
Abstract
Background
Evidence is increasingly indicating that circular RNAs (circRNAs) are closely involved in tumorigenesis and cancer progression. However, the function of circRNAs in gastric cancer (GC) are still unknown. Here, we aimed to determine the regulatory mechanism of circRNAs in GC.
Methods
Expression profiles of circRNAs were downloaded from four Gene Expression Omnibus (GEO) microarray datasets. Expression profiles of miRNAs and mRNAs were collected from The Cancer Genome Atlas (TCGA) database. We used the robust rank aggregation method to identify differentially expressed circRNAs (DEcircRNAs) and a ceRNA network was constructed based on circRNA–miRNA pairs and miRNA–mRNA pairs. Functional and pathway enrichment analyses were performed and interactions between proteins were predicted using Cytoscape. Aa subnetwork regulatory module was built using the MCODE plugin.
Results
A total of eight DEcircRNAs, 240 DEmiRNAs, and 4578 DEmRNAs were identified. The circRNA–miRNA–mRNA network was constructed based on seven circRNAs, 33 miRNAs, 69 mRNAs in GC. GO and KEGG pathway analysis indicated DEmRNAs might be associated with GC onset and progression. A PPI network was established and four hub genes (MCM4, KIF23, MCM8, and NCAPD2) were determined from the network. Then a circRNA–miRNA-hub gene subnetwork was constructed based on the four DEcircRNAs, three DEmiRNAs, and four DEmRNAs.
Conclusions
Our findings provide a deeper understanding the circRNA-related competing endogenous RNA regulatory mechanism in GC pathogenesis.
Similar content being viewed by others

Background
Gastric cancer (GC) is one of the most common malignancies and the second leading cause of cancer-related deaths worldwide [1, 2]. Despite advances in surgical techniques and combined chemotherapy strategies, the 5-year overall survival (OS) of GC remains poor [3]. The lack of improvement in OS is largely due to a low early diagnostic rate and a high frequency of recurrence and metastasis [4, 5]. Therefore, elucidation of the molecular mechanisms underlying GC is imperative for the development of effective diagnostic and therapeutic targets.
Circular RNA (circRNA), an emerging class of non-coding RNA, has a covalently closed loop structure in which the 3′ and 5′ ends are linked in a non-collinear way by a process termed “back-splicing” [6, 7]. The lack of 5′ caps and 3′ tails makes circRNAs resistant to exonucleases and more stable than linear RNA [8]. CircRNA is structurally stable in certain tissue, time and disease specificity [9, 10]. Therefore, circRNAs have become new hotspots.
An increasing number of studies have demonstrated that circRNAs play important regulatory roles in the development of cancers. CircRNA can decrease the cytoplasmic levels of target microRNAs (miRNAs) by absorbing miRNAs and, thus, liberate mRNA transcripts that are targeted by the miRNAs. For example, Gao et al. found that has_circ_101280 was upregulated in hepatocellular carcinoma (HCC) tissues and cell lines. In their study, overexpression of hsa_circ_101280 inhibited the expression of tumor suppressor miRNA miR-375 and increased expression of the miR-124 target gene, JAK2, which leads to cancer cell proliferation. Further experiments showed that knockdown of hsa_circ_101280 inhibited growth of HCC xenografts in nude mice, which also showed downregulation of JAK2. This study demonstrated conclusively that hsa_circ_101280 interacts with JAK2 by sponging miR-1261 in HCC [11]. Similarly, circZFR was shown to interact with C8orf4 through the sponging of miR-1261 in papillary thyroid carcinoma [12].
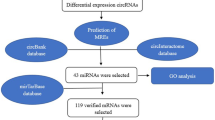
In the current study, we collected the expression profiles of circRNAs, miRNAs, and mRNAs in GC tissues and adjacent normal gastric tissues from Gene Expression Omnibus (GEO) datasets and the TCGA database. Differentially expressed circRNAs (DECs) were identified by the RobustRankAggreg package in R. After predicting sponging of miRNAs by circRNA and miRNA target genes, we constructed a circRNA–miRNA–mRNA network. To evaluate the main functional pathways of GC, DEmRNAs of the competitive endogenous RNA (ceRNA) network were assessed by gene ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. We then established a protein–protein interaction (PPI) network and extracted hub genes from the PPI network. To better understand the pathogenesis of GC, a circRNA–miRNA-hub gene subnetwork regulation module was also constructed. The flowchart for this procedure is shown in Fig. 1.

Flow chart of the approach utilized in the present study
Materials and methods
Microarray data and RNA sequencing data
The microarray data used in this study were retrieved from the GEO database. Four circRNA expression profiles were obtained from GSE78092, GSE83521, GSE93541, and GSE100170. The RNA-sequencing (RNA-seq) data were downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/). The miRNA sequencing data included 410 gastric cancer tissues and 42 adjacent normal gastric tissues, the mRNA sequencing data included 343 GC tissues and 30 adjacent normal tissues. No ethical approval nor informed consent was required in this study due to the public availability of data in the GEO and TCGA databases.
Differentially expression analysis
Raw microarray data were normalized and log2-transformed. The Bioconductor Limma package was used to identify differentially expressed circRNA (DEcircRNA) in each dataset. We then integrated and ranked all of the DEGs using the robust rank aggregation method [13]. Additionally, the edgeR package was used to screen differentially expressed miRNA (DEmiRNA) and mRNA (DEmRNA) with thresholds of |log 2 (fold change [FC])| > 1 and adjusted P‐value < 0.05.
Prediction of miRNA binding sites
The Circular RNA Interactome (CircInteractome) (https://circinteractome.nia.nih.gov/) and Cancer-Specific CircRNA (CSCD) (https://gb.whu.edu.cn/CSCD/) were used to predict miRNA binding sites (MREs). Overlapping miRNAs in the two databases were considered as potential target miRNAs of the DEcircRNAs. These target miRNAs were further screened by DEmiRNA based on The Cancer Genome Atlas (TCGA).
Prediction of miRNA target genes
Interactions between miRNA and mRNA were predicted based on the TargetScan, miRTarBase, and miRDB databases [14,15,16]. Only mRNAs recognized by all three databases were considered as candidate mRNAs and intersections with the DEmRNAs were identified to screen out the DEmRNAs targeted by the DEmiRNAs.
Construction of the ceRNA network
The circRNA–miRNA–mRNA regulatory network was constructed using a combination of circRNA–miRNA pairs and miRNA–mRNA pairs. Finally, the regulatory network was visualized using Cytoscape 3.6.1.
Gene ontology and pathway enrichment analysis
To assess the main functional pathways of GC, DEmRNAs in the ceRNA network were assessed by GO annotation and KEGG pathway analyses with the clusterProfiler package [17] in R. A P-value of less than 0.05 was set as the cut-off criterion.
Construction PPI network and module analysis
Based on the DEmRNAs identified, the Search Tool for the Tetrieval of Interacting Genes (STRING) database was used to construct a protein–protein interaction (PPI) network. Visualization was performed using Cytoscape 3.6.1. The Molecular Complex Detection (MCODE) app was used to screen modules of hub genes from the PPI network [18].
Results
Identification of DEGs in GC
Expression of circRNAs in GC and control tissues was evaluated in four microarray datasets (GSE78092, GSE83521, GSE93541, and GSE100170), the basic information of which are listed in Table 1. A total of 112 DEcircRNAs, 23 upregulated and 89 downregulated, were identified in the GSE78092 dataset. A total of 73 DEcircRNAs, 43 upregulated and 30 downregulated, were identified in the GSE83521 dataset. A total of 306 DEcircRNAs, 146 upregulated genes and 160 downregulated, were identified in the GSE93541 dataset. A total of 1414 DEcircRNAs, 537 upregulated and 877 downregulated, were identified in the GSE100170 dataset. The DEcircRNAs from each of the four datasets were ranked and a total of eight DEcircRNAs, three upregulated and five downregulated, were present in the top rank (P < 0.05) (Fig. 2). The basic characteristics of the eight circRNAs are listed in Table 2. Their basic structural patterns are in shown Fig. 3. A total of 240 DEmiRNAs, 180 upregulated and 60 downregulated, and 4578 DEmRNAs, 2403 upregulated and 2176 downregulated, DEmiRNAs, were identified in the TCGA database (P < 0.05 and logFC > 1).

Heatmap of the eight differentially expressed circRNAs in the four microarray datasets

Structural patterns of the seven circRNAs: a hsa_circ_0076092, b hsa_circ_0002019, c hsa_circ_0005699, d hsa_circ_0005777, e hsa_circ_0007991, f hsa_circ_0067934, g hsa_circ_0006896
Construction of the ceRNA network
To better understand the role of circRNAs and miRNAs in the ceRNA network of GC tissues, we established a circRNA–miRNA–mRNA (ceRNA) network. We retrieved data relating to the eight top-ranked DEcircRNAs identified from the microarray datasets from the CSCD and CircInteractome online databases and identified 349 pairs of interacting circRNAs and miRNAs. After intersecting with the DEmiRNAs, only 35 circRNA–miRNA pairs, including seven circRNAs and 33 DEmiRNAs, remained. We then identified mRNAs targeted by these 33 DEmiRNAs in three databases (miRDB, miRTarBase, and TargetScan). These targeted mRNAs were cross-checked against the DEmRNAs retrieved from TCGA database. These results indicated that 69 DEmRNAs were involved in the ceRNA network. Finally, we constructed a ceRNA network based on seven circRNA nodes, 33 miRNA nodes, and 69 mRNA nodes in GC (Fig. 4).

The ceRNA network of circRNA–miRNA–mRNA in GC. Diamonds indicate circRNAs, rounded rectangles indicate miRNA, and ellipses indicate mRNA. The nodes highlighted in red and blue represent up-regulation and down-regulation, respectively
Functional and pathway enrichment analyses
Gene ontology analysis revealed that the 69 DEmRNAs were enriched in 203 GO terms (P < 0.05). The top 10 enriched GO terms are shown in Fig. 5, and include “extracellular matrix structural constituent”, “platelet-derived growth factor binding”, and “adenylate cyclase binding”. The top 10 pathways associated with DEmRNAs according to KEGG analysis included “AGE-RAGE signaling pathway in diabetic complications,” “Relaxin signaling pathway,” and “PI3K-Akt signaling pathway” (Fig. 6).

Ten most enriched GO, in terms of DEmRNAs, involved in the ceRNA network

Ten most enriched KEGG pathways, in terms of DEmRNAs, involved in the ceRNA network
Construction of PPI network and module analysis
In total, 26 nodes and 33 edges were mapped in the PPI network (Fig. 7a). The MCODE approach in Cytoscape was used to identify hub genes in the PPI network. With the k-core = 2, a significant module containing four nodes and six edges was identified. These highest-scoring nodes were screened as hub genes: MCM4, KIF23, MCM8, and NCAPD2 (Fig. 7b). We constructed a circRNA–miRNA-hub gene subnetwork based on five circRNA–miRNA–mRNA regulatory modules (hsa_circ_0002019/hsa-mir-1301/KIF23, hsa_circ_0005699/hsa-mir-504/MCM8, hsa_circ_0005699/hsa-mir-504/NCAPD2, hsa_circ_0006896/hsa-mir-373/MCM4, and hsa_circ_0007991/hsa-mir-373/ MCM4).

Identification of hub genes from the PPI network with the MCODE algorithm. a PPI network of 69 genes. b PPI network of four hub genes that extracted from the PPI network
Discussion
CircRNAs are an enigmatic type of stable, non-coding RNA that often exhibit tissue or developmental stage specific expression, although the functions of circRNAs in mammalian cells remain mostly unclear [19, 20]. The high stability of circRNAs, imparted by their cyclic structures, makes these molecules potentially valuable as novel tumor biomarkers [21, 22]. Several studies have shown that circRNAs have important influence on many complicated human diseases, including malignant tumors [23, 24]. Recently, studies have unveiled how circRNAs participate in regulation of malignant biological processes [25, 26]. Other evidence has revealed that circRNAs contain multiple MREs and can bind to miRNAs, often termed “miRNA sponges,” decreasing cytoplasmic levels of miRNAs and liberating their respective downstream target mRNAs [27,28,29]. However, the exact role of circRNAs in GC remains largely unknown. To identify whether circRNAs function as ceRNAs in GC, we first performed microarray data analysis to examine DEGs in GC samples and normal samples using a robust rank aggregation method. We constructed a circRNA–miRNA–mRNA regulatory network based on biological predictions and developed a model PPI network of DEmRNAs. We also constructed a circRNA–miRNA-hub gene subnetwork based on regulatory modules identified in the circRNA–miRNA–mRNA network.
Numerous studies have shown that expression of circRNA is dysregulated in GC and that this dysregulation is associated with pathogenesis and prognosis, suggesting that circRNAs could be used as tumor-associated biomarkers [30]. Fang et al. revealed that circFAT1 was downregulated in GC tissues and cell lines and was associated with OS. In vitro, overexpression of circFAT1 reduced cell proliferation, migration and invasion. That same study also found that circFAT1(e2) regulates expression of Y-box binding protein-1 (YBX1) of the nucleus by cytoplasmic sponging of miR-548g [30,31,32]. Similarly, high circ-SFMBT2 was observed in GC tissues and was correlated with higher stages of tumors in GC. Knockdown of circ-SFMBT2 significantly inhibited proliferation of GC cells. The authors concluded that circ-SFMBT2 participates in development and progression of GC through sponge miR-182-5p that targets CREB1 [31]. Liu [32] identified that the circular RNA-ZFR inhibited cell proliferation and promoted GC apoptosis via sponging of miR-130a/miR-107 and regulating PTEN.
In the current study, seven cicRNAs (hsa_circ_0000788, hsa_circ_0002019, hsa_circ_0005699, hsa_circ_0005777, hsa_circ_0006896, hsa_circ_0007991, and hsa_circ_0067934) were identified to be involved in the ceRNA network. One of these, hsa_circ_0067934, was identified previously by Xia et al., who analyzed 51 esophageal squamous cell carcinoma (ESCC) samples and normal samples, finding that hsa_circ_0067934 was significantly up-regulated in ESCC tissues and was associated with poor differentiation, I–II T stage, and I–II TNM stage. Knockdown of hsa_circ_0067934 in vitro by siRNA can inhibit proliferation and migration of ESCC cells and blocks cell cycle progression [33]. No relevant studies have reported involvement of hsa_circ_0000788, hsa_circ_0002019, hsa_circ_0005699, hsa_circ_0005777, hsa_circ_0006896, or hsa_circ_0007991 in cancer.
It is well known that miRNA-mediated pathways are essential to tumorigenesis; miRNAs can regulate cell proliferation, differentiation, apoptosis, and migration [34]. In the current study we identified a total of 69 DEmRNAs and 33 DEmiRNAs involved in the ceRNA network, some of which have been found as a biomarker for diagnosis and prognosis. To further identify the key circRNAs participating in the regulatory network we established a PPI network, screening four hub genes, including MCM4, MCM8, NCAPD2, and KIF23. Previous work has identified two genes, E2F1 and KIF23, that play important roles in the carcinogenesis and development of GC. It is thought that E2F1 has an important role in the cell cycle pathway by regulating MCM3, which may interact with MCM4 [35]. KIF23 is highly expressed in GC tissue, and its expression is associated with advanced TNM stage, recurrence, and poor prognosis. In vitro and in vivo experiments confirmed that inhibition of KIF23 inhibits proliferation of GC cells, leading the authors to conclude that KIF23 might serve as a potential therapeutic target for GC treatment [36]. However, there are no reports linking MCM8 and NCAPD2 with GC, nor of their association with circRNAs. Here, we identified five circRNA–miRNA-hub gene axes, indicating competitive regulatory relationships of four circRNAs with the four genes in GC. However, given that these results are based solely on bioinformatics models, further in-depth studies are critical to verifying the possible role of these four axes in GC.
Conclusions
We screened differentially expressed circRNAs, miRNAs, and mRNAs from publicly available microarray data to construct a circRNA-associated ceRNA network. The circRNA–miRNA-hub genes regulatory subnetwork uncovered four important circRNAs that might be involved in carcinogenesis, providing new insight into the pathogenesis of GC and suggesting potential therapeutic targets that warrant further investigation.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Xi HQ, Zhang KC, Li JY, Cui JX, Gao YH, Wei B, Huang D, Chen L. RNAi-mediated inhibition of Lgr5 leads to decreased angiogenesis in gastric cancer. Oncotarget. 2017;8(19):31581–91.
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–403.
Dong HM, Wang Q, Wang WL, Wang G, Li XK, Li GD, Chen J. A clinical analysis of systemic chemotherapy combined with radiotherapy for advanced gastric cancer. Medicine. 2018;97(23):e10786.
Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015;385(9972):977–1010.
Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H. Circular RNA: a new star of noncoding RNAs. Cancer Lett. 2015;365(2):141–8.
Petkovic S, Muller S. RNA circularization strategies in vivo and in vitro. Nucleic Acids Res. 2015;43(4):2454–65.
Li J, Yang J, Zhou P, Le Y, Zhou C, Wang S, Xu D, Lin HK, Gong Z. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res. 2015;5(2):472–80.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57.
Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–85.
Cao S, Wang G, Wang J, Li C, Zhang L. Hsa_circ_101280 promotes hepatocellular carcinoma by regulating miR-375/JAK2. Immunol Cell Biol. 2018;97:218–28.
Wei H, Pan L, Tao D, Li R. Circular RNA circZFR contributes to papillary thyroid cancer cell proliferation and invasion by sponging miR-1261 and facilitating C8orf4 expression. Biochem Biophys Res Commun. 2018;503(1):56–61.
Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28(4):573–80.
Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–152.
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44(D1):D239–247.
Fromm B, Billipp T, Peck LE, Johansen M, Tarver JE, King BL, Newcomb JM, Sempere LF, Flatmark K, Hovig E, et al. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu Rev Genet. 2015;49:213–42.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Bandettini WP, Kellman P, Mancini C, Booker OJ, Vasu S, Leung SW, Wilson JR, Shanbhag SM, Chen MY, Arai AE. MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: a clinical validation study. J Cardiovasc Magn Reson. 2012;14:83.
Chen BJ, Huang S, Janitz M. Changes in circular RNA expression patterns during human foetal brain development. Genomics. 2018. https://doi.org/10.1016/j.ygeno.2018.04.015.
Shao Y, Li J, Lu R, Li T, Yang Y, Xiao B, Guo J. Global circular RNA expression profile of human gastric cancer and its clinical significance. Cancer Med. 2017;6(6):1173–80.
Meng S, Zhou H, Feng Z, Xu Z, Tang Y, Li P, Wu M. CircRNA: functions and properties of a novel potential biomarker for cancer. Mol Cancer. 2017;16(1):94.
Jiang XM, Li ZL, Li JL, Xu Y, Leng KM, Cui YF, Sun DJ. A novel prognostic biomarker for cholangiocarcinoma: circRNA Cdr1as. Eur Rev Med Pharmacol Sci. 2018;22(2):365–71.
Li S, Sun X, Miao S, Lu T, Wang Y, Liu J, Jiao W. hsa_circ_0000729, a potential prognostic biomarker in lung adenocarcinoma. Thorac Cancer. 2018;9(8):924–30.
Li P, Yang X, Yuan W, Yang C, Zhang X, Han J, Wang J, Deng X, Yang H, Li P, et al. CircRNA-Cdr1as exerts anti-oncogenic functions in bladder cancer by sponging MicroRNA-135a. Cell Physiol Biochem. 2018;46(4):1606–16.
Zhou B, Zheng P, Li Z. CircPCNXL2 sponges miR-153 to promote the proliferation and invasion of renal cancer cells through upregulating ZEB2. J Transl Med. 2018;17(23):2644–54.
Lv C, Sun L, Guo Z, Li H, Kong D, Xu B, Lin L, Liu T, Guo D, Zhou J, et al. Circular RNA regulatory network reveals cell–cell crosstalk in acute myeloid leukemia extramedullary infiltration. J Transl Med. 2018;16(1):361.
Bai N, Peng E, Qiu X, Lyu N, Zhang Z, Tao Y, Li X, Wang Z. circFBLIM1 act as a ceRNA to promote hepatocellular cancer progression by sponging miR-346. J Exp Clin Cancer Res. 2018;37(1):172.
Wang WL, Yang Z, Zhang YJ, Lu P, Ni YK, Sun CF, Liu FY. Competing endogenous RNA analysis reveals the regulatory potency of circRNA_036186 in HNSCC. Int J Oncol. 2018;53(4):1529–43.
He R, Liu P, Xie X, Zhou Y, Liao Q, Xiong W, Li X, Li G, Zeng Z, Tang H. circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J Exp Clin Cancer Res. 2017;36(1):145.
Fang J, Hong H, Xue X, Zhu X, Jiang L, Qin M, Liang H, Gao L. A novel circular RNA, circFAT1(e2), inhibits gastric cancer progression by targeting miR-548g in the cytoplasm and interacting with YBX1 in the nucleus. Cancer Lett. 2018;442:222–32.
Sun H, Xi P, Sun Z, Wang Q, Zhu B, Zhou J, Jin H, Zheng W, Tang W, Cao H, et al. Circ-SFMBT2 promotes the proliferation of gastric cancer cells through sponging miR-182-5p to enhance CREB1 expression. Cancer Manag Res. 2018;10:5725–34.
Liu T, Liu S, Xu Y, Shu R, Wang F, Chen C, Zeng Y, Luo H. Circular RNA-ZFR inhibited cell proliferation and promoted apoptosis in gastric cancer by sponging miR-130a/miR-107 and modulating PTEN. Cancer Res Treat. 2018;50(4):1396–417.
Xia W, Qiu M, Chen R, Wang S, Leng X, Wang J, Xu Y, Hu J, Dong G, Xu PL, et al. Circular RNA has_circ_0067934 is upregulated in esophageal squamous cell carcinoma and promoted proliferation. Sci Rep. 2016;6:35576.
Liu C, Liu R, Zhang D, Deng Q, Liu B, Chao HP, Rycaj K, Takata Y, Lin K, Lu Y, et al. MicroRNA-141 suppresses prostate cancer stem cells and metastasis by targeting a cohort of pro-metastasis genes. Nat Commun. 2017;8:14270.
Jian T, Chen Y. Regulatory mechanisms of transcription factors and target genes on gastric cancer by bioinformatics method. Hepatogastroenterology. 2015;62(138):524–8.
Li XL, Ji YM, Song R, Li XN, Guo LS. KIF23 promotes gastric cancer by stimulating cell proliferation. Dis Markers. 2019;2019:9751923.
Acknowledgements
Not applicable.
Funding
This work was supported by the Suzhou Youth Science and Technology Foundation (KJXW2018032); the Suzhou team introduction project (No. SZYJTD201712).
Author information
Authors and Affiliations
Contributions
YJG and JYM carried out data analysis. YJG and WS participated in study design and data collection. All authors drafted the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethical approval nor informed consent was required in this study due to the public-availability of the data used.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guan, Yj., Ma, Jy. & Song, W. Identification of circRNA–miRNA–mRNA regulatory network in gastric cancer by analysis of microarray data. Cancer Cell Int 19, 183 (2019). https://doi.org/10.1186/s12935-019-0905-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-019-0905-z