
Abstract
Background
To contribute to the discovery of new microbial strains with metabolic and physiological robustness and develop them into successful chasses, Paracoccus pantotrophus DSM 2944, a Gram-negative bacterium from the phylum Alphaproteobacteria and the family Rhodobacteraceae, was chosen. The strain possesses an innate ability to tolerate high salt concentrations. It utilizes diverse substrates, including cheap and renewable feedstocks, such as C1 and C2 compounds. Also, it can consume short-chain alkanes, predominately found in hydrocarbon-rich environments, making it a potential bioremediation agent. The demonstrated metabolic versatility, coupled with the synthesis of the biodegradable polymer polyhydroxyalkanoate, positions this microbial strain as a noteworthy candidate for advancing the principles of a circular bioeconomy.
Results
The study aims to follow the chassis roadmap, as depicted by Calero and Nikel, and de Lorenzo, to transform wild-type P. pantotrophus DSM 2944 into a proficient SynBio (Synthetic Biology) chassis. The initial findings highlight the antibiotic resistance profile of this prospective SynBio chassis. Subsequently, the best origin of replication (ori) was identified as RK2. In contrast, the non-replicative ori R6K was selected for the development of a suicide plasmid necessary for genome integration or gene deletion. Moreover, when assessing the most effective method for gene transfer, it was observed that conjugation had superior efficiency compared to electroporation, while transformation by heat shock was ineffective. Robust host fitness was demonstrated by stable plasmid maintenance, while standardized gene expression using an array of synthetic promoters could be shown. pEMG-based scarless gene deletion was successfully adapted, allowing gene deletion and integration. The successful integration of a gene cassette for terephthalic acid degradation is showcased. The resulting strain can grow on both monomers of polyethylene terephthalate (PET), with an increased growth rate achieved through adaptive laboratory evolution.
Conclusion
The chassis roadmap for the development of P. pantotrophus DSM 2944 into a proficient SynBio chassis was implemented. The presented genetic toolkit allows genome editing and therewith the possibility to exploit Paracoccus for a myriad of applications.
Similar content being viewed by others


Background
The concept of a microbial chassis captures the true sense of a genetically engineered microbe possessing genetically, and metabolically relevant properties to serve functions in the light of applied microbiology. The term chassis started gaining popularity in the early 2000s when microbiologists engineered genetically modified strains for à la carte applications [1]. Although there are several widely used microbial chassis organisms currently in use, e.g., Escherichia coli as the flagship bacterium [2], Bacillus subtilis as a robust workhorse for heterologous protein production [3, 4], Saccharomyces cerevisiae for bioethanol production [5, 6], and many more [7], the search for new and improved microbial hosts is far from being over. It has been long since argued that a handful of microbes, although having immense potential and being amenable to genetic modification, cannot be the solution to every challenge encountered in using microbes in a circular bioeconomy. There is a clear need to establish further chassis organisms with robust physiology (e.g., against temperature and osmotic stresses) and versatile metabolism [8], including the ability to harness cost-effective and sustainable resources while concurrently facilitating the synthesis of bio-based products [9]. With the help of Cultivarium (www.cultivarium.org) and other initiatives, technological improvements to access previously untouched microbes become more accessible.
Based on the above-mentioned arguments, Calero and Nikel described six major laboratory research steps required to develop wild-type bacteria into a chassis [7]. These milestones include (1) genome sequencing with high-quality annotations, (2) development of a robust genetic toolbox (comprised of replicative and suicide plasmids, promoters, and other precise genetic engineering tools), (3) in silico metabolic model backend with experimental validation, (4) in-depth physiological characterization, (5) construction of genome reduced strains for increased biomass or product formation by curbing unnecessary reactions, and finally (6) a mutant strains with improved functions (as shown in Fig. 1) [7].

Roadmap highlighting the development of a SynBio chassis. The figure is derived from the proposals of Calero and Nikel [7] and de Lorenzo [1], with the latter suggesting a defined nomenclature (wild-type, recombinant DNA host, SynBio chassis, and standardized SynBio chassis) aiding in regulatory guidelines. The check marks shown in the figure signify milestones that have already been reached for P. pantotrophus DSM 2944 in previous studies [19, 24]
Furthermore, the concept of constructing a new chassis strain was advanced with the help of a more recent roadmap developed by de Lorenzo et al. [1]. This roadmap elucidated a step-by-step guide for constructing a novel chassis microorganism created with the aid of synthetic biology (SynBio), keeping in mind the industrial and regulatory acceptance guidelines [10]. Thus, de Lorenzo aptly coined the term SynBio chassis. Unlike Calero’s approach, this updated approach although comprising laboratory-based research goals, was further supported with a definite nomenclature signifying the development of the chassis from one stage to the next. The following four stages depict the promotion of an environmental isolate to a fully-fledged standardized SynBio chassis.
-
1)
Wild type or isolate: selection of a non-virulent wild-type strain possessing metabolic or physiological advantage.
-
2)
Recombinant DNA (rDNA) host: conversion of the isolate into a reusable biological platform organism, characterized by the absence of virulence genes, the ability of genetic modification, and the availability of a robust genetic toolbox.
-
3)
SynBio chassis: the rDNA host is additionally characterized by high-quality genome sequencing, factors concerning stress tolerance, and defined energy metabolism. Also, information regarding antibiotic sensitivity and the capability of horizontal gene transfer (HGT) should be available, and finally, the genome should be edited for improved efficiency in targeted approaches [1].
-
4)
Standardized SynBio chassis: finally, the engineered SynBio chassis undergoes comprehensive analysis, and traceability is established through the incorporation of unique gene identifiers. This is subsequently followed by attaining the designation of being generally recognized as safe (GRAS) status [11] and accompanied by a qualified presumption of safety (QPS) certification, which ultimately gets applied using the environmental risk assessment (ERA) norms [10]. These evaluations collectively render the strain eligible for expedited approval in industrial and food-based applications [1]. Additionally, this step is also reinforced in chassis development by Adams [12], with an additional characteristic of having a host-vector biosafety (HVB) certification [13] for a smooth transition of the laboratory-constructed SynBio chassis to varied industrial applications.
In 1983, Paracoccus pantotrophus DSM 2944 (then known as Thiosphaera pantotropha) was isolated from a denitrifying, sulfide-oxidizing effluent treatment plant located in Delft, The Netherlands, possessing the ability to grow aerobically and anaerobically on reduced sulfur compounds and hydrogen while fixing carbon dioxide [14,15,16], and tolerating short-chain alkanes as carbon source, promoting bioremediation [17]. Salient features also include efficient energy management due to the absence of flagella and related energy sinks [18] and the presence of a complete and extremely versatile electron-transport chain. Furthermore, out of over one hundred Paracoccus species, only one reported strain, Paracoccus yeei CCUG 32053, which is phylogenetically distant from our selected chassis strain, P. pantotrophus DSM 2944 [19], was declared to be an opportunistic human pathogen [20, 21]. All these features render P. pantotrophus DSM 2944 a promising candidate for exploration as a production host for industrial applications. Moreover, in-depth physiological characterization revealed the organism’s resistance against high salinity (> 10% NaCl) and good thermotolerance (up to 45°C), thereby promoting auto sterility (these extreme conditions are unsuitable for the growth of contaminating microbes) and economically feasible industrial applications [22]. P. pantotrophus also possesses a versatile metabolic arsenal, including the ability to utilize the C1 compound formate and C2 compound ethylene glycol (EG), organic acids, and alcohols, all coupled with the production of short-chained biopolymer, polyhydroxybutyrate [19, 23].
This study presents the development of P. pantotrophus DSM 2944 into an rDNA host. The genetic toolbox comprises now essential genetic tools (e.g., plasmids, including origins of replication, promoters, information on plasmid stability, antibiotic resistance profiling, and genome editing through gene deletion and integration). Importantly, the strain can be manipulated by exogenous DNA uptake through transformation and conjugation methods. Backed by previous studies reporting whole-genome sequencing and annotation [24] and in-depth studies of physiology and tolerance against abiotic stressors [19], and here presented adaptive laboratory evolution experiments, the newly developed rDNA host was further promoted following the chassis roadmap. The tools available are showcased to engineer P. pantotrophus DSM 2944 to grow on the PET monomers ethylene glycol and terephthalic acid at a high rate. In summary, P. pantotrophus DSM 2944 is developed into a SynBio chassis (Fig. 2), further paving the path to becoming a host for future industrial applications [7].

Completed milestones in this study. While the check marks in black represent the starting point of the development of P. pantotrophus as a chassis strain (see also Fig. 1), the check marks in green signify the advancements that have been made in this scientific quest for promoting P. pantotrophus DSM 2944 from a wild-type organism with interesting properties to a novel SynBio chassis
Results
Designing a suitable plasmid for efficient genetic engineering
To develop P. pantotrophus DSM 2944 into a SynBio chassis, a genetic engineering toolbox was constructed. Special focus was given to the curation of a replicative plasmid (including oris, promoters, selective, and counter-selective markers) and a non-replicative plasmid (which can be utilized for gene integration and deletion). Studies involving plasmid stability and DNA transfer techniques (transformation/conjugation) conclude the toolbox construction.
Antibiotic resistance profiling for suitable marker selection
To determine the antibiotic resistance profile of P. pantotrophus, the cells were grown in the presence of seven different antibiotics with varying concentrations; namely kanamycin (5–100 mg/L), gentamycin (15–200 mg/L), ampicillin (10–100 mg/L), chloramphenicol (2.5–20 mg/L), streptomycin (50–250 mg/L), tetracycline (5–25 mg/L), and spectinomycin (50–250 mg/L). The range of the different antibiotics was selected to cover the standard concentrations as described by the Barrick Lab [25]. The minimum inhibitory concentration (MIC) for all antibiotics specific to P. pantotrophus is determined based on the resulting growth rates (Fig. 3).

Antibiotic screening. The heatmap depicts the growth rate (h−1) of P. pantotrophus DSM 2944 towards seven different antibiotics with varying concentrations. The intensity of the color is directly proportional to higher growth. The experiment was performed in triplicates in 96-well plates with lysogeny broth (LB) and the respective antibiotic concentrations using the Growth Profiler
P. pantotrophus has very high resistance to streptomycin and gentamycin (200 and 250 mg/L respectively), which makes these antibiotics suitable as selection markers, e.g., during conjugation, against the growth of other microbes such as E. coli. On the other hand, at 25 mg/L and 5 mg/L for kanamycin and chloramphenicol, respectively, the MIC is reached. Finally, no growth was obtained at any ampicillin, tetracycline, and spectinomycin concentration, suggesting high sensitivities and rendering these antibiotics suitable reporter genes for gene transfer technologies. Thus, MICs of only two antibiotics (kanamycin and chloramphenicol) were determined. These findings regarding the specific antibiotic resistance profile of P. pantotrophus DSM 2944, aid in the one-step selection of transconjugants with kanamycin (50 mg/L) as selective and streptomycin (50 mg/L) as a counter-selective marker. With suitable antibiotics in hand, a plasmid could be developed.
Finding the best origin of replication
It is critical for a genetic toolbox to identify two types of replication origins. One that can replicate in P. pantotrophus DSM 2944 and another one that is incapable of replication and thus is suitable for application in a suicide vector.
Nine plasmids were selected from the Standard European Vector Architecture (SEVA) platform (http://seva.cnb.csic.es) [26] with similar backbones containing a multiple cloning site and a kanamycin resistance gene. The only difference between the different SEVA plasmids was the varying oris. The nine oris were R6K [27], RK2 [28], pBBR1 [29], pRO1600/ColE1 [26], RFS1010 [26], p15A [30], pSC101 [31], pUC [32], and pBR322/ROP [33].
The P. pantotrophus cells were treated to be electrically competent and a constant volume of 100 µL of these electrocompetent cells was then transformed with a fixed concentration of 100 ng of the above-mentioned SEVA plasmids using electroporation [34]. After two days, single colonies were observed on LB plates containing 50 mg/L kanamycin, which were counted to assess the functionality of the oris. Given the fixed quantities of plasmid, electrocompetent cells, and the antibiotic, the assumption was made that ori compatibility exhibited a direct proportionality to the number of transformed colonies observed.
The results (Table 1) show the low copy number, broad host range ori R2K [35] as the best replicative origin in P. pantotrophus DSM 2944 (possessing the highest number of transformants and was thus deemed as the most compatible ori). Whereas high copy-number plasmid oris pRO1600/ColE1, (in the case of E. coli) [36, 37] and RFS1010 (Pseudomonas) [38] when transformed resulted only in a few colonies, hinting that they possess low compatibility with P. pantotrophus DSM 2944 as the host. This can stem from the fact that higher copy number plasmids generally cause metabolic burden to the host [39]. No transformants were obtained with any of the other origins, thereby acting as potential oris for suicide vectors. Out of which, ori R6K was chosen as the basis for a suicide plasmid for future genome-based editing.
After the selection of a suitable ori, endeavors were undertaken to determine the most favorable method for exogenous DNA uptake (transformation/conjugation), further aiding in the development of P. pantotrophus DSM 2944 from wild-type to an rDNA host.
Conjugation rather than electroporation is the method of choice for plasmid transfer
Several exogenous DNA uptake methods were tested to find the best approach for genetic engineering in P. pantotrophus. The strategies included heat shock [40], electroporation, and conjugation [34].
P. pantotrophus DSM 2944 cells were made chemically and electrically competent for heat shock and electroporation, respectively. The transformation process involved utilizing a fixed quantity of competent cells, which were then transformed with 100 ng plasmid DNA. Five distinct SEVA plasmids were chosen for this experiment, each carrying P. pantotrophus-sensitive antibiotics as the selection markers. These antibiotics included standard concentrations of the following antibiotics; ampicillin (100 mg/L), kanamycin (50 mg/L), chloramphenicol (10 mg/L), spectinomycin (100 mg/L), and tetracycline (20 mg/L). It is noteworthy that all the constructs shared common features, including a multiple cloning site and the ori RK2, with the sole variable being the antibiotic marker.
To delve deeper into potential genetic engineering techniques, we examined conjugation efficiency through direct cell-to-cell contact [41] facilitated by pili. The previous ori and antibiotic screening are pivotal as they determine suitable parameters for genetic engineering using conjugation. Conjugation was carried out in the presence and absence of a helper strain transformed with plasmid pRK600 carrying the F or fertility factor [42] and the plasmids pSEVA121 to 521 donor strains. Conjugation was performed using patch mating [43] (streaking all strains on top of each other). After one day, this mixed culture was plated on selective agar plates containing the respective pSEVA plasmid encoding antibiotic marker, supplemented with 50 mg/L streptomycin as a counterselection marker (only P. pantotrophus DSM 2944 can grow). The utilization of this dual antibiotic strategy demonstrated a straightforward and greatly efficient approach to selectively isolate recombinant P. pantotrophus (able to grow on both streptomycin 50 mg/l as well as pSEVA harbored antibiotic). The recombinants possessed inherent resistance to streptomycin (50 mg/L), alongside acquired resistance to the specific antibiotic present in the pSEVA plasmid used, thereby eliminating the growth of both other strains involved in the conjugation. Following 48 h, the colonies of transconjugants were quantified, serving as an assessment of the efficacy of gene transfer facilitated by conjugation into P. pantotrophus DSM 2944.
After 48 h of incubation at 37 °C on LB plates with respective antibiotics (Table 2), it was observed that no colonies were formed in any of the plates inoculated with chemically transformed bacteria. This suggests that a standard protocol for gram-negative bacteria is not suitable for P. pantotrophus. On the contrary, electroporation yielded successful transformation as P. pantotrophus showed newly gained resistance on both 50 mg/L kanamycin and 100 mg/L spectinomycin. This finding also confirms that these antibiotics can be used as selection markers.
Compared to electroporation and chemical transformation, conjugation was shown to be the best method for obtaining a maximum number of transconjugants. Although P. pantotrophus DSM 2944 was found to be capable of performing conjugation without the helper plasmid, the number of recombinant clones increased 100-fold with the aid of the helper plasmid pRK600 in the case of plasmid pSEVA421.
Since transforming P. pantotrophus DSM 2944 was found to be most effective with conjugation [1], which is a key step in the construction of a novel SynBio chassis, all subsequent DNA modification experiments were conducted using this method.
Post establishment of successful gene transfer techniques, the following investigations were conducted to depict the strain's fitness or stability as a suitable host for maintaining a specific plasmid under non-selective conditions. This marks the completion of P. pantotrophus as a rDNA host and marks the beginning of becoming a successful SynBio chassis.
Novel chassis P. pantotrophus DSM 2944 is capable of plasmid maintenance
Plasmid stability in P. pantotrophus DSM 2944 under non-selective conditions was tested [44]. Low copy number RK2-based vector pSEVA221 was selected to determine host fitness. As a control, E. coli DH5α containing pSEVA221 was used [26]. Both strains were grown in antibiotic-free LB medium in shake flasks and passaged on to the next shake flask using the same conditions daily. An aliquot was plated on LB-agar containing the selective marker (kanamycin 50 mg/L) during each passaging. For every sub-culturing, the number of transferred cells was kept constant, and sub-culturing was performed after the cells ceased to divide.
The comparative analysis between the established chassis E. coli and the emerging chassis P. pantotrophus (Fig. 4) demonstrates similar plasmid maintenance using pSEVA221 in the absence of antibiotic selection (kanamycin 50 mg/L). In the case of E. coli, the cfu/mL decreases at a steady rate from generation 22 (3.9*105 cells) to 44 (3.6*105 cells). On the other hand, P. pantotrophus experiences a more rapid reduction, with a substantial decline of 200 cfu/mL occurring between generations 14 and 22. Nonetheless, when considering the overall decrease of host cells carrying the pSEVA221 plasmid throughout 60 generations, the difference in cfu/mL between E. coli (3.9*105 to 3.4*105) and P. pantotrophus (3.1*105 to 2.7*105) is merely 100 cfu/mL. This inference shows that P. pantotrophus DSM 2944 can maintain plasmids [45, 46], and thus can be employed for varied plasmid-based applications. After ensuring the establishment of plasmid stability, an examination of synthetic promoters on gene expression was conducted.

Test of plasmid maintenance. P. pantotrophus DSM 2944 and E. coli DH5α were tested for stable plasmid maintenance without selection pressure
Synthetic promoters for tailored gene expression
The mini-Tn7-based promoter transposon system originally constructed for P. putida KT2440, consisting of the synthetic constitutive promoters BG14x (x = b, c, d, e, f, and g) and BG13 [47, 48], was used. Although the Tn7-based system possesses an advantage over plasmid-based systems, the former can prove inadequate due to the significant variation in translation efficiency contingent upon the non-coding 5′ region sequence of the gene of interest (GOI) [49, 50]. This challenge can be addressed by implementing translational couplers in a bicistronic design as described in [51, 52]. The constructs containing a gene coding for superfolder green fluorescent protein (msfGFP) [53] under the control of the different promoters with kanamycin as the selection marker were maintained as vectors in E. coli PIR2 (Table 3) with RK6 origin that cannot replicate in P. pantotrophus. Hence the target promoter-containing construct is chromosomally integrated into the attTn7 site (Fig. 5).

Characterization of seven synthetic promoters in P. pantotrophus DSM 2944 in comparison to P. putida KT2440. Triplicates of every strain were cultured in the BioLector (Bechmann Coulter GmbH, Aachen, Germany) in MSM medium with 20 mM glucose in a 96-well plate. Promoter activity was calculated from the slope of GFP fluorescence to optical density during the exponential phase. Error bars indicate the deviation from the mean of the three replicates
The resulting P. pantotrophus DSM 2944 strains, each regulated by distinct promoters controlling msfGFP expression, exhibited a consistent pattern in terms of promoter strength. Comparative analysis with the P. putida KT2440 control, specifically in the context of the BG14x system, revealed a hierarchy of expression levels among the promoters. Notably, the pBG14b promoter demonstrated the weakest expression, while the pBG14g promoter displayed the strongest, resulting in an overall approximately three-fold reduction in strength (46 vs. 15, respectively). Interestingly, promoter BG13, which exhibited the lowest activity in P. putida KT2440, had a nearly twofold increase in promoter activity when it was introduced into P. pantotrophus DSM 2944. The above promoter expression profile demonstrates the application of cross-species regulatory elements (promoters) as tools for genetic engineering. Thereby showcasing the applicability of transcription tuning with the help of synthetic promoters originally designed for P. putida KT2440 and bicistronic design as a promising strategy to overcome expression rate challenges, adjust enzyme concentrations, modulate metabolic fluxes [54], and improve production of recombinant proteins [55]. Thus, through the incorporation of regulatory elements within P. pantotrophus DSM 2944, the strain’s transition from an rDNA host to a SynBio chassis was achieved.
Gene deletion protocol that facilitates the elimination of PHB production
The technique of precise gene deletion using the pEMG platform, developed by Martinez-et al. for P. putida KT2440 was adapted to P. pantotrophus DSM 2994 [56]. Here, the PHB operon was selected as proof-of-concept gene deletion. The pEMG vector was constructed with the I-SceI sites flanking 1,000 bp each upstream and downstream of the PHB cassette consisting of the four genes phaR, phaP, phaC, and phaZ [24].
Strain P. pantotrophus DSM 2944 ∆PHB produced no PHB, while the WT consisted of 25% PHB (Fig. 6), thus confirming successful gene deletion (with a success rate of 98%). This concluded the successful application of the pEMG-based deletion technique in P. pantotrophus DSM 2944 and asked for the introduction of heterologous genes into the genome.

Polyhydroxybutyrate (PHB) cassette deletion. Both P. pantotrophus DSM 2944 WT and the gene deletion strain ∆PHB were grown in MSM medium supplemented with 40 mM of glucose and nitrogen limitation. Cultivation conditions included growth in shake flasks at 37 °C with 200 rpm. Harvested and extracted biomass was used for PHB content [% CDW] quantification after 16 h. The experiment was done in triplicates and error bars indicate deviation from the mean
Gene integration enables the metabolization of PET monomers
To advance the genetic toolbox of the SynBio chassis, a conscious effort was made to align these endeavors with sustainability objectives. P. pantotrophus DSM 2944, known for its broad substrate utilization, naturally possesses the ability to utilize the PET monomer ethylene glycol as the sole carbon source [19]. Here, terephthalic acid (TA) catabolism was engineered to complement the native ability.
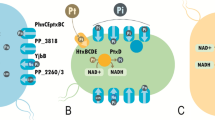
Fortunately, the strain could already grow on protocatechuic acid (PCA) or 3,4-dihydroxybenzoic acid, an intermediate molecule for terephthalic acid degradation (Fig. 7) [57]. Henceforth, the introduction of the tph cassette (rDNA) comprising genes encoding the proteins PcaR, TphA2, A3, A1, B, and PcaK (encoding transporter for TA) was performed using the Tn7-based plasmid pBG_pcaRtphA2A3BA1pcaK [58].

Schematic representation of terephthalic acid metabolism. The proteins marked in green (TphA1A2A3 and TphB) are heterologous and encoded on the plasmid while the strain’s native pathway is marked in blue. Moreover, heterologous proteins, PcaR encoding regulator function, and PcaK encoding cytoplasmic TA transporter are not marked in the above picture
The resulting recombinant P. pantotrophus DSM 2944 tph was able to grow on both monomers of PET (EG and TA) (Fig. 8), thus displaying the metabolic versatility of P. pantotrophus DSM 2944 and its applicability as a chassis microorganism for biotechnology. These experiments conclude the successful genome engineering (deletion and integration) as part of the genetic toolbox and represent establishing P. pantotrophus DSM 2944 as a novel chassis organism. To further streamline and improve the substate uptake of PET monomers, the newly developed SynBio chassis was subjected to adaptive laboratory evolution, thereby showcasing ALE as another tool for metabolic engineering (Fig. 8).
Adaptive laboratory evolution to enhance growth and utilization of PET monomers
ALE was performed with the recombinant strain P. pantotrophus DSM 2944 tph. Two types of ALE strategies were carried out, comprising static (adaptation using one substrate) and dynamic (adaptation using alternating substrates) ALE [59]. Ultimately, the strain with the highest growth rate was tested for substrate consumption (Fig. 8).

Adaptive laboratory evolution results on PET monomers. Comparison of the wild type versus non-evolved and evolved strains of P. pantotrophus DSM 2944 tph on A being 40 mM ethylene glycol (EG) and B being 20 mM of terephthalic acid (TA) as the sole carbon source. The Growth Profiler was used for online biomass measurements. The experiment was conducted in triplicates, and the error bars represent the deviation from the mean
P. pantotrophus DSM 2944 showed enhanced growth on the PET monomers after 120 generations of ALE. The isolated mutants grow faster than the wild-type or non-evolved counterparts. The growth rate observed when using EG as the exclusive carbon source reveals intriguing outcomes, particularly showcasing an ascending trend in growth rates: starting from the wild-type P. pantotrophus DSM 2944 (0.07 h−1), advancing to the evolved variant P. pantotrophus DSM 2944 EG static tph (cultured solely in EG) (0.15 h−1), and culminating in the highest growth rate with P. pantotrophus DSM 2944 dynamic tph (evolved under alternating EG and TA conditions) (0.21 h−1). The incorporation of the heterologous tph cassette appeared to negatively influence the growth rate on EG, as indicated by the slower growth rate of P. pantotrophus DSM 2944 tph (0.04 h−1) when contrasted with the wild-type strain. In the context of utilizing TA as the sole carbon source, a comparable pattern in growth rate emerges. The non-adapted P. pantotrophus DSM 2944 tph strain exhibits the lowest growth rate (0.04 h−1), succeeded by TA static tph (cultivated exclusively in TA) (0.13 h−1), while the dynamic tph strain demonstrates the highest growth rate (0.17 h−1). Despite 20 mM TA having four times the carbon per mole compared to EG, the observed lowered growth rates in TA compared to EG strongly indicate that the strains did not fully utilize the entire substrate during growth. These results motivated us to perform a more comprehensive investigation to determine the assimilation of PET monomers by the adapted P. pantotrophus DSM 2944 dynamic tph, featuring enhanced growth rates on both monomers (Fig. 8). The strain was cultivated utilizing equimolar concentrations (20 mM) of EG and TA, as carbon sources (Fig. 9).

PET monomer utilization. The line graph depicts the growth (OD600) of P. pantotrophus DSM 2944 dynamic tph on both ethylene glycol (EG) and terephthalic acid (TA). The cultivation was performed for 34 h in MSM medium supplemented with equimolar concentrations of EG and TA (20 mM) in polypropylene square 24-deep well microplates at 37 °C with 2 mL of medium (20% v/v) for optimal oxygen transfer rate [60]
It was evident that EG was fully consumed within 30 h. In contrast, the concentration of TA decreased by approximately 4 mM, with the remaining portion of the substrate persisting in the medium. This can arise from the fact that the engineered strain prefers to consume EG over TA, being the substrate, it can degrade naturally.
In conclusion, the outcomes highlight the application of the genetically modified P. pantotrophus DSM 2944 dynamic tph strain in utilizing both PET monomers, as carbon sources and highlighting a successful application of the genetic toolbox for the development of the novel SynBio chassis.
Discussion
This study is centered on transforming P. pantotrophus DSM 2944, an environmental isolate from waste effluent treatment, into a metabolically and physiologically resilient SynBio chassis. The following discussion highlights existing studies that revolve around the genus Paracoccus and is used to compare the findings from this study to provide a comprehensive overview.
As previously mentioned, genetic robustness in a chassis necessitates freedom from reliance solely on strain-specific plasmids. An instance of such strain specificity is evident in plasmid pWKS1 for P. pantotrophus DSM 11073 [61]. Characterized as a cryptic plasmid, conveying no overt phenotype to the host cell and harboring genes with unidentified attributes, it constrains the applicability of cross-species gene transfer techniques. Whereas the novel chassis, belonging to the above species, possesses the capability to use plasmids having different oris, thus removing the dependency of strain-specific plasmids and making genetic manipulations on P. pantotrophus DSM 2944 simpler.
Taking an alternative viewpoint, the utilization of frequently employed plasmids like pUC19, known for its capacity to generate a high copy number, demonstrated effective expression of cloned genes in Paracoccus sp. SY [62]. But on the contrary, the novel chassis P. pantotrophus DSM 2944 was unable to replicate pUC19. It was found that low copy number ori RK2 (broad-range plasmid affiliated with the incP incompatibility group) [63] to be the most compatible ori. This is in agreement with previous results from P. denitrificans [64], thus showcasing the similarity and dissimilarities in plasmid preferences between members of the genus Paracoccus. Preference for low copy number oris can be explained by the metabolic burden where the cell prioritizes high amounts of protein production over essential metabolic reactions crucial to the host cell [65, 69].
In the light of efficient gene transfer techniques, where it was shown that strain P. denitrificans is capable of undergoing electroporation [66], this study provides further information through testing of several high- and low-copy number plasmids and their related transformation efficiencies. Interestingly, although no tra genes (coding for conjugational transfer) were reported in P. pantotrophus DSM 2944, colony formation was observed when spectinomycin at a concentration of 100 mg/L, was used as a selection marker for the selection of transconjugants. This phenomenon can be explained with the help of a previous study that portrays gene aadA (GeneID: 1,252,782) [67] encoding protein aminoglycoside (3'') (9) adenylyltransferase. This enzyme was found to be responsible for providing resistance to aminoglycoside antibiotics including streptomycin, and spectinomycin was found in P. pantotrophus DSM 2944, with an 80% (BLAST) nucleotide similarity. This can justify the gained resistance to spectinomycin when the strains were subjected to the antibiotic, thereby explaining the observation of positive P. pantotrophus clones in the selection plate containing 100 mg/L spectinomycin. A previous study highlighted that both Rhodobacter sphaeroides and P. denitrificans, belonging to the same family (Rhodobacteraceae), were capable of replicating the same plasmids [68]. Based on these findings, this study expands the concept further by showcasing the application of several different plasmids (both SEVA and Tn7-based) in different classes of microorganisms: The Alphaproteobacterium P. pantotrophus DSM 2944 and Gammaproteobacteria (E. coli, P. putida). This showcases the broad applicability of the selected plasmids and the genetic robustness of the chosen chassis.
The gene deletion system pEMG was originally developed for P. putida KT2440 and successfully applied in related species, such as Pseudomonas protegens [69], Pseudomonas aeruginosa [70], P. umsogensis, P. taiwanensis, and others. It was here used successfully to delete the genes coding for PHB-producing enzymes. This pivotal accomplishment holds notable importance, marking the first application of the pEMG system within the Paracoccus genus and opening doors for further genome reduction in Paracoccus.
Existing studies involving the application of a Tn5-based transposon system [71], as a genetic tool for gene insertion were already established in P. denitrificans PD1222 [72]. One of the key factors using transposon-based molecular applications is the uniform distribution of transposon insertion [73]. Tn5 transposon is found to randomly integrate into the target genome [74]. To expand the metabolic versatility of P. pantotrophus DSM 2944 the introduction of the terephthalic acid catabolic pathway by the encoding operon was performed using the Tn7 system. Although the insertion of this operon resulted in the slight consumption of TA, P. pantotrophus DSM 2944 tph was further expedited for growth on TA using adaptive laboratory evolution. Thus, to make the novel chassis P. pantotrophus DSM 2944 reach high growth rates such as P. putida TDM461 (0.72 h−1 on 10 mM TA as carbon source) [75], further studies need to be performed. One hint could be the potential issue with TA permeability across the cell membrane. To address this limitation and enhance TA consumption, the solution could be the incorporation of the tphC gene. This gene encodes the solute-binding protein TphC, which is responsible for binding TA and facilitating its transport via the transmembrane transport proteins TpiA and TpiC [76].
Conclusion
Here, P. pantotrophus DSM 2944 was promoted from a wild-type strain with interesting metabolic properties to a SynBio chassis by following the chassis construction guidelines laid out by Calero and Nickel [7] and de Lorenzo [1].
Key findings included the identification of broad-range ori RK2 as a convenient tool for plasmid-based gene expression. Notably, the strain exhibited promising plasmid stability without antibiotics. Transformation methods were explored, highlighting the efficiency of electric pulse-based transformation and the ineffectiveness of calcium chloride-based methods. Conjugation emerged as the most effective method, further improved by a helper plasmid (pRK600). The toolbox extended to targeted scar-less gene deletion (P. pantotrophus DSM 2944 ΔPHB) [56] and Tn7-based GFP-tagged promoter systems for gene expression [51]. Native resistance was investigated employing a dual antibiotic-based combination, comprising kanamycin and streptomycin, to facilitate uncomplicated selection during conjugational gene transfer. Beyond the double-antibiotic selection approach, alternative selection parameters can be explored by leveraging the extensive metabolic repertoire of P. pantotrophus DSM 2944. This repertoire encompasses diverse carbon sources such as formate, acetate, and ethylene glycol, along with nitrogen sources encompassing various amino acids. Notably, l-histidine emerges as a prominent nitrogen source, exhibiting a growth rate of 0.73 h−1.
Furthermore, the introduction of the TA operon led to the creation of strain P. pantotrophus DSM 2944 tph and this strain demonstrated growth on both EG (complete consumption) and TA (low consumption) as carbon sources, highlighting PET consumption. Lastly aided with an adaptive evolution strategy, enhanced growth rates on both PET monomers were shown to be possible, signifying the capability of this new chassis strain toward genetic modifications and improved functionality. The culmination of this work not only expands P. pantotrophus DSM 2944 as a new SynBio but beckons further research to propel it into a Standardized SynBio chassis [1], in the future. The contributions shown here may motivate researchers to choose the metabolically interesting genus Paracoccus for their endeavors.
Materials and methods
Bacterial strains and growth media
The chemicals utilized in this study were acquired from Carl Roth (Karlsruhe, Germany), Sigma-Aldrich (St. Louis, MO, USA), or Merck (Darmstadt, Germany) unless specified otherwise. The wild-type Paracoccus pantotrophus DSM 2944 [24] was procured from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Except for experiments utilizing specific carbon sources, both the wild-type P. pantotrophus and genetically modified strains were cultivated in Lysogeny broth (LB) consisting of 10 g tryptone, 5 g yeast extract, and 5 g of NaCl per liter of deionized water, serving as a complex medium, incubated at 37°C. Agar at a concentration of 2% was added to media requiring solidification. Furthermore, both the wild-type as well as genetically modified strains of P. pantotrophus were also cultivated in delft mineral salt medium (MSM) containing 3.88 g of K2HPO4, 1.63 g of NaH2PO4, 2.00 g of (NH4)2SO4, 0.1 g of MgCl2 × 6 H2O, 10 mg of EDTA, 2 mg of ZnSO4 × 7 H2O, 1 mg of CaCl2 × 2 H2O, 5 mg of FeSO4 × 7 H2O, 0.2 mg of Na2MoO4 × 2 H2O, 0.2 mg of CuSO4 × 5 H2O, 0.4 mg of CoCl2 × 6H2O, and 1 mg of MnCl2 × 2 H2O per liter of water to evaluate growth on a sole carbon source.
Bacterial growth quantification and rate determination
The optical density was measured using an Ultrospec 10-cell density meter (Amersham Biosciences, UK) at 600 nm. An OD600 of 1.0 corresponded with a cell dry weight of 366 mg L−1.
Furthermore, to enable real-time and automated monitoring of bacterial growth and growth rate [85], the Growth Profiler 960 (System Duetz, EnzyScreen BV, Heemstede, The Netherlands) was employed. Throughout the cultivations in the Growth Profiler, a constant temperature of 30°C and a shaking speed set to 250 rpm (revolutions per minute) were maintained. The cultivation was carried out in 96-well MTP plates (CR1496dg: polystyrene white square 96-half-deep well microtiter plates), which were covered with sandwich covers (CR1396: universal sandwich cover for 96-well MTPs). The entire run spanned 48 h.
The Growth Profiler recorded online green values, which were subsequently converted to OD600 values, depicting the growth of the bacteria [86]. To calculate growth rates, a MATLAB-based script was utilized in the analysis of the recorded data, which portrays exponential growth [87].
Cloning and strain engineering
The plasmids were constructed using Gibson assembly [88] with the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs GmBH, Frankfurt, Germany). All the designed primers used in this study were manufactured as unmodified DNA oligonucleotides from Eurofins Genomics (Ebersberg, Germany). Q5 High-Fidelity DNA Polymerase was used for all Polymerase Chain Reaction (PCR) products over 2000 bp, while smaller fragments were amplified using One-Taq DNA Polymerase (New England Biolabs GmBH, Frankfurt, Germany). Whereas in fragments with high GC content, Q5 High GC Enhancer (New England Biolabs GmBH, Frankfurt, Germany) was supplemented with the PCR reaction mix. In-depth information about used strains and plasmids can be found in Table 3 and about oligonucleotides in Table 4.
The newly assembled vector constructs were transformed into chemically competent E. coli PIR2 cells using a heat shock protocol [79]. Gene transfer in P. pantotrophus was conducted using either transformation using electroporation or conjugation as described by Wynands et al. [34]. The efficiency of transformation (cfu/µg) was calculated as described in (https://www.edvotek.com/how-to-calculate-transformation-efficiency). To assess P. pantotrophus DSM 2944's capability for direct cell-to-cell gene transfer via conjugation, mating experiments were conducted under two conditions: one with a helper strain carrying plasmid pRK600, facilitating the conjugational transfer, and another without. Donor strains carried plasmids (pSEVA121, 221, 231, 241, 251) with specific antibiotics as selection markers. Transconjugants were identified by positive growth on antibiotic plates with the requisite markers.
The pEMG genomic deletion was executed through homologous recombination involving plasmid p∆PHB and P. pantotrophus DSM 2944, facilitated by plasmid pSW2 expressing the I-SceI enzyme via conjugation. The selection of desired recombinant clones was accomplished utilizing the appropriate TS1 and TS2 primers (refer to Table 3). The resulting deleted strain, denoted as P. pantotrophus DSM 2944 ∆PHB, exhibited the absence of polyhydroxybutyrate (PHB) production, as achieved through the pEMG deletion method detailed in [56]. In contrast, heterologous gene integration into the genome was achieved using a Tn7 transposon-based system, as illustrated in [81], positioning the requisite genes downstream of the glmS gene [89] in P. pantotrophus DSM 2944. Conjugation was performed by the E. coli donor strain holding the respective pBG-plasmid, the helper strain E. coli HB101 pRK600, E. coli DH5α λpir pTnS1 providing the required transposase, and the recipient, P. pantotrophus DSM 2944.
The amplified PCR products were gel-extracted with a DNA Gel Extraction kit (New England Biolabs, Ipswich, Massachusetts, USA). The concentration of purified fragments was measured with a NanoDrop One (Thermo Scientific, Waltham, Massachusetts, USA). Colony PCR was performed to either amplify DNA fragments from the genome or verify the recombinant strain using 16S sequencing. Finally, all required fragments were sequenced using the Mix2Seq service from Eurofins Genomics (Ebersberg, Germany).
Fluorescent measurement and determination of promoter activity
The activity of the synthetic promoters was determined by the intensity of the msfGFP [90]. Fluorescence was quantified using the Biolector (M2P Labs, Baesweiler, Germany) where the excitation and the emission wavelength were set to 488 and 520 nm respectively, along with a gain of 40. Tested strains included P. putida KT2440 [47, 48] as the control and newly constructed P. pantotrophus DSM 2944 both having expression cassettes integrated into the genome. The cells were grown at 30°C with a shaking speed of 200 rpm on MSM media supplemented with 20 mM glucose as the sole carbon source. To omit the influence of varying cell numbers on fluorescence measurement, the starting OD600 of all the cells for the experiment was set to 0.01. A calibration between OD600 and fluorescence was conducted for both strains. Finally, the promoter activity is determined by calculating the slope of GFP fluorescence to optical density during the exponential phase [47].
Adaptive laboratory evolution
Adaptive laboratory evolution was performed on P. pantotrophus DSM 2944 tph, to obtain phenotypes with improved substrate-utilizing capabilities. Two adaptive laboratory evolution techniques were employed: static ALE involved continuous sub-culturing with a fixed carbon source (EG or TA), while in dynamic ALE alternated substrates [59] were used. Delft minimal medium supplemented with 40 mM EG or 20 mM TA was used for all experiments. The strains were grown in polypropylene square 24-deep well microplates at 37°C. OD600 was measured every 24 h for cells grown in EG and 48 h in terephthalic acid and the cells were sequentially transferred to a fresh medium with a starting OD600 of 0.03. Sub-culturing was carried out for 21 days. After this period, the adapted strains were streaked out on 20 mM TA plates to obtain single isolates, which were subsequently tested for improved growth in the Growth Profiler.
Polyhydroxyalkanoate quantification
Pre-weighed 10 mg of lyophilized cell biomass of the required strain were mixed with a (1:1) ratio of acidified methanol and chloroform in heat-resistant Pyrex tubes. To this mixture, 10 μl of tridecanoic acid (in 20 g L−1 in ethanol) was added as the internal standard. The mixture was vortexed, incubated at 100 °C for 2 h, and then cooled. After cooling, autoclaved distilled water was added and centrifuged, and finally, the organic phase was extracted for quantification.
1 mL of this organic phase was filled into a gas chromatography vial and injected in the Thermo Scientific Trace GC Ultra (Thermo Scientific, Waltham, MA, USA), combined with a flame ionization detector (FID). After derivatization, the fatty acid methyl esters obtained from lyophilized cells were separated on a Zebron ZB-WAX column (30 m length, 0.25 mm inner diameter, 0.25 μm film thickness, Phenomenex, Torrance, USA). The split ratio was set to 1:10, and the injection volume was 1 μL. The column oven temperature was kept constant for 5 min at 120 °C and then increased to 180 °C for 20 min. The temperature was then kept constant for 10 min and further increased to 250 °C for 11 min followed by a 2 min hold. The temperature of the FID was set to 290 °C. C4 to C24 even carbon-saturated fatty acid methyl esters (FAMEs) were used for quantification and peak identification.
Quantification of PET monomers using High-Performance Liquid Chromatography (HPLC)
Ethylene glycol measurements were conducted through HPLC-WVD-RI using an UltiMate 3000 HPLC system. This system comprised the TCC-3000SD column compartment, a WPS-3000SL autosampler, an ISO-3100SD pump, a WVD-3100 variable Wavelength Detector set at 210nm, and the SHODEX RI-101 refractive index detector sourced from Showa Denko Europe GmbH in Munich, Germany. Ethylene glycol (EG) elution was accomplished using a Metab-ACC ion exchange column with dimensions of 300 × 7.8 mm and a particle size of 10 μm, procured from ISERA in Düren, Germany. In the isocratic method, a mobile phase of 5 mM H2SO4 was employed, with a consistent flow rate of 0.6 mL min−1. The column oven temperature was maintained at 60 °C. Each injection contained a volume of 5 μl.
A set of standards was utilized to calibrate EG concentrations, including concentrations of 1.25 mM, 2.5 mM, 5 mM, 10 mM, 20 mM, and 40 mM.
Terephthalic acid was quantified through HPLC using the UltiMate 3000 HPLC system. This system was comprised of the TCC-3000SD column, a WPS-3000TSL analytical autosampler, an HPG-3400SD pump, and the MWD-3000 Multiple Wavelength Detector, which was set to wavelengths of 254 nm and 280 nm for detection. Terephthalic acid was eluted using an ISApher 100–5 C18 BDS gravity column (250 × 4.0 mm, particle size 5 μm; ISERA, Düren, Germany). The elution process employed a binary gradient comprising 90% formic acid (0.1%, v/v, in ultrapure water) and 10% acetonitrile (ACN). From the 2nd to the 14th minute, the gradient linearly transitioned from 90% formic acid and 10% ACN to 100% ACN, and this composition was maintained for an additional 2 min. Returning to the initial state, the period from the 16th to the 18th minute saw the restoration of 10% ACN and 90% formic acid, maintained until the measurement's end.
A sample injection volume of 1 μl was utilized, while the flow rate was set at 0.8 mL min−1. The column oven temperature was maintained at 40°C. Calibration of TA relied on a series of standards with concentrations ranging from 0.5 mM to 40 mM, including concentrations of 1.25 mM, 2.5 mM, 5 mM, 10 mM, 20 mM, and 40 mM.
Availability of data and materials
All data generated in the course of this study has been incorporated into the published article.
References
de Lorenzo V, Krasnogor N, Schmidt M. For the sake of the Bioeconomy: define what a Synthetic Biology Chassis is! N Biotechnol. 2021;60:44–51.
Ruiz N, Silhavy TJ. How Escherichia coli became the flagship bacterium of molecular biology. J Bacteriol. 2022;204(9): e0023022.
Liu Y, et al. Synthetic biology toolbox and chassis development in Bacillus subtilis. Trends Biotechnol. 2019;37(5):548–62.
Cui W, et al. Exploitation of Bacillus subtilis as a robust workhorse for production of heterologous proteins and beyond. World J Microbiol Biotechnol. 2018;34(10):145.
Jiang L, et al. Metabolic engineering tools for Saccharomyces cerevisiae. Sheng Wu Gong Cheng Xue Bao. 2021;37(5):1578–602.
Li H, et al. Evaluation of industrial Saccharomyces cerevisiae strains as the chassis cell for second-generation bioethanol production. Microb Biotechnol. 2015;8(2):266–74.
Calero P, Nikel PI. Chasing bacterial chassis for metabolic engineering: a perspective review from classical to non-traditional microorganisms. Microb Biotechnol. 2019;12(1):98–124.
Nikel PI, de Lorenzo V. Pseudomonas putida as a functional chassis for industrial biocatalysis: From native biochemistry to trans-metabolism. Metab Eng. 2018;50:142–55.
Patermann C, Aguilar A. The origins of the bioeconomy in the European Union. N Biotechnol. 2018;40(Pt A):20–4.
Herman L, et al. The qualified presumption of safety assessment and its role in EFSA risk evaluations: 15 years past. FEMS Microbiol Lett. 2019;366(1):89.
Burdock GA, Carabin IG. Generally recognized as safe (GRAS): history and description. Toxicol Lett. 2004;150(1):3–18.
Adams BL. The next generation of synthetic biology chassis: moving synthetic biology from the laboratory to the field. ACS Synth Biol. 2016;5(12):1328–30.
Meyer EL, Jenkins C, Rengarajan K. NIH Guidelines April 2019. Appl Biosaf. 2019;24(4):179–81.
Robertson LA, Kuenen JG. Thiosphaera pantotropha gen. nov. sp. Nov., a facultatively anaerobic, facultatively autotrophic sulfur bacterium. J Gen Microbiol. 1983;129(9):2847–55.
Friedrich CG, et al. Novel genes coding for lithotrophic sulfur oxidation of Paracoccus pantotrophus GB17. J Bacteriol. 2000;182(17):4677–87.
van Spanning RJ, et al., Paracoccus. Bergey's Manual of Systematics of Archaea and Bacteria, 2015: p. 1–14.
Wang J, et al. Nitrate stimulation of N-Methylpyrrolidone biodegradation by Paracoccus pantotrophus: Metabolite mechanism and Genomic characterization. Bioresour Technol. 2019;294: 122185.
Schavemaker PE, Lynch M. Flagellar energy costs across the tree of life. Elife. 2022;11:89.
Bachmann D, et al. C-, N-, S-, and P-Substrate Spectra in and the Impact of Abiotic Factors on Assessing the Biotechnological Potential of Paracoccus pantotrophus. Appl Microbiol. 2023;3(1):175–98.
Health F.I.f.O.S.a., Classification of Prokaryotes (Bacteria and Archaea) into Risk Groups. 2010(GMBl 2010, No. 68–80 of 06.12.2010, pp. 1428–1667).
Lasek R, et al. Genome Structure of the Opportunistic Pathogen Paracoccus yeei (Alphaproteobacteria) and Identification of Putative Virulence Factors. Front Microbiol. 2018;9:2553.
Cardoso VM, et al. Cost analysis based on bioreactor cultivation conditions: Production of a soluble recombinant protein using Escherichia coli BL21(DE3). Biotechnol Rep (Amst). 2020;26: e00441.
Jia K, et al. Study of Class I and Class III Polyhydroxyalkanoate (PHA) Synthases with Substrates Containing a Modified Side Chain. Biomacromol. 2016;17(4):1477–85.
Bockwoldt JA, et al. Complete Genome Sequence and Annotation of the Paracoccus pantotrophus Type Strain DSM 2944. Microbiol Resour Announc. 2020;9(1):89.
Barrick Jeffrey BC, Dasgupta A, Leonard S, Deatherage D, Suarez G, Monk J, Rodriguez A, Leon Dacia, Elston Kate, Alton S, Ashraf Z, Robinson E, Mishler D, Renda B. Antibiotic concentrations and stock solutions, Making_stock_antibiotics_and_other_reagents. 2019.
Silva-Rocha R, et al. The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2013;41:D666–75.
Rakowski SA, Filutowicz M. Plasmid R6K replication control. Plasmid. 2013;69(3):231–42.
Figurski DH, Helinski DR. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A. 1979;76(4):1648–52.
Poschel L, Gehr E, Buchhaupt M. A pBBR1-based vector with IncP group plasmid compatibility for Methylorubrum extorquens. Microbiologyopen. 2022;11(5): e1325.
Selzer G, et al. The origin of replication of plasmid p15A and comparative studies on the nucleotide sequences around the origin of related plasmids. Cell. 1983;32(1):119–29.
Yamaguchi K, Yamaguchi M. The replication origin of pSC101: the nucleotide sequence and replication functions of the ori region. Gene. 1984;29(1–2):211–9.
Green MR, Sambrook J. Screening Bacterial Colonies Using X-Gal and IPTG: alpha-Complementation. Cold Spring Harb Protoc. 2019;2019(12):78.
Balbas P, Bolivar F. Back to basics: pBR322 and protein expression systems in E. coli. Methods Mol Biol. 2004;267:77–90.
Wynands B, et al. Metabolic engineering of Pseudomonas taiwanensis VLB120 with minimal genomic modifications for high-yield phenol production. Metab Eng. 2018;47:121–33.
Friehs K. Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol. 2004;86:47–82.
Lin-Chao S, Chen WT, Wong TT. High copy number of the pUC plasmid results from a Rom/Rop-suppressible point mutation in RNA II. Mol Microbiol. 1992;6(22):3385–93.
Lee CL, Ow DS, Oh SK. Quantitative real-time polymerase chain reaction for determination of plasmid copy number in bacteria. J Microbiol Methods. 2006;65(2):258–67.
Bagdasarian M, et al. Specific-purpose plasmid cloning vector. II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene. 1981;16(13):237–47.
Bentley WE, et al. Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng. 1990;35(7):668–81.
Froger A, Hall JE. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp. 2007;6:253.
Holmes RK, Jobling MG. Genetics, in Medical Microbiology, S. Baron, Editor. 1996: Galveston (TX).
Lawley TD, et al. F factor conjugation is a true type IV secretion system. FEMS Microbiol Lett. 2003;224(1):1–15.
Silbert J, Lorenzo V, Aparicio T. Refactoring the Conjugation Machinery of Promiscuous Plasmid RP4 into a Device for Conversion of Gram-Negative Isolates to Hfr Strains. ACS Synth Biol. 2021;10(4):690–7.
Wein T, et al. Emergence of plasmid stability under non-selective conditions maintains antibiotic resistance. Nat Commun. 2019;10(1):2595.
Turner PE, Cooper VS, Lenski RE. Tradeoff between horizontal and vertical modes of transmission in bacterial plasmids. Evolution. 1998;52(2):315–29.
Lee H, Ko KS. Effect of multiple, compatible plasmids on the fitness of the bacterial host by inducing transcriptional changes. J Antimicrob Chemother. 2021;76(10):2528–37.
Ackermann YS, et al. Engineering adipic acid metabolism in Pseudomonas putida. Metab Eng. 2021;67:29–40.
Kobbing S, Blank LM, Wierckx N. Characterization of context-dependent effects on synthetic promoters. Front Bioeng Biotechnol. 2020;8:551.
Kammerer W, et al. Functional dissection of Escherichia coli promoters: information in the transcribed region is involved in late steps of the overall process. EMBO J. 1986;5(11):2995–3000.
Kosuri S, et al. Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc Natl Acad Sci U S A. 2013;110(34):14024–9.
Zobel S, et al. Tn7-based device for calibrated heterologous gene expression in Pseudomonas putida. ACS Synth Biol. 2015;4(12):1341–51.
Neves D, et al. Cross-species synthetic promoter library: finding common ground between Pseudomonas taiwanensis VLB120 and Escherichia coli. ACS Synth Biol. 2023;12(7):2029–40.
Pedelacq JD, et al. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24(1):79–88.
Solem C, et al. The las enzymes control pyruvate metabolism in Lactococcus lactis during growth on maltose. J Bacteriol. 2007;189(18):6727–30.
Marschall L, Sagmeister P, Herwig C. Tunable recombinant protein expression in E. coli: promoter systems and genetic constraints. Appl Microbiol Biotechnol. 2017;101(2):501–12.
Martinez-Garcia E, de Lorenzo V. Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ Microbiol. 2011;13(10):2702–16.
Narancic T, et al. Genome analysis of the metabolically versatile Pseudomonas umsongensis GO16: the genetic basis for PET monomer upcycling into polyhydroxyalkanoates. Microb Biotechnol. 2021;14(6):2463–80.
Utomo RNC. Upcycling of plastic monomers by mixed microbial cultures. 2022: p. 148.
Sandberg TE, et al. Laboratory Evolution to Alternating Substrate Environments Yields Distinct Phenotypic and Genetic Adaptive Strategies. Appl Environ Microbiol. 2017;83(13):8.
Garcia-Ochoa F, Gomez E. Bioreactor scale-up and oxygen transfer rate in microbial processes: an overview. Biotechnol Adv. 2009;27(2):153–76.
Bartosik D, Sochacka M, Baj J. Identification and characterization of transposable elements of Paracoccus pantotrophus. J Bacteriol. 2003;185(13):3753–63.
Attere SA, et al. The Role for the Small Cryptic Plasmids As Moldable Vectors for Genetic Innovation in Aeromonas salmonicida subsp. salmonicida. Front Genet. 2017;8:211.
Stinchcombe A, Hammond NG, Hopper S. Changes in executive function in the Canadian longitudinal study on aging over 3-years: A focus on social determinants of health. Front Psychol. 2023;14:1060178.
de Vries GE, et al. Isolation and characterization of Paracoccus denitrificans mutants with increased conjugation frequencies and pleiotropic loss of a (nGATCn) DNA-modifying property. Arch Microbiol. 1989;152(1):52–7.
Glick BR. Metabolic load and heterologous gene expression. Biotechnol Adv. 1995;13(2):247–61.
Nobrega MP, Nobrega FG, Tzagoloff A. COX10 codes for a protein homologous to the ORF1 product of Paracoccus denitrificans and is required for the synthesis of yeast cytochrome oxidase. J Biol Chem. 1990;265(24):14220–6.
Fling ME, Kopf J, Richards C. Nucleotide sequence of the transposon Tn7 gene encoding an aminoglycoside-modifying enzyme, 3"(9)-O-nucleotidyltransferase. Nucleic Acids Res. 1985;13(19):7095–106.
Ind AC, et al. Inducible-expression plasmid for Rhodobacter sphaeroides and Paracoccus denitrificans. Appl Environ Microbiol. 2009;75(20):6613–5.
Kupferschmied P, et al. Domain shuffling in a sensor protein contributed to the evolution of insect pathogenicity in plant-beneficial Pseudomonas protegens. PLoS Pathog. 2014;10(2): e1003964.
Drabinska J, et al. Individual Nudix hydrolases affect diverse features of Pseudomonas aeruginosa. Microbiologyopen. 2020;9(8): e1052.
Reznikoff WS. The Tn5 transposon. Annu Rev Microbiol. 1993;47:945–63.
Ghozlan HA, et al. Genetic tools for Paracoccus denitrificans. FEMS Microbiol Lett. 1991;82(3):303–5.
Green B, et al. Insertion site preference of Mu, Tn5, and Tn7 transposons. Mob DNA. 2012;3(1):3.
Li N, et al. Tn5 Transposase Applied in Genomics Research. Int J Mol Sci. 2020;21(21):8329.
Werner AZ, et al. Tandem chemical deconstruction and biological upcycling of poly(ethylene terephthalate) to beta-ketoadipic acid by Pseudomonas putida KT2440. Metab Eng. 2021;67:250–61.
Gautom T, et al. Structural basis of terephthalate recognition by solute binding protein TphC. Nat Commun. 2021;12(1):6244.
Boyer HW, Roulland-Dussoix D. A complementation analysis of the restriction and modification of DNA in Escherichia coli. J Mol Biol. 1969;41(3):459–72.
Yang XW, Jian HH, Wang FP. pSW2, a novel low-temperature-inducible gene expression vector based on a filamentous phage of the deep-sea bacterium Shewanella piezotolerans WP3. Appl Environ Microbiol. 2015;81(16):5519–26.
Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166(4):557–80.
Platt R, et al. Genetic system for reversible integration of DNA constructs and lacZ gene fusions into the Escherichia coli chromosome. Plasmid. 2000;43(1):12–23.
Choi KH, et al. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2(6):443–8.
Nelson KE, et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol. 2002;4(12):799–808.
Eden PA, et al. Phylogenetic analysis of Aquaspirillum magnetotacticum using polymerase chain reaction-amplified 16S rRNA-specific DNA. Int J Syst Bacteriol. 1991;41(2):324–5.
Jiang H, et al. Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China. Appl Environ Microbiol. 2006;72(6):3832–45.
Marr AG. Growth rate of Escherichia coli. Microbiol Rev. 1991;55(2):316–33.
Duetz WA, et al. Methods for intense aeration, growth, storage, and replication of bacterial strains in microtiter plates. Appl Environ Microbiol. 2000;66(6):2641–6.
Hemmerich J, Wiechert W, Oldiges M. Automated growth rate determination in high-throughput microbioreactor systems. BMC Res Notes. 2017;10(1):617.
Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–5.
Ferre-D’Amare AR. The glmS ribozyme: use of a small molecule coenzyme by a gene-regulatory RNA. Q Rev Biophys. 2010;43(4):423–47.
Valbuena FM, et al. A photostable monomeric superfolder green fluorescent protein. Traffic. 2020;21(8):534–44.
Acknowledgements
The authors thank Armin Quentmeier from TU Dortmund University for valuable insights derived from decades of research with Paracoccus.
Funding
Open Access funding enabled and organized by Projekt DEAL. Funding for this research was provided by the Federal Ministry of Education and Research (BMBF; Germany) under the ParaCoquette project (FKZ 031B0854) and from the Excellence Initiative of the German Federal and State Governments as part of the SeedFund PapaBio project. Additionally, the laboratory of L.M.B. receives partial funding from the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) as part of Germany’s Excellence Strategy within the Cluster of Excellence FSC 2186, "The Fuel Science Center."
Author information
Authors and Affiliations
Contributions
UP conducted most of the experiments and drafted the manuscript. DB and CP conducted some experiments and provided experimental help to UP. JC helped conceptualize some experiments. TT conceptualized the manuscript and wrote parts of the manuscript. LMB and TT provided vital feedback and revised the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests. The funders played no role in the study's design, data collection, analysis, interpretation, manuscript writing, or the decision to publish the findings.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pal, U., Bachmann, D., Pelzer, C. et al. A genetic toolbox to empower Paracoccus pantotrophus DSM 2944 as a metabolically versatile SynBio chassis. Microb Cell Fact 23, 53 (2024). https://doi.org/10.1186/s12934-024-02325-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-024-02325-0