
Abstract
Background
Methanol, synthesized from CO2, is a potentially sustainable one-carbon (C1) resource for biomanufacturing. The use of methanol as a feedstock to produce single cell protein (SCP) has been investigated for decades as an alternative to alleviate the high global demand for animal-derived proteins. The methylotrophic yeast Pichia pastoris is an ideal host for methanol-based SCP synthesis due to its natural methanol assimilation ability. However, improving methanol utilization, tolerance to higher temperature, and the protein content of P. pastoris are also current challenges, which are of great significance to the economical industrial application using methanol as a feedstock for SCP production.
Results
In the present work, adaptive laboratory evolution (ALE) has been employed to overcome the low methanol utilization efficiency and intolerance to a higher temperature of 33 °C in P. pastoris, associated with reduced carbon loss due to the lessened detoxification of intracellular formaldehyde through the dissimilation pathway and cell wall rearrangement to temperature stress resistance following long-term evolution as revealed by transcriptomic and phenotypic analysis. By strengthening nitrogen metabolism and impairing cell wall synthesis, metabolic engineering further increased protein content. Finally, the engineered strain via multi-strategy produced high levels of SCP from methanol in a pilot-scale fed-batch culture at 33 °C with a biomass of 63.37 g DCW/L, methanol conversion rate of 0.43 g DCW/g, and protein content of 0.506 g/g DCW. SCP obtained from P. pastoris contains a higher percentage of protein compared to conventional foods like soy, fish, meat, whole milk, and is a source of essential amino acids, including methionine, lysine, and branched-chain amino acids (BCAAs: valine, isoleucine, leucine).
Conclusions
This study clarified the unique mechanism of P. pastoris for efficient methanol utilization, higher temperature resistance, and high protein synthesis, providing a P. pastoris cell factory for SCP production with environmental, economic, and nutritional benefits.
Similar content being viewed by others


Background
To meet the global demand for animal-derived proteins, the world will need to produce 1,250 million tons of meat and dairy products annually. However, due to the low efficiency in converting feed to meat and dairy products, the growing human demand for proteins will not be met sustainably by increasing meat and dairy production [1, 2]. Although artificial meat and plant-based meat substitutes are currently developing rapidly, higher production costs and poorer taste make them less competitive than animal-based proteins produced through traditional livestock farming [3, 4]. Therefore, it is still necessary to find and develop new protein resources. Single cell protein (SCP), which is produced in algae (protein content is approximately 60–70%), fungi (protein content is approximately 30–50%) and bacteria (protein content is approximately 50–80%) cells, is a potential option [5,6,7]. The unique substrate (such as CO2 or methane) utilization ability and high growth rate of these microorganisms make the production process of SCP more efficient and sustainable than traditional agriculture [8].
The concentration of CO2 in the global atmosphere is increasing at an alarming rate, and the climate and greenhouse effect have affected human survival and development. The growing environmental crisis has exacerbated the need for a shift away from the traditional petrochemical economy towards sustainable feedstocks. One-carbon (C1) compound assimilation by microorganisms has emerged as a promising approach in abating climate change [9,10,11]. Methanol is regarded as an ideal C1 resource for chemical industry and biomanufacturing, since it can be produced not only from low-quality coal, but also from CO2 and hydrogen (electrolyzed water) in large quantities by electrocatalysis or photocatalysis, which is considered as one of the most potential ways to achieve global carbon neutrality. As a liquid, methanol is easier to transport and store compared to those gaseous C1 compounds [12, 13]. Furthermore, methanol has a high degree of reduction that can provide more driving force for product biosynthesis [14]. Therefore, the cheap and abundant sources, flexible production processes, compatibility with current transportation and fermentation infrastructure, and high reduction potential make methanol an attractive substrate for SCP biosynthesis (Fig. 1).

The concept of methanol-based SCP production in P. pastoris. Methanol can be produced from low-quality coal and CO2 hydrogenation in large quantities, representing a sustainable production mode using methanol as a feedstock for future biomanufacturing and one of the potential ways to achieve global carbon neutrality
In recent years, researchers have tried to construct a methanol assimilation pathway by constructing heterologous and artificial utilization pathways, and achieved the conversion of methanol to central metabolism in model microorganisms such as Escherichia coli, Saccharomyces cerevisiae and Corynebacterium glutamicum [15,16,17]. However, these engineered strains still cannot grow use methanol as sole carbon source. Recently, Chen et al. realized the growth of E. coli using methanol as the sole carbon through the combination of metabolic engineering and adaptive laboratory evolution (ALE). However, the doubling time of this strain is up to 8.5 h, and the maximum optical density (OD600) is only about 2, which greatly limits the methanol metabolism rate and product synthesis efficiency [11]. Notably, native methylotrophic yeast, Pichia pastoris (also known as Komagataella phaffii), might be an ideal host for methanol biotransformation given its natural capacity in methanol assimilation system. Particularly, P. pastoris is an important industrial methanol yeast with GRAS (Generally Recognized as Safe) certification by the U.S. Food and Drug Administration (FDA), which is widely used in the production of recombinant proteins [18, 19]. With the whole genome sequencing and subsequent annotation of commonly used strains CBS7435 and GS115, as well as the development of more accurate genome editing system, the research on obtaining target products through engineering P. pastoris based on methanol has become a hot spot [20,21,22]. So far, some chemicals, such as lactic acid, non-animal chondroitin sulfate, malic acid and free fatty acids etc., have been synthesized from methanol in P. pastoris after metabolic rewiring [23,24,25,26]. However, the complex methanol metabolism pathway and the toxicity of intracellular formaldehyde led to the low methanol utilization efficiency in natural P. pastoris, which is not conducive to the synthesis of target products [27, 28].
Since decades, P. pastoris has been using methanol as a feedstock for producing SCP [29], but improving the conversion of methanol into biomass has always been problematic. Further, since the fermentation temperature of P. pastoris is actually higher than its growth temperature (28–30 °C), intolerance to higher temperature usually reduces substrate conversion efficiency and increases cooling costs in the industrial production process [30]. Thus, using a higher-temperature-resistant P. pastoris for SCP biosynthesis is a potential way to reduce production costs. Importantly, the nutritional value of SCP is closely related to the protein content of P. pastoris, a P. pastoris strain with high-protein content will be more competitive with other protein sources [1, 2]. As a result, we can conclude that one of the key considerations of economic production of SCP based on methanol is to develop a P. pastoris cell factory capable of utilizing methanol efficiently, tolerating higher temperature, and producing high amounts of protein.
In the current study, ALE was used to obtain a P. pastoris strain with high methanol utilization efficiency and tolerance to a higher temperature of 33 °C (this temperature was selected in consideration of both biomass and protein content). Transcriptome and intracellular formaldehyde analyses showed that less formaldehyde was detoxified through the dissimilation pathway, ultimately resulting in a reduction in carbon loss in the evolved strain. It was also found that effective induction of cell wall remodeling was necessary to protect P. pastoris cells from temperature stress during evolution. Following that, metabolic engineering was used to improve protein content in this evolved host by improving nitrogen metabolism and impairing cell wall synthesis, with the best strain achieving high-level production of SCP from sole methanol in a pilot-scale fed-batch culture at 33 °C. In addition to high protein content, SCP from P. pastoris is also a source of essential amino acids. This study successfully elucidated the unique mechanism of methanol utilization efficiently, higher temperature tolerance and high protein synthesis in P. pastoris, providing a P. pastoris cell factory for economical industrial application using methanol as a substrate for SCP production.
Materials and methods
Strains, media and cultivation
All strains and plasmids used in this study are listed in Table 1. The plasmid pPICZ-Cas9-gGUT1 [22] was kindly offered by Prof. Yongjin Zhou from Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences, and other P. pastoris strains and plasmids were stored in our lab or constructed in this study. E. coli DH5α used as the host for plasmid construction were cultivated at 37 °C in a rotatory shaker at 220 rpm in Luria-Bertani (LB) consisting of 5 g/L yeast extract, 10 g/L tryptone, and 10 g/L NaCl with 50 µg/mL zeocin to maintain plasmids. Unless otherwise specified, P. pastoris strains were cultivated in YPD medium at 30 or 33 °C in a rotatory shaker at 220 rpm consisting of 20 g/L glucose, 20 g/L peptone, and 10 g/L yeast extract. And 100 µg/mL zeocin was added to YPD medium for screening transformants. The composition of the Delft basic salt medium used for cell cultivation with methanol as single carbon source were 14.4 g/L KH2PO4, 7.5 g/L (NH4)2SO4, 0.5 g/L MgSO4•7H2O, 1 ml/L vitamin solution, and 2 ml/L trace metal solution [22, 31].
Plasmids and strains construction
The method used for genetic manipulation in the chromosome of P. pastoris was based on the utilization of CRISPR-Cas9 meditated genome editing system as described previously [22]. To construct gRNA expression plasmids, 20 bp target sequences of gRNAs for genome targeting were designed using a user-friendly online web tool (CRISPR RGEN Tools, https://www.rgenome.net/). As for construction of plasmid pPICZ-Cas9-gPNSII-3, the Cas part and gRNA part were obtained by PCR amplification with primers gPNSII-3-1 F/gPNSII-3-1R and gPNSII-3-2 F/gPNSII-3-2R using the plasmid pPICZ-Cas9-gGUT1(containing Cas9 and GUT1-gRNA under the control of bidirectional promoter PHTX1) as a template. The two parts were fused by Gibson assembly to yield plasmid pPICZ-Cas9-gPNSII-3. Transformations were screened on LB agar plate with 50 µg/mL Zeocin. The plasmids pPICZ-Cas9-gPNSI-2 and pPICZ-Cas9-gPAS_chr4_0305 were constructed based on the same method using primers gPNSI-2-1 F/gPNSI-2-1R, gPNSI-2-2 F/gPNSI-2-2R, gPAS_chr4_0305-1 F/gPAS_chr4_0305-1R and gPAS_chr4_0305-2 F/gPAS_chr4_0305-2R, respectively.
To construct GDH1-donor DNA, the upstream part, promoter part, GDH1 part, terminator part and downstream part, were amplified from the P. pastoris genome using primers PNSII-3-up-F/PNSII-3-up-R, PAOX1-F/PAOX1-R, GDH1-F/GDH1-R, TADH2 (GDH1)-F/TADH2 (GDH1)-R and PNSII-3-down-F/PNSII-3-down-R, respectively. These five fragments were then fused by fusion PCR to obtain GDH1-donor DNA cassettes. As for GLN1-donor DNA, the upstream part, promoter part, GLN1 part, terminator part and downstream part, were amplified from the P. pastoris genome using primers PNSI-2-up-F/PNSI-2-up-R, PDAS2-F/PDAS2-R, GLN1-F/GLN1-R, TADH2 (GLN1)-F/TADH2 (GLN1)-R and PNSI-2-down-F/PNSI-2-down-R, respectively. The five fragments were then fused by fusion PCR to yield GLN1-donor DNA cassettes. To construct PAS_chr4_0305-donor DNA, fragments upstream and downstream of the PAS_chr4_0305 gene were amplified from the P. pastoris genome using primers PAS_chr4_0305-up-F/PAS_chr4_0305-up-R and PAS_chr4_0305-down-F/PAS_chr4_0305-down-R, respectively. The upstream and downstream fragments were then fused and amplified by fusion PCR to create PAS_chr4_0305-donor cassettes.
Finally, the gRNA expression plasmid and the corresponding donor DNA were co-transformed into the competent cells of P. pastoris, respectively, and the transformed cells were grown for three days on YPD plates containing 100 µg/mL Zeocin. The mutants were verified by PCR and further confirmed by gene sequencing. All primers used for strains and plasmids construction are listed in Supplementary Table 1.
Adaptive laboratory evolution
To avoid methanol evaporation, ALE was performed in the shake flasks covered with a sealing membrane. The process of ALE is graphically explained in Fig. 2a. Briefly, parent strain P. pastoris X-33 was spotted on a YPD agar plate and three independent colonies were cultivated in a 10 ml glass tube with 3 ml of Delft basic salt medium containing 2% glycerol for 24 h. These three strains were transferred to a 250 ml shake flask with 100 ml of Delft basic salt medium containing 0.5% methanol with an initial OD600 of 0.25. After 24 h of cultivation at 33 °C in a rotatory shaker at 220 rpm, the strains were transferred to the fresh medium with the same initial OD600, which were then incubated under the same conditions. Repeated the above steps about 100 times until a high density of cells were obtained. The final evolved colonies were streaked on YPD plates, and three colonies were picked from each group for further cell growth analysis.

ALE enhanced final biomass and tolerance to 33 °C of P. pastoris in 0.5% methanol minimal medium. a, The process of ALE; b, Procedure of ALE that was illustrated by enhanced cell growth; c, Automatic growth curves of the starting strain X-33 at 30 or 33 °C and the evolved strains at 33 °C; d, % maximum OD600 of the starting strain X-33 and the evolved strains at 33 °C relative to the starting strain X-33 at 30 °C. Three independent colonies of X-33 were transferred to a 250 ml shake flask with 100 mL of 0.5% methanol minimal medium with an initial OD600 of 0.25 at 33 °C at 220 rpm. Transferred the evolved strains to fresh growth medium every 24 h, until a high density of cells were obtained. Final evolved strains were spotted on YPD plates for further growth analysis using an automatic microbial growth curve analyzer. Data are average values and standard deviations of triplicate experiments
Automatic microbial growth curve analysis
The cell growth of P. pastoris was evaluated using an automatic microbial growth curve analyzer. Briefly, overnight cultures of strains were inoculated into a 48-well plate containing 1 ml Delft basic salt medium containing 0.5% methanol with an initial OD600 of 0.25 per well at 30 or 33 °C at 800 rpm. Cell growth was measured every hour to plot the growth curve and calculate final biomass. Three replicates per condition were used.
Transcriptome analysis
The control strain X-33 and the evolved strain HTX-33 were grown in Delft basic salt medium containing 0.5% methanol at 30 and 33 °C, respectively. The total RNA was extracted from the cells under these two culture conditions when grown to the exponential phase using TRIzol (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s protocols. RNA with the integrity of more than 6.5 that was detected by Agilent 2100 Nano (Agilent Technologies) was adopted to perform library construction and sequencing. The complementary DNA libraries were constructed and sequenced using the Illumina HiSeq 2000 platform at the Meiji Biotechnology (Shanghai) Co., Ltd. RESM software (https://deweylab.github.io/RSEM/) was used to quantify gene expression levels, yielding transcripts per million reads (TPM). The differential expression between the two samples was analyzed by the DEGseq (https://bioinfo.au.tsinghua.edu.cn/software/degseq), and genes with |log2FC|≥1 and P < 0.05 were identified as significantly differentially expressed genes (DEGs). The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed using the KOBAS (https://kobas.cbi.pku.edu.cn/home.do).
Formaldehyde quantification
Cells grown to the mid-log phase from 48-well plates were harvested by centrifugation at 5000×g for 10 min, washed three times and resuspended in 1 mL of PBS buffer solution (pH = 7.4). Added equal volume of cell lysate and then incubated at 37 °C for 30 min. After centrifugation, the supernatants were collected for formaldehyde detection using the method as described previously [32]. Briefly, 50 µL of supernatant collected after the cultivation was added into a 96-well plate. 150 µL of NASH reagent (5 M ammonium acetate, 50 mM acetyl acetone and 135 mM acetic acid) were then added into each well. The plate was kept on ice until all samples had been added. Incubated the plate at 37 °C for 10 min and read the absorbance at 414 nm. The formaldehyde concentrations were expressed as µM/OD.
Validation of genes by real-time quantitative polymerase chain reaction (RT-qPCR)
Twenty-one DEGs were selected to test the reliability of the RNA-Seq data by RT-qPCR. Total RNA was extracted from X-33 and HTX-33 strains grown to the exponential phase, respectively, using the TransZol Up Plus RNA Kit (TransGen, Beijing, China). The extracted RNA was then tested for its quality and concentration using a Nanodrop 2000c (Thermo). And cDNA was synthesized from 200 ng RNA using Quant Reverse Transcriptase in the presence of random primers (TransGen, Beijing, China). RT-qPCR was carried out using Real Master Mix (SYBR Green) and specific primers (Supplementary Table 2) in a Light Cycler 480 II (Roche, Basel, Switzerland) under the following cycling conditions: 5 min at 50 °C, 30 s at 94 °C, 45 cycles of 5 s at 94 °C, and 30 s at 60 °C. The ACT1 gene was used as a reference gene for normalization and all reactions were carried out in triplicate. Relative transcription levels were analyzed using the 2−ΔΔCt method described previously [33].
Cell morphology analysis
Scanning electron microscopy (SEM) was used to visualize the changes in the cell wall surface of P. pastoris. Transmission electron microscopy (TEM) was used to check the thickness of the cell wall of P. pastoris. Cells grown to the mid-log phase were harvested by centrifugation at 5000×g for 10 min, washed three times, and resuspended in PBS buffer solution (pH = 7.4). The suspension was premixed with an equal volume of 2.5% glutaraldehyde for 12 h at 4◦C and subsequently dehydrated with 25, 50, 70, 80, 95, and 100% ethanol. For SEM, the dehydrated samples were air-dried immediately, followed by smearing on SEM stubs and gold covering. The micrographs of the cell wall surface were obtained using a SEM (Hitachi SU8010, Tokyo, Japan). For TEM, pure ethanol was changed to propylene oxide and specimens were gradually infiltrated with increasing concentrations (30, 50, 70, and 100%) of Agar 100 epoxy resin mixed with propylene oxide for a minimum of 3 h per step. Samples were embedded in pure, fresh Agar 100 epoxy resin and polymerized at 60 °C for 48 h. Ultrathin sections were stained for 3 min with lead citrate and viewed with a JEM-1230 electron microscope. As for observation of colony morphology, P. pastoris strains stored at -80◦C were streaked on YPD agar plates. After the strain was cultured at 30 or 33 °C for 72 h, the morphology of the colonies were observed and photographed with a digital camera.
Biomass and total protein determination
Overnight cultures of P. pastoris strains were inoculated into a 48-well plate containing 1 mL 0.5% methanol minimal medium with an initial OD600 of 0.25 at 30 or 33 °C at 800 rpm. Cell pellets from 3 mL of mid-log phase culture were washed with sterile water three times and resuspended in 1 mL of sterile water. The tubes containing centrifuged cells were dried at 105 °C until a constant weight was maintained. The total nitrogen content was measured using the Kjeldahl method [34]. A conversion factor of 6.25 was used to convert nitrogen content to protein content. Total nitrogen content and protein content are proportional to yeast dry mass (%).
Pilot-scale fed-batch fermentation and nutritional benefit assessment
Pilot-scale fed-batch fermentation was performed in a 500 L automatic bioreactor system (Shanghai Bailun Biotechnology Co., Ltd). The initial batch fermentation was carried out in Delft + 2% glycerol with 300 L working volume. The initial inoculum was 28 vials of overnight culture strain HTX-33-GLN1-ΔPAS_chr4_0305 in Delft + 2% glycerol. Temperature and pH were set at 33 °C and 5.6, respectively. Initial agitation was set at 100 rpm/min and increased to a maximum of 300 rpm/min depending on the air volume level. Air volume was initially provided at 0.55 vvm and increased to a maximum of 1.5 vvm. Fed-batch cultivation started immediately after the glycerol in the batch medium was exhausted and methanol was pumped into the vessel. During fermentation, residual methanol was monitored to control the feeding rates, and biomass and methanol consumption were recorded every 2 h. After fermentation, all cells were centrifuged, washed and dried. The finished products were used for subsequent total protein determination as mentioned above and amino acids profile analysis through an automatic amino acid analyzer. Total protein content and amino acids content are related to yeast dry mass (%).
Statistical analysis
One-way analysis of variance was implemented in SPSS for Windows 10.0 (SPSS, Inc., Chicago, IL, United States).
Results
Adaptive laboratory evolution towards growth on methanol and higher temperature
ALE is recognized as a powerful strategy for achieving desirable phenotypes via causal mutations [11, 17, 35], and was thus used in this study to promote the growth of X-33 strain in methanol minimal medium at higher temperature. ALE was performed in Delft basic salt medium containing 0.5% methanol at 33 °C at 220 rpm, and three independent colonies of the X-33 strain were adopted as the starting strain, which was subsequently transferred to fresh growth medium in 24-h intervals for 100 days, yielding a total of approximately 350 generations. As shown in Fig. 2b, all three groups acquired higher cell growth in a minimal medium containing 0.5% methanol at 33 °C. Three colonies were randomly selected from each group to characterize their growth performance using an automatic microbial growth curve analyzer. As can be seen in Fig. 2c, the final biomass of strain X-33 at 33 °C (up to OD600 of 3.05) was 91.59% of that at 30 °C (up to OD600 of 3.33), indicating that higher temperature inhibited the cell growth of P. pastoris. All of nine clones from the three evolved populations showed increased final biomass on this growth medium, of which the highest final biomass (up to OD600 of 4.78) accounted for 1.43 times that of the starting strain X-33 cultured at 30 °C (Fig. 2d). Finally, this superior-performance strain with high conversion efficiency of methanol to biomass and 33 °C tolerance was named HTX-33.
Changes in cell wall and protein content of the evolved strain HTX-33
The cell wall is the toughest outer layer of yeast cells and is important for supporting cell structure and protecting cells from chemical and physical damage [36]. Since the cell wall is the first line of defense against external stress, cell wall strength and efficient cell wall remodeling are essential to withstand higher temperature stress. Studies have shown that genes involved in cell wall biogenesis were altered after cells evolved in a high temperature environment, resulting in a strengthened cell wall [37]. Consistent with this finding, the cell wall thickness of the evolved strain HTX-33 was increased after adaptation at 33 °C (Fig. 3a). Furthermore, SEM results showed that the cell wall surface of HTX-33 strain had obvious wrinkles, which was in strong contrast with the relatively smooth surface of X-33 (Fig. 3b). These changes in cell wall structure eventually led to the irregular round shape of the HTX-33 colonies (Fig. 3c). In addition, it was found that the total nitrogen (g/g DCW) and protein content (g/g DCW) of the X-33 strain at 33 °C (0.064 and 0.4, respectively) were 10% higher than those at 30 °C (0.058 and 0.364, respectively) (Table 2), which might be beneficial for tolerance to higher temperature due to increased protein synthesis. However, the total nitrogen and protein content of evolved strain HTX-33 were decreased to 0.059 and 0.368, respectively (Table 2), after long-term evolution at 33 °C. Our results indicated that the cell wall was strengthened in response to higher temperature stress, but the protein content was not further improved in the evolved strain HTX-33 compared to the starting strain X-33 at 30 °C.

Changes in cell wall and colony morphology of starting strain X-33 at 30 °C and the evolved strain HTX-33 at 33 °C in 0.5% methanol minimal medium. a, Representative TEM images of cell wall thickness; b, Representative SEM images of cell wall surface. The surface of the cell wall is partially magnified; c, Changes in colony morphology. Representative images are from three independent experiments
Transcriptomics analysis and intracellular formaldehyde determination of the evolved strain HTX-33
Subsequently, the whole-transcriptome analysis was conducted to identify essential DEGs related to methanol utilization and higher temperature stress in the evolved strain HTX-33 at 33 °C relative to the parent strain X-33 at 30 °C in minimal medium containing 0.5% methanol. The RNA-Seq data revealed that for the evolved strain HTX-33, 592 genes were significantly differentially expressed, of which 293 were upregulated and 299 were downregulated (Supplementary Table 3). The KEGG enrichment analysis showed that a total of 87 pathways had been enriched, and only methane metabolism (also regarded as methanol metabolism) pathway was significantly enriched (P < 0.05) (Supplementary Table 4). Unexpectedly, genes in methanol oxidation including PAS_chr4_0821 (encoding AOX1, alcohol oxidase 1) and PAS_chr4_0152 (encoding AOX2, alcohol oxidase 2), formaldehyde assimilation including PAS_chr3_0841 (encoding Dhak, dihydroxyacetone kinase), PAS_chr1-1_0319 (encoding Fba, fructose 1,6-bisphosphate aldolase), PAS_chr3_0834 (encoding DAS1, dihydroxyacetone synthase 1), PAS_chr3_0832 (encoding DAS2, dihydroxyacetone synthase 2) and PAS_chr4_0212 (encoding RpiA, ribose-5-phosphate ketol-isomerase), and formaldehyde dissimilation including PAS_chr3_1028 (encoding Fld1, S-(hydroxymethyl) glutathione dehydrogenase), PAS_chr3_0867 (encoding Fgh1, S-formylglutathione hydrolase) and PAS_chr3_0932 (encoding Fdh1, NAD+-dependent formate dehydrogenase) were greatly downregulated for the higher growth of evolved strain HTX-33 in methanol minimal medium at 33 °C (Fig. 4a). Since formaldehyde toxicity is supposed to be one of the primary obstacles limiting the carbon loss encountered by P. pastoris [27], the intracellular formaldehyde accumulation was characterized in the control strain X-33 and the evolved strain HTX-33. As expected, the HTX-33 strain accumulated a low level of formaldehyde at 214 µM/OD, compared to a relatively high level of formaldehyde in X-33 cells up to 531 µM/OD within 24 h (Fig. 4b).

DEGs and intracellular formaldehyde accumulation analyses of the evolved strain HTX-33 at 33 °C compared to the starting strain X-33 at 30 °C in 0.5% methanol minimal medium. a, Cluster analysis related to DEGs from methanol metabolism, PPP, EMP, TCA and cell wall synthesis; b, Changes in intracellular formaldehyde accumulation (µM per OD); c, Validation of selected DEGs by RT-qPCR; d, Correlation of the RNA-Seq data (X-axis) and the RT-qPCR data (Y-axis). Data are average values and standard deviations of triplicate experiments. An asterisk indicates a significant difference with ***P < 0.001; e, Sketch of DEGs in methanol metabolism. Genes in green showed significant down-regulation in expression. Bold border represented enhanced cell wall. Enzymes: Aox, alcohol oxidase; Fld1, formaldehyde dehydrogenase; Fgh1, S-formylglutathione hydrolase; Fdh1, formate dehydrogenase; Das, dihydroxyacetone synthase; Dhak, dihydroxyacetone kinase; Fba, fructosebisphosphate aldolase/sedoheptulose-bisphosphate aldolase; Fbp, fructose bisphosphatase; Sbp, sedoheptulose bisphosphatase; Rpe, ribulose-phosphate 3-epimerase; RpiA, ribose-5-phosphate isomerase; Metabolites: Xu5P, xylulose-5-phosphate; Ru5P, ribulose-5-phosphate; R5P, ribose-5-phosphate; DHA, dihydroxyacetone; DHAP, dihydroxyacetone phosphate; F16dP, fructose-1,6-bisphosphate; F6P, fructose-6-phosphate; G3P, glyceraldehyde-3-phosphate; E4P, erythrose-4-phosphate; S7P, sedoheptulose-7-phosphate; S17dP, sedoheptulose-1,7-bisphosphate
In addition, genes involved in pentose phosphate pathway (PPP) including PAS_chr3_0277 (encoding Gnd, 6-phosphogluconate dehydrogenase) and PAS_chr2-2_0338 (encoding TalB, transaldolase), glycolysis pathway (EMP) including PAS_chr1-4_0669 (encoding GntK, gluconokinase), PAS_chr2-1_0771 (encoding Pgm, phosphoglucomutase), PAS_chr1-4_0042 (encoding GPE, glucose-6-phosphate 1-epimerase) and PAS_chr3_0693 (encoding PGAM, tetrameric phosphoglycerate mutase), TCA cycle including PAS_chr4_0815 (encoding MDH2, mitochondrial malate dehydrogenase) and PAS_chr2-1_0580 (encoding IDH, cytosolic NADP-specific isocitrate dehydrogenase) were also downregulated (Fig. 4a).
In contrast to most downregulated genes in methanol and central carbon metabolism, genes participated in cell wall synthesis including PAS_chr4_0559 (encoding CHT, putative chitin transglycosidase, cell wall protein), PAS_chr1-1_0393 (encoding CHS1, Chitin synthase I) and PAS_chr2-1_0065 (encoding CHS III, Chitin synthase III, catalyzes the transfer of N-acetylglucosamine to chitin) were upregulated (Fig. 4a), which may help to explain the changes in cell wall of the evolved strain HTX-33, thereby endowing the strain with higher temperature tolerance.
Based on the above analysis, seven sets of genes related to methanol oxidation, formaldehyde assimilation, formaldehyde dissimilation, PPP, EMP, TCA and cell wall synthesis were then selected for RT-qPCR analysis to verify the accuracy and reproducibility of RNA-Seq data. As expected, the expression trends of the 21 candidate DEGs were consistent with those detected by RNA-seq (Fig. 4c), and the correlation coefficient between RNA-Seq and RT-qPCR data was 0.9617 (Fig. 4d), indicating that the RNA-Seq data are accurate and reliable. In summary, although methanol and central carbon metabolism were slowed down, the reduction in carbon loss due to the lessened detoxification of intracellular formaldehyde through the dissimilation pathway ultimately increased methanol utilization efficiency (Fig. 4e). Further, it was also found that the ability to effectively induce cell wall strengthening is necessary for protecting cells against higher temperature stress in the evolved strain HTX-33 (Fig. 4e).
Enhancement of protein content by regulating nitrogen metabolism
Even though ALE solved the problem of low methanol utilization efficiency and intolerance to higher temperature, the protein content of the evolved strain HTX-33 remains low, which requires further regulation. Nitrogen metabolism has been shown to be critical for the catabolism and anabolism of proteins. In yeast, nitrogenous substances for amino acid biosynthesis are mainly converted from glutamate and glutamine. Amino nitrogen from glutamate and amide from glutamine account for 85% and 15% of total cellular nitrogen, respectively [38]. Glutamate is synthesized mainly by a reaction between-ketoglutarate and ammonia, which is catalyzed by NADPH-dependent glutamate dehydrogenase (GDH1) [39], and glutamate could further be used to synthesize glutamine with ammonia, which is catalyzed by glutamine synthase (GLN1) [40, 41]. Glutamate and glutamine, together with α-ketoglutarate, link the TCA cycle and nitrogen metabolism [42]. This study attempted to improve the protein content of strain HTX-33 by regulating nitrogen metabolism through overexpression of GDH1 and GLN1. It was found that overexpression of GDH1 in HTX-33 reduced final biomass to 4.49 (Fig. 5a), while the total nitrogen and protein content was increased to 0.063 and 0.394 (representing 8% increase compared with X-33 at 30 °C) (Table 2), respectively, when cultured in 0.5% methanol minimal medium at 33 °C. Notably, overexpression of GLN1 in HTX-33 increased total nitrogen to 0.066 and protein content to 0.414 (representing 14% increase compared with X-33 at 30 °C) (Table 2), respectively, without reducing final biomass (Fig. 5a). However, simultaneous overexpression of GDH1 and GLN1 resulted in a decrease in final biomass (Fig. 5a), and the increase in total nitrogen and protein content did not have a significant advantage over overexpression of GLN1 alone (Table 2). The above results indicate that overexpression of GDH1 and GLN1 in the nitrogen metabolism pathway could increase the protein content in the evolved strain HTX-33 at 33 °C, and that the conversion of methanol to biomass remains the same as in HTX-33 with GLN1 overexpression alone.

Effects of regulating nitrogen metabolism and engineering cell wall on cell growth of P. pastoris in 0.5% methanol minimal medium at 33 °C. a, Effects of overexpression of GDH1 and GLN1 on cell growth of strain HTX-33; b, Effect of deletion of PAS_chr4_0305 on cell growth of strain HTX-33; c, Effect of deletion of PAS_chr4_0305 on cell growth of strain HTX-33-GLN1. Data are average values and standard deviations of triplicate experiments
Enhancement of protein content by impairing cell wall synthesis
In a previous study, we discovered that as methanol concentration increased (from 0.5 to 3%), the cell wall thickened, resulting in a significant decrease in protein content in P. pastoris. Further RNA-Seq results showed that the upregulation of the gene PAS_chr4_0305, encoding the protein involved in O-glycosylation required for cell wall stability, predominated in promoting cell wall synthesis in response to the increase in methanol concentration (data not shown). These results led us to infer that impairing cell wall synthesis by knocking out the gene PAS_chr4_0305 may be an efficient engineering strategy to increase the protein content of P. pastoris. Here, deletion of gene PAS_chr4_0305 was performed in the evolved strain HTX-33 at 33 °C. In response to this modification, the cell wall thickness was reduced (Fig. 6a), and correspondingly, the total nitrogen and the protein content were increased to 0.071 and 0.444, respectively, which increased by 22% compared with X-33 at 30 °C (Table 2); however, the final biomass was slightly reduced (Fig. 5b). These results indicate that although the inactivation of gene PAS_chr4_0305 increased the sensitivity of the strain to higher temperature, resulting in a slight decrease in the conversion of methanol to biomass at 33 °C, the disruption of cell wall synthesis could still improve the protein content of evolved strain HTX-33 under this culture condition.

Effects of deletion of PAS_chr4_0305 on the cell wall thickness of P. pastoris in 0.5% methanol minimal medium at 33 °C. a, Representative TEM images after deletion of PAS_chr4_0305 in strain HTX-33; b, Representative TEM images after deletion of PAS_chr4_0305 in strain HTX-33-GLN1. Representative images are from three independent experiments
Further, the gene PAS_chr4_0305 was deleted in strain HTX-33-GLN1, resulting in strain HTX-33-GLN1-ΔPAS_chr4_0305. Likewise, the cell wall thickness was decreased (Fig. 6b), and correspondingly, the total nitrogen and protein content of strain HTX-33-GLN1-ΔPAS_chr4_0305 at 33 °C reached 0.073 and 0.456, respectively, which were 25% higher than those of the wild-type strain X-33 at 30 °C (Table 2). In addition, although the deletion of the gene PAS_chr4_0305 reduced the conversion of methanol to biomass to a certain extent, the final biomass of the strain HTX-33-GLN1-ΔPAS_chr4_0305 could reach 4.54 at 33 °C, which was 1.36 times that of the wild-type strain X-33 at 30 °C (Figs. 2c and 5c). Thus, a P. pastoris strain HTX-33-GLN1-ΔPAS_chr4_0305 with high methanol utilization, tolerance to 33 °C and high protein synthesis was obtained through the combination of ALE, strengthening nitrogen metabolism and impairing cell wall synthesis.
Pilot-scale fed-batch fermentation
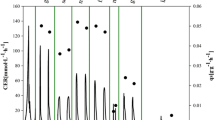
To test the industrial potential of our engineered strain to produce SCP from methanol at 33 °C, we performed fed-batch fermentation in a 500 L (the filling volume was 300 L) bioreactor using the best strain HTX-33-GLN1-ΔPAS_chr4_0305. Seed liquor was obtained by fed-batch fermentation with 2% glycerol in the initial batch stage. The methanol feed started at 33 h, the methanol induction stage was at 33 to 35 h, and the fermentation stage was at 35 to 72 h. The bioreactor process stopped at 72 h due to the large amount of heat generated during P. pastoris growth. In the fermentation process, the highest levels of biomass (DCW) reached 63.37 g/L with a methanol conversion rate of 0.43 g DCW/g, amounting to 86% of the maximum theoretical yield (0.5 g DCW/g). Interestingly, the total nitrogen and the protein content from bioreactor-cultured cells reached 0.081 g/g DCW and 0.506 g/g DCW, respectively, which were much higher than those of well plate cells due to the more abundant supply of oxygen in the bioreactor (Fig. 7). Based on the above data and the cost accounting (Supplementary Table 5), the conversion rate of methanol to protein can be converted to 0.22 g/g and the cost of SCP was caculated as 29.24 CNY/kg DCW in the bioreactor. All the above findings indicate the outstanding potential of our engineered strain for economical industrial application using methanol as a feedstock for SCP production at 33 °C.

Pilot-scale fed-batch fermentation of strain HTX-33-GLN1-ΔPAS_chr4_0305 in bioreactor. a, Changes in biomass and methanol consumption. Pilot-scale fed-batch fermentation was performed in a 500 L automatic bioreactor system with 300 L working volume. Temperature and pH were set at 33 °C and 5.6, respectively. Fed-batch fermentation was carried out with 2% glycerol in the initial batch stage. Methanol feeding was started at 33 h, achieving the maximum biomass at 72 h; b, The total nitrogen, the protein content and the conversion rate of methanol to biomass of strain HTX-33-GLN1-ΔPAS_chr4_0305 in the bioreactor; c, The pilot-scale bioreactor used in this experiment; d, P. pastoris SCP sample after centrifugation, washing and drying. Data are average values and standard deviations of triplicate experiments
Nutritional benefits of SCP from P. pastoris
In addition to protein content, the amino acid composition is also a key determinant of SCP quality [2]. In this study, the amino acids profile of SCP produced by strain HTX-33-GLN1-ΔPAS_chr4_0305 cultured in the bioreactor were quantified. As shown in Fig. 8, a total of 17 amino acids were detected, accounting for 41.78% of DCW, of which 21.74% were essential amino acids, including 2.38% isoleucine, 3.49% leucine, 3.5% lysine, 1.25% methionine, 2.89% phenylalanine, 2.36% threonine, 2.31% arginine, 2.16% valine and 1.4% histidine. It should be noting that SCP from P. pastoris is a source of the limiting amino acids lysine and methionine, of which the former is deficient in cereals, and the latter is relatively deficient in soy, peanut, milk and meat proteins [2, 43,44,45], suggesting that SCP from P. pastoris could be mixed to obtain a more nutritive food. Another interesting finding is that the total amount of branched-chain amino acids (BCAAs: valine, isoleucine, leucine) is up to 8.03%. BCAAs are important energy source in the body, which could increase ATP production by promoting glucose uptake [46]. BCAAs are also involved in the regulation of body lipid metabolism, protein synthesis and immune response etc., [47, 48]. Thus, SCP from P. pastoris is also a source of BCAAs supplements in low-protein foods. In summary, SCP from P. pastoris (50.6% in this report) generally contains a higher percentage of protein compared to soy (38.6%), fish (17.8%), meat (21.2%) and whole milk (3.28%) [2]. SCP from P. pastoris is a source of essential amino acids, including methionine, lysine, and BCAAs.

The amino acids profile of SCP produced by strain HTX-33-GLN1-ΔPAS_chr4_0305 cultured in the bioreactor. Ile, isoleucine; Leu, leucine; Lys, lysine; Met, methionine; Phe, phenylalanine; Thr, threonine; Arg, arginine; Val, valine; His, histidine; Asp, aspartic acid; Ser, serine; Glu, glutamic acid; Gly, glycine; Ala, alanine; Cys, cystine acid; Tyr, tyrosine; Pro, proline. Data are average values and standard deviations of triplicate experiments
Discussion
SCP biosynthesis from P. pastoris using C1 feedstock offers an attractive alternative to animal-derived proteins due to its rapid production rate, lower space requirements, independence of climate or seasons and more sustainable production process [1, 2, 8]. Nowadays, some companies (Such as Calysta, DeepBranch etc.) have successfully produced SCP by CO2 and methane, but most of these processes are anaerobic fermentation, which is inefficient and ultimately low in biomass. On the contrary, the production of SCP by P. pastoris is an aerobic fermentation process with high production efficiency and high biomass. Methanol is an ideal C1 feedstock for biomanufacturing due to its high reduction potential and extensive production sources [12,13,14]. At present, the methanol production by CO2 and hydrogen has been widely recognized [49]. The cost of the methanol production via CO2 and hydrogen is determined by the cost of electricity. As electricity gets cheaper due to the development of renewable energy, the methanol is also getting cheaper. In order to achieve the carbon neutrality goal for the entire planet, large-scale biomanufacturing based on methanol may contribute to the creation of process chains leading to SCP with a (nearly) zero CO2 footprint. However, the complexity of methanol metabolism pathway and the toxicity of intracellular formaldehyde hinder the efficient utilization of methanol in P. pastoris [27, 28]. Improving methanol utilization is of great significance to the economical industrial application using methanol as a feedstock for SCP production. It is true that using P. pastoris to SCP production at higher temperature could be not only adapt to the industrial environment in summer, but could also decrease the cooling water consumption. However, the cell wall will be strengthened in response to higher temperature stress [37], which is not conducive to the synthesis of protein in P. pastoris as found in this study. After all, the nutritional value of SCP is closely related to the protein content of P. pastoris, a P. pastoris strain with high-protein content will be more competitive with other protein sources [2]. Furthermore, higher temperature could inhibit the cell growth of P. pastoris in methanol minimal medium. Our preliminary research shows that P. pastoris can grow well at 28 and 30 °C. The cell growth was lightly repressed at 33 °C but strongly repressed at 35 °C. Notly, P. pastoris can not grow at 37 °C in 0.5% methanol minimal medium (Supplementary Fig. 1) [50]. In consideration of both biomass and protein content, we finally choose a higher temperature of 33 °C as an evolution pressure. Using P. pastoris to SCP production at 33 °C still have the potential in reducing production costs. In this study, the optimal evolved strain HTX-33 achieved the highest final biomass in 0.5% methanol minimal medium cultured at 33 °C, representing 1.43 times that of the parent strain X-33 cultured at 30 °C, indicating that both the low methanol utilization efficiency and the inability to tolerate higher temperature were solved through adaptive evolution.
Furthermore, transcriptomic analysis revealed that the majority of genes involved in methanol metabolism, such as alcohol oxidase genes AOX1 and AOX2, formaldehyde assimilation key genes DAS1 and DAS2, and formaldehyde dissimilation genes Fld1, Fgh1 and Fdh1, were all significantly downregulated in the evolved strain HTX-33. A long-term evolution of strain HTX-33 resulted in a reduced accumulation of intracellular formaldehyde. In fact, we performed genetic sequencing of all DEGs involved in methanol metabolism and discovered that only the 1257th base of AOX1 was mutated (base C to base A), resulting in the 419th amino acid of AOX1 changing from F to L (date not shown). AOX1 is the first enzyme in the methanol utilization pathway, so it is not surprising that this key enzyme represents a potential selective target during the adaptation towards growth on methanol. The same mutation was also found in a previous report that long-term adaptation resulted in the selection of clones with mutations in the AOX1 gene, which subsequently reduced alcohol oxidase activity and increased growth rates as compared with the ancestor [35]. Until now, the relationship among AOX1 activity, intracellular formaldehyde toxicity and methanol utilization efficiency has not been clearly elucidated. By combining the results of this study with previous findings, we concluded that reduced AOX1 activity decreased intracellular formaldehyde accumulation, which ultimately resulted in a reduction in carbon loss. Taken together, although the methanol and central carbon metabolism were slowed down after long-term evolution, the reduced carbon loss through the dissimilation pathway ultimately promoted the conversion of methanol to biomass in the evolved strain HTX-33. In addition, our results also revealed the effects of higher temperature on inducing cell wall stress. Consistent with previous findings [37], the cell wall thickening was observed after adaptive evolution at 33 °C, this may be a direct result of up-regulation of genes involved in cell wall synthesis. According to these results, P. pastoris cells require the ability to induce cell wall remodeling to withstand the stress associated with higher temperature during fermentation.
Besides being able to withstand higher temperature and reduce carbon loss, a P. pastoris strain with high protein content will necessarily be able to produce methanol-based SCP at a competitive level [2]. This study identified that overexpression of GDH1 or GLN1 in the nitrogen metabolism pathway could promote protein content in the evolved strain HTX-33 at 33 °C. Overexpression of GDH1 increased the protein content at the expense of reduced biomass, which may be a consequence of the disruption of the reducing equivalent of NADPH following overexpression of NADPH-dependent GDH1. Fortunately, overexpression of GLN1 increased protein content without reducing biomass. However, simultaneous overexpression of GDH1 and GLN1 did not have significant advantage over overexpression of GLN1 alone in terms of cell biomass and protein content in evolved strain HTX-33 at 33 °C. Knowledge gained from P. pastoris nitrogen metabolism pathway engineering indicates that overexpressing glutamine synthase GLN1 is a potential strategy to increase protein content of P. pastoris in methanol medium. So far, there was few studies adapted P. pastoris cell wall engineering for enhancing protein content. In yeast, most of the cell wall components are polysaccharides, and only a very small part is protein [36]. Consequently, it is reasonable to infer that impairing cell wall synthesis could be an effective strategy to reduce the C/N ratio, resulting in increase in protein content of P. pastoris. In this study, PAS_chr4_0305, a gene encoding the O-glycosylation related protein of cell wall, was found to be a key target for improving the protein content of P. pastoris on methanol as the sole carbon source. Although the knockout of gene PAS_chr4_0305 increased the sensitivity of P. pastoris to higher temperature, the cell wall engineering by deletion of gene PAS_chr4_0305 still increased the protein content of the evolved strain HTX-33 at 33 °C.
Finally, both overexpression of GLN1 and knockout of gene PAS_chr4_0305 were implemented into the evolved strain HTX-33, creating the best strain HTX-33-GLN1-ΔPAS_chr4_0305, which achieved high-level production of SCP from sole methanol with a biomass of 63.37 g DCW/L, methanol conversion rate of 0.43 g DCW/g, and protein content of 0.506 g/g DCW in pilot-scale fed-batch culture at 33 °C. Indeed, the Phillips Petroleum Company has reported the 130 g/L of P. pastoris biomass on methanol during the 1970s. However, this high biomass was obtained based on the utilization of high-cell density cultivation method [29]. As we all known, the P. pastoris cells will grow slowly and most of methanol will be dissipated rather than utilized by the end of fermentation under high-density culture mode, which is not conducive to the economic production of SCP from methanol. Our research focuses on creating a P. pastoris strain with high methanol utilization, higher temperature resistance and high protein content, showing outstanding potential for economical industrial application using methanol as a feedstock for SCP production in the continuous fermentation mode. In this mode, we can continuously release the fermentation broth and feed at the stage of the highest methanol conversion rate, thus achieving continuous production of SCP without impairing the specific growth rate of P. pastoris strain. The protein content of SCP from P. pastoris reported here (50.6%) is much higher than that of soy (38.6%), fish (17.8%), meat (21.2%) and whole milk (3.28%) [2, 51]. In addition, compared with other SCP from low-value raw materials (Table 3), the protein content of SCP from P. pastoris reported here was almost equivalent to that of other fungi (11–63%) [52,53,54,55,56] and algae (42-64.9%) [5, 44, 57, 58], and slightly inferior to that of bacteria (41–83%) [59,60,61,62]. Further amino acid analysis showed that SCP from P. pastoris is a source of limiting amino acids, including lysine, methionine, and BCAAs supplements, indicating that SCP from P. pastoris could be mixed to obtain a more nutritive food. Future research could focus on fine-tuning the ratio of certain amino acids to increase the nutritional value of SCP from P. pastoris.
Conclusions
In summary, a P. pastoris strain with high methanol utilization efficiency, tolerance to 33 °C and high protein content was obtained through a combination of ALE, strengthening nitrogen metabolism and engineering the cell wall, showing outstanding potential for economical industrial application using methanol as a substrate for SCP production. SCP obtained from P. pastoris can have a higher protein content compared to conventional foods like soy, meat, milk and fish, and is a source of essential amino acids. SCP from P. pastoris using C1 feedstock is a promising alternative to obtain protein resource with environmental, economic and nutritional benefits, which is expected to become a sustainable production mode in the future.
Data Availability
The data supporting the conclusions of this article are all available in the manuscript and supplementary.
Abbreviations
- C1:
-
One-carbon
- SCP:
-
Single cell protein
- ALE:
-
Adaptive laboratory evolution
- DCW:
-
Dry cell weight
- BCAAs:
-
Branched-chain amino acids
- GRAS:
-
Generally recognized as safe
- DEGs:
-
Differentially expressed genes
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- SEM:
-
Scanning electron microscopy
- TEM:
-
Transmission electron microscopy
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
- TCA:
-
Reductive tricarboxylic acid
- PPP:
-
Pentose phosphate pathway
- EMP:
-
Glycolysis pathway
- Aox:
-
Alcohol oxidase
- Fld1:
-
Formaldehyde dehydrogenase
- Fgh1:
-
S-formylglutathione hydrolase
- Fdh1:
-
Formate dehydrogenase
- Das:
-
Dihydroxyacetone synthase
- Dhak:
-
Dihydroxyacetone kinase
- Fba:
-
Fructosebisphosphate aldolase/sedoheptulose-bisphosphate aldolase
- Fbp:
-
Fructose bisphosphatase
- Sbp:
-
Sedoheptulose bisphosphatase
- Rpe:
-
Ribulose-phosphate 3-epimerase
- RpiA:
-
Ribose-5-phosphate isomerase
- Xu5P:
-
Xylulose-5-phosphate
- Ru5P:
-
Ribulose-5-phosphate
- R5P:
-
Ribose-5-phosphate
- DHA:
-
Dihydroxyacetone
- DHAP:
-
Dihydroxyacetone phosphate
- F16dP:
-
Fructose-1,6-bisphosphate
- F6P:
-
Fructose-6-phosphate
- G3P:
-
Glyceraldehyde-3-phosphate
- E4P:
-
Erythrose-4-phosphate
- S7P:
-
Sedoheptulose-7-phosphate
- S17dP:
-
Sedoheptulose-1,7-bisphosphate
- Ile:
-
Isoleucine
- Leu:
-
Leucine
- Lys:
-
Lysine
- Met:
-
Methionine
- Phe:
-
Phenylalanine
- Thr:
-
Threonine
- Arg:
-
Arginine
- Val:
-
Valine
- His:
-
Histidine
- Asp:
-
Aspartic acid
- Ser:
-
Serine
- Glu:
-
Glutamic acid
- Gly:
-
Glycine
- Ala:
-
Alanine
- Cys:
-
Cystine acid
- Tyr:
-
Tyrosine
- Pro:
-
Proline
References
Ritala A, Hakkinen ST, Toivari M, Wiebe MG. Single cell protein-state-of-the-art, industrial landscape and patents 2001–2016. Front Microbiol. 2017;8:2009.
Salazar-Lopez NJ, Barco-Mendoza GA, Zuniga-Martinez BS, Dominguez-Avila JA, Robles-Sanchez RM, Ochoa MAV, Gonzalez-Aguilar GA. Single-cell protein production as a strategy to reincorporate food waste and agro by-products back into the processing chain. Bioeng. 2022;9:623.
Zhang L, Hu YY, Badar IH, Xia XF, Kong BH, Chen Q. Prospects of artificial meat: opportunities and challenges around consumer acceptance. Trends Food Sci Tech. 2021;116:434–44.
Szenderak J, Frona D, Rakos M. Consumer acceptance of plant-based meat substitutes: a narrative review. Foods. 2022;11:1274.
Waghmare AG, Salve MK, LeBlanc JG, Arya SS. Concentration and characterization of microalgae proteins from Chlorella pyrenoidosa. Bioresour Bioprocess. 2016;3:16.
Kurbanoglu EB, Algur OF. Single-cell protein production from ram horn hydrolysate by bacteria. Bioresour Technol. 2002;85:125–9.
Nasseri AT, Rasoul-Amini S, Morowvat MH, Ghasemi Y. Single cell protein: production and process. Am J Food Technol. 2011;6:103–16.
Overland M, Tauson AH, Shearer K, Skrede A. Evaluation of methane-utilising bacteria products as feed ingredients for monogastric animals. Arch Anim Nutr. 2010;64:171–89.
Cotton CA, Claassens NJ, Benito-Vaquerizo S, Bar-Even A. Renewable methanol and formate as microbial feedstocks. Curr Opin Biotechnol. 2020;62:168–80.
Gassler T, Sauer M, Gasser B, Egermeier M, Troyer C, Causon T, Hann S, Mattanovich D, Steiger MG. The industrial yeast Pichia pastoris is converted from a heterotroph into an autotroph capable of growth on CO2. Nat Biotechnol. 2020;38:210–7.
Chen FYH, Jung HW, Tsuei CY, Liao JC. Converting Escherichia coli to a synthetic methylotroph growing solely on methanol. Cell. 2020;182:933–46.
Kuk SK, Singh RK, Nam DH, Singh R, Lee JK, Park CB. Photoelectrochemical reduction of carbon dioxide to methanol through a highly efficient enzyme cascade. Angew Chem Int Ed Engl. 2017;56:3827–32.
Patel SKS, Jeon MS, Gupta RK, Jeon Y, Kalia VC, Kim SC, Cho BK, Kim DR, Lee JK. Hierarchical macroporous particles for efficient whole-cell immobilization: application in bioconversion of greenhouse gases to methanol. ACS Appl Mater Interface. 2019;11:18968–77.
Zhou YJ, Kerkhoven EJ, Nielsen J. Barriers and opportunities in bio-based production of hydrocarbons. Nat Energy. 2018;3:925–35.
Muller JEN, Meyer F, Litsanov B, Kiefer P, Potthoff E, Heux S, Quax WJ, Wendisch VF, Brautaset T, Portais JC, Vorholt JA. Engineering Escherichia coli for methanol conversion. Metab Eng. 2015;28:190–201.
Dai Z, Gu H, Zhang S, Xin FX, Zhang WM, Dong WL, Ma JF, Jia HH, Jiang M. Metabolic construction strategies for direct methanol utilization in Saccharomyces cerevisiae. Bioresour Technol. 2017;245:1407–12.
Tuyishime P, Wang Y, Fan LW, Zhang QQ, Li QG, Zheng P, Sun JB, Ma YH. Engineering Corynebacterium glutamicum for methanol dependent growth and glutamate production. Metab Eng. 2018;49:220–31.
Schwarzhans JP, Luttermann T, Geier M, Kalinowski J, Friehs K. Towards systems metabolic engineering in Pichia pastoris. Biotechnol Adv. 2017;35:681–710.
Zhu W, Gong GH, Pan J, Han S, Zhang W, Hu YJ, Xie LP. High level expression and purification of recombinant human serum albumin in Pichia pastoris. Protein Expr Purif. 2018;147:59–66.
De Schutter K, Lin YC, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouze P, Van de Peer Y, Callewaert N. Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol. 2009;27:561–6.
Valli M, Tatto NE, Peymann A, Gruber C, Landes N, Ekker H, Thallinger GG, Mattanovich D, Gasser B, Graf AB. Curation of the genome annotation of Pichia pastoris (Komagataella phaffii) CBS7435 from gene level to protein function. FEMS Yeast Res. 2016;16:fow051.
Cai P, Duan XP, Wu XY, Gao LH, Ye M, Zhou YJ. Recombination machinery engineering facilitates metabolic engineering of the industrial yeast Pichia pastoris. Nucleic Acids Res. 2021;49:7791–805.
Yamada R, Ogura K, Kimoto Y, Ogino H. Toward the construction of a technology platform for chemicals production from methanol: d-lactic acid production from methanol by an engineered yeast Pichia pastoris. World J Microbiol Biotechnol. 2019;35:37.
Jin XR, Zhang WJ, Wang Y, Sheng JY, Xu RR, Li JH, Du GC, Kang Z. Biosynthesis of non-animal chondroitin sulfate from methanol using genetically engineered Pichia pastoris. Green Chem. 2021;23:4365–74.
Guo F, Dai ZX, Peng WF, Zhang SJ, Zhou J, Ma JF, Dong WL, Xin FX, Zhang WM, Jiang M. Metabolic engineering of Pichia pastoris for malic acid production from methanol. Biotechnol Bioeng. 2021;118:357–71.
Cai P, Wu XY, Deng J, Gao LH, Shen YW, Yao L, Zhou YJ. 2022. Methanol biotransformation toward high-level production of fatty acid derivatives by engineering the industrial yeast Pichia pastoris. P Natl Acad Sci USA 119, e2201711119.
Guo YK, Liao Y, Wang J, Ma C, Qin JL, Feng J, Li Y, Wang X, Chen KQ. Methylotrophy of pichia pastoris: current advances, applications, and future perspectives for methanol-based biomanufacturing. ACS Sustainable Chem Eng. 2022;10:1741–52.
Gao JQ, Li YX, Yu W, Zhou YJ. Rescuing yeast from cell death enables overproduction of fatty acids from sole methanol. Nat Metab. 2022;4:932–43.
Cereghino JL, Cregg JM. Heterologous protein expression in the methylotropic yeast Pichia pastoris. FEMS Microbiol Rev. 1999;24(2000):45–66.
Abdel-Banat BM, Hoshida H, Ano A, Nonklang S, Akada R. High-temperature fermentation: how can processes for ethanol production at high temperatures become superior to the traditional process using mesophilic yeast? Appl Microbiol Biotechnol. 2010;85:861–7.
Zhou YJ, Buijs NA, Zhu Z, Qin J, Siewers V, Nielsen J. Production of fatty acid-derived oleochemicals and biofuels by synthetic yeast cell factories. Nat Commun. 2016;7:11709.
Woolston BM, Roth T, Kohale I, Liu DR, Stephanopoulos G. Development of a formaldehyde biosensor with application to synthetic methylotrophy. Biotechnol Bioeng. 2018;115:206–15.
Meng J, Wang BY, Liu DY, Tao C, Wang ZW, Zhao XM. High-yield anaerobic succinate production by strategically regulating multiple metabolic pathways based on stoichiometric maximum in Escherichia coli. Microb Cell Fact. 2016;15:141.
Marco A, Rubio R, Compano R, Casals I. Comparison of the Kjeldahl method and a combustion method for total nitrogen determination in animal feed. Talanta. 2002;57:1019–26.
Moser JW, Prielhofer R, Gerner SM, Graf AB, Wilson IBH, Mattanovich D, Dragosits M. Implications of evolutionary engineering for growth and recombinant protein production in methanol-based growth media in the yeast Pichia pastoris. Microb Cell Fact. 2017;16:49.
Klis FM, Boorsma A, De Groot PW. Cell wall construction in Saccharomyces cerevisiae. Yeast. 2006;23:185–202.
Kitichantaropas Y, Boonchird C, Sugiyama M, Kaneko Y, Harashima S, Auesukaree C. Cellular mechanisms contributing to multiple stress tolerance in Saccharomyces cerevisiae strains with potential use in high-temperature ethanol fermentation. AMB Express. 2016;6:107.
Magasanik B, Kaiser CA. Nitrogen regulation in Saccharomyces cerevisiae. Gene. 2002;290:1–18.
Grenson M, Dubois E, Piotrowska M, Drillien R, Aigle M. Ammonia assimilation in Saccharomyces cerevisiae as mediated by the two glutamate dehydrogenases. Evidence for the gdhA locus being a structural gene for the NADP-dependent glutamate dehydrogenase. Mol Gen Genet. 1974;128:73–85.
Mitchell AP, Magasanik B. Purification and properties of glutamine synthetase from Saccharomyces cerevisiae. J Biol Chem. 1983;258:119–24.
Mitchell AP. The GLN1 locus of Saccharomyces cerevisiae encodes glutamine synthetase. Genet. 1985;111:243–58.
Zhang WP, Du GC, Zhou JW, Chen J. 2018. Regulation of sensing, transportation, and catabolism of nitrogen sources in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 82, e00040.
Pires JF, Ferreira GMR, Reis KC, Schwan RF, Silva CF. Mixed yeasts inocula for simultaneous production of SCP and treatment of vinasse to reduce soil and fresh water pollution. J Environ Manag. 2016;182:455–63.
Yap SM, Lan JCW, Kee PE, Ng HS, Yim HS. Enhancement of protein production using synthetic brewery wastewater by Haematococcus pluvialis. J Biotechnol. 2022;350:1–10.
FAO. Amino-Acid Content of Foods and Biological Data on Proteins. Rome, Italy: FAO; 1981. ISBN 92-5-001102-4.
Iwai S, Hasegawa T, Ikeda HO, Tsujikawa A. Branched chain amino acids promote ATP production via translocation of glucose transporters. Invest Ophth Vis Sci. 2022;63:7.
Zhang LY, Li FN, Guo QP, Duan YH, Wang WL, Yang YH, Yin YJ, Gong SM, Han MM, Yin YL. Different proportions of branched-chain amino acids modulate lipid metabolism in a finishing pig model. J Agric Food Chem. 2021;69:7037–48.
Ahmad I, Ahmed I, Fatma S, Peres H. Role of branched-chain amino acids on growth, physiology and metabolism of different fish species: a review. Aquacult Nutr. 2021;27:1270–89.
Shih CF, Zhang T, Li J, Bai C. 2018. Powering the future with liquid sunshine. Joule. 2018, 2(10).
Han MH, Wang WX, Gong X, Zhou JL, Xu CB, Li YF. Increased expression of recombinant chitosanase by co-expression of hac1p in the yeast pichia pastoris. Protein Pept Lett. 2021;28:1434–41.
USDA. FoodData Central. Available online: https://fdc.nal.usda.gov/index.html (accessed on 1 September 2022).
Valentino MJG, Ganado LS, Undan JR. Single cell protein potential of endophytic fungi associated with bamboo using rice bran as substrate. Adv Appl Sci Res. 2016;7:68–72.
Tropea A, Ferracane A, Albergamo A, Potortì AG, Lo Turco V, Di Bella G. Single cell protein production through multi food-waste substrate fermentation. Fermentation. 2022;8(3):91.
Khan MKI, Asif M, Razzaq ZU, Nazir A, Maan AA. Sustainable food industrial waste management through single cell protein production and characterization of protein enriched bread. Food Biosci. 2022;46:101406.
Chiou PWS, Chiu SW, Chen CR. Value of Aspergillus niger fermentation product as a dietary ingredient for broiler chickens. Anim Feed Sci Technol. 2001;91:171–82.
Ahmadi AR, Ghoorchian H, Hajihosaini R, Khanifar J. Detremination of the amount of protein and amino acids extracted from the microbial protein (SCP) of lignocellulosic wastes. Pakistan J Biol Sci. 2010;13:355–61.
Zepka LQ, Jacob-Lopez E, Goldbeck R, Souza-Soares LA, Queiroz MI. Nutritional evaluation of single-cell protein produced by Aphanothece microscopica Nägeli. Bioresour Technol. 2010;101:7107–11.
Putri D, Ulhidayati A, Musthofa IA, Wardani AK. Single cell protein production of Chlorella sp using food processing waste as a cultivation medium. IOP Conf Ser Earth Environ Sci. 2018;131:012052.
Zha X, Tsapekos P, Zhu XY, Khoshnevisan B, Lu XW, Angelidaki I. Bioconversion of wastewater to single cell protein by methanotrophic bacteria. Bioresour Technol. 2021;320:124351.
Jiang YF, Yang XY, Zeng DF, Su YY, Zhang YF. Microbial conversion of syngas to single cell protein: the role of carbon monoxide. Chem Eng J. 2022;450:138041.
Saejung C, Salasook P. Recycling of sugar industry wastewater for single-cell protein production with supplemental carotenoids. Environ Technol. 2020;41:59–70.
Lin XF, Zou FQ, Mo ZP, Song QK, Tong SH, Li CY. The production of bacterial proteins from ethanol mash by fermentation of iron and steel industry tail gas. Chem Enterp Manage. 2022;11:143–5.
Acknowledgements
None.
Funding
This work was supported by the National key R&D program of China (2021YFD1301002), Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-CXRC-031) and China Postdoctoral Science Foundation-funded project (2021M703439).
Author information
Authors and Affiliations
Contributions
M.J. and L.S.F. performed the experiments under the guidance of W.X. M.J., L.S.F., and G.L. analyzed the experimental data and drafted the manuscript. G.L. and H.K. made substantial contributions to conception, interpretation of data and revised the manuscript. W.X., M.J., and L.S.G. developed the idea for the study and designed the research. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Meng, J., Liu, S., Gao, L. et al. Economical production of Pichia pastoris single cell protein from methanol at industrial pilot scale. Microb Cell Fact 22, 198 (2023). https://doi.org/10.1186/s12934-023-02198-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-023-02198-9