrate that elevated TNFRSF4 expression contributes to predict the poor clinical outcome of patients with non-M3 AML. This study provides a rationale for further functional and mechanistic studies aiming to understand the role of TNFRSF4 in non-M3 AML.
Availability of data and materials
The 200 AML patient datasets obtained from the Cancer Genome Atlas (TCGA) at cBioPortal (https://www.cbioportal.org). The clinical patient datasets for the current study are not publicly accessible in accordance with local health research ethics protocols; however, it may be available from the corresponding author.
Abbreviations
- AML:
-
Acute myeloid leukemia
- TNFRSF4:
-
Tumor necrosis factor receptor superfamily member 4
- CD8+:
-
CD8-positive
- HDs:
-
Healthy donors
- MDS:
-
Myelodysplastic syndrome
- EB:
-
Excess blasts
- DEGs:
-
Differentially expressed genes
- TCGA:
-
The Cancer Genome Atlas
- FAB:
-
French–American–British
- NCCN:
-
National Comprehensive Cancer Network
- log2FC:
-
log2FoldChange
- KEGG:
-
The Kyoto Encyclopedia of Genes and Genome
- GO:
-
Gene Ontology
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- FDR:
-
False discovery rate
- GSEA:
-
Gene set enrichment analysis
- MSigDB:
-
Molecular Signatures Database
- MCC:
-
Maximal Clique Centrality
- RT-qPCR:
-
Reverse transcribed quantitative PCR
- BMCs:
-
Bone marrow cells
- OS:
-
Overall survival
- DFS:
-
Disease free survival
- PPI:
-
Protein–protein interaction
- CTLA4:
-
Anti-cytotoxic T-lymphocyte antigen 4
- PD1:
-
Anti-programmed death 1
- PD-L1:
-
Programmed death-ligand 1
- FOXP3:
-
Forkhead box P3
References
Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty JD, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Lane DP. Cancer p53, guardian of the genome. Nature. 1992;358(6381):15–6.
Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med. 2016;6(5):a026070.
Oren M, Rotter V. Introduction: p53–the first twenty years. Cell Mol Life Sci. 1999;55(1):9–11.
Kadia TM, Jain P, Ravandi F, Garcia-Manero G, Andreef M, Takahashi K, Borthakur G, Jabbour E, Konopleva M, Daver NG, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016;122(22):3484–91.
Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Peng J, Tang G, Goswami M, Young KH, Singh R, Medeiros LJ, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015;8:45.
Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer. 2005;43(3):227–38.
Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–21.
Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. 2009;229(1):173–91.
Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem. 1998;273(10):5808–14.
Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68.
Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
Gough MJ, Crittenden MR, Sarff M, Pang P, Seung SK, Vetto JT, Hu HM, Redmond WL, Holland J, Weinberg AD. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33(8):798–809.
Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50–66.
Liu Z, Jiang Z, Gao Y, Wang L, Chen C, Wang X. TP53 mutations promote immunogenic activity in breast cancer. J Oncol. 2019;2019:5952836.
Williams P, Basu S, Garcia-Manero G, Hourigan CS, Oetjen KA, Cortes JE, Ravandi F, Jabbour EJ, Al-Hamal Z, Konopleva M, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer. 2019;125(9):1470–81.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Pashaiefar H, Yaghmaie M, Tavakkoly-Bazzaz J, Ghaffari SH, Alimoghaddam K, Momeny M, Izadi P, Izadifard M, Kasaeian A, Ghavamzadeh A. PARP-1 overexpression as an independent prognostic factor in adult non-M3 acute myeloid leukemia. Genet Test Mol Biomarkers. 2018;22(6):343–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Ito K, Murphy D. Application of ggplot2 to pharmacometric graphics. CPT Pharmacomet Syst Pharmacol. 2013;2:e79.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–50.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR, Traer E, Abel M, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526–31.
Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342(6165):1432–3.
Zitvogel L, Kroemer G. A p53-regulated immune checkpoint relevant to cancer. Science. 2015;349(6247):476–7.
Kow NY, Mak A. Costimulatory pathways: physiology and potential therapeutic manipulation in systemic lupus erythematosus. Clin Dev Immunol. 2013;2013:245928.
Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68(13):5206–15.
Pardee AD, McCurry D, Alber S, Hu P, Epstein AL, Storkus WJ. A therapeutic OX40 agonist dynamically alters dendritic, endothelial, and T cell subsets within the established tumor microenvironment. Cancer Res. 2010;70(22):9041–52.
Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R, Mukhopadhyay R, Vanura K, Blazar BR, Karp JE, et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight. 2018;3(21):e120974.
Pierson BA, Miller JS. CD56 + bright and CD56 + dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood. 1996;88(6):2279–87.
Nuebling T, Schumacher CE, Hofmann M, Hagelstein I, Schmiedel BJ, Maurer S, Federmann B, Rothfelder K, Roerden M, Dorfel D, et al. The immune checkpoint modulator OX40 and its ligand OX40L in NK-cell immunosurveillance and acute myeloid leukemia. Cancer Immunol Res. 2018;6(2):209–21.
Acknowledgements
We would like show sincere appreciation to the colleagues for their assistance on this study.
Funding
This work was supported in part by The National Natural Science Foundation of China (81770172); Jiangsu Provincial Special Program of Medical Science (BE2017747); Jiangsu Province “333” project (BRA2019103); The Fundamental Research Funds for the Central Universities (2242019K3DZ02); Milstein Medical Asian American Partnership (MMAAP) Foundation Research Project Award in Hematology (2017); Key Medical of Jiangsu Province (ZDXKB2016020).
Author information
Authors and Affiliations
Contributions
SG designed and analyzed data; JZ and QH analyzed data; ZG and CS designed and supervised data analysis; SG and ZG wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The written informed consents were provided by all the patients in accordance with the Declaration of Helsinki before enrollment in the study. The Institutional Review Board of the Nanjing Medical University and Zhongda Hospital Southeast University, Nanjing, China, approved the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
The statistical methods used in present research. Table S2. The top 15 genes with the highest score of each pathway through the Cytoscape “cytoHubba” module analysis.
Additional file 2: Figure S1.
(A) Wayne diagram with 5 overlapping DEGs from cytokine–cytokine receptor interaction pathway, immune response pathway and the IL15 signaling pathway. (B) The box plot from GEPIA2 matched TCGA normal and GTEx AML data with log2(TPM + 1) for log-scale. *p < 0.05. TNFRSF4 Z-score and mRNA expression distribution. (C) Histogram of TNFRSF4 Log2 transformed mRNA expression; (D) TNFRSF4 mRNA expression Z score; (E) Scatterplot of mRNA Z-score vs mRNA log2 mRNA expression. Relative TNFRSF4 log2 mRNA expression categorized by (F) FAB classification and (G) NCCN risk stratification. (H) TNFRSF4 expression in relapsed vs. de novo non-M3 AML samples from the BEAT AML. **p < 0.01, ***p < 0.001.
Additional file 3: Figure S2.
Survival analysis of patients with respect to TNFRSF4 expression after stratification based on TP53, NPM1, FLT3 and RUNX1 mutation status. Disease-free survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) among patients with (A)TP53, (B) NPM1, (C) FLT3 and (D) RUNX1 wild-type gene. Survival analysis of patients with respect to TNFRSF4 expression. (E) Disease-free survival of patients with TNFRSF4 Z score ≥ 0 and TNFRSF4 Z score < 0. (F) Overall survival of patients with TNFRSF4 Z score ≥ 0.5 and TNFRSF4 Z score < 0.5. (G) Overall survival of patients that dichotomized based on TNFRSF4 median mRNA expression into TNFRSF4 high and TNFRSF4 low according to the log2 median-centered expression.
Additional file 4: Figure S3.
Survival analysis of patients with respect to TNFRSF4 expression after stratification based on FAB classification. Overall survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) among patients with (A) M0, (B) M1, (C) M2 and (D) M4 classification. (E) Disease-free survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) among patients with M1 classification.
Additional file 5: Figure S4.
Survival analysis of patients with respect to TNFRSF4 expression based on patient risk stratification. (A) Disease-free survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) in patients with poor risk stratification. Survival analysis of patients with respect to TNFRSF4 expression after stratification based on patient transplant status. (B) Overall survival and (C) disease-free survival of patients with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0) in patients who did not received a transplant. Survival analysis of patients with respect to TNFRSF4 expression based on age. (D) Disease-free survival of patients < 60 years of age with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0). (E) Overall survival and (F) disease-free survival of patients ≥ 60 years of age with TNFRSF4 high (Z score ≥ 0) versus TNFRSF4 low (Z score < 0).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Gu, S., Zi, J., Han, Q. et al. Elevated TNFRSF4 gene expression is a predictor of poor prognosis in non-M3 acute myeloid leukemia. Cancer Cell Int 20, 146 (2020). https://doi.org/10.1186/s12935-020-01213-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01213-y