
Abstract
Escherichia coli is a useful platform for producing valuable materials through the implementation of synthetic gene(s) derived from other organisms. The production of lactate (LA)-based polyester poly[LA-co-3-hydroxybutyrate (3HB)] was carried out in E. coli using a set of five other species-derived genes: Pseudomonas sp. 61-3-derived phaC1STQK (for polymerization), Cupriavidus necator-derived phaAB (for 3HB-CoA generation), and Megasphaera elsdenii-derived pct (for LA-CoA generation) cloned into pTV118NpctphaC1ps(ST/QK)AB. Here, we aimed to optimize the expression level and timing of these genes to improve the production of P(LA-co-3HB) and to manipulate the LA fraction by replacing the promoters with various promoters in E. coli. Evaluation of the effects of 21 promoter replacement plasmids revealed that the phaC1STQK-AB operon is critical for the stationary phase for P(LA-co-3HB) production. Interestingly, the effects of the promoters depended on the composition of the medium. In glucose-supplemented LB medium, the dps promoter replacement plasmid resulted in the greatest effect, increasing the accumulation to 8.8 g/L and an LA fraction of 14.1 mol% of P(LA-co-3HB), compared to 2.7 g/L and 8.1 mol% with the original plasmid. In xylose-supplemented LB medium, the yliH promoter replacement plasmid resulted in the greatest effect, with production of 5.6 g/L and an LA fraction of 40.2 mol% compared to 3.6 g/L and 22.6 mol% with the original plasmid. These results suggest that the selection of an appropriate promoter for expression of the phaC1STQK-AB operon could improve the production and LA fraction of P(LA-co-3HB). Here, we propose that the selection of cell-growth phase-dependent promoters is a versatile biotechnological strategy for effective intracellular production of polymeric materials such as P(LA-co-3HB), in combination with the selection of sugar-based carbon sources.
Similar content being viewed by others

Introduction
Plastics derived from renewable biomass are of interest because they are a potential solution to dwindling and increasingly expensive petroleum resources and have the capacity to reduce carbon dioxide emissions. Polylactide (PLA), a representative bio-based plastic, can be chemically synthesized in multiple steps from renewable carbon sources. PLA has a wide range of applications, such as biomedical materials and food-related items. However, the stiffness and brittleness of PLA limit its competitive market expansion. In contrast, P(lactate-co-3-hydroxybutyrate) [P(LA-co-3HB)], an LA-based polyester copolymerized with 3HB, which is a typical constituent of polyhydroxyalkanoates (PHAs), can be processed into flexible and transparent plastic materials and can be used in a broad range of applications (Fig. 1) [29, 30]. Such copolymers are known to exhibit various mechanical and thermal properties depending on their monomer fraction, and thus can be used in a wide range of applications [24, 28]. In addition, P(LA-co-3HB) is likely to have certain beneficial properties performed by both homopolymers, such as the high glass transition temperature of PLA and the excellent biodegradability of P(3HB) [23, 30]. Especially, even the highly LA-enriched P(LA-co-3HB) exhibits excellent biodegradability in soil environments, and its biodegradation mechanism has been investigated from the viewpoint of enzymes and polymer products [4, 25, 26].

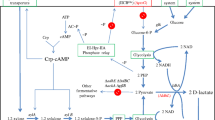
Biosynthetic pathway scheme of P(LA-co-3HB) production in E. coli used in this study. Two sugars, glucose and xylose, derived from biomass were used as carbon sources for production of P(LA-co-3HB) in Escherichia coli. LDH: lactate dehydrogenase; PCT: propionyl-CoA transferase; LPE: lactate-polymerizing enzyme [29] engineered from PHA synthase of Pseudomonas sp. 61-3; PhaA: β-ketothiolase; PhaB: NADPH-dependent acetoacetyl-CoA reductase
In 2008, Taguchi et al. succeeded in creating a microbial biosynthetic system for P(LA-co-3HB) using Escherichia coli as a platform (Fig. 1) [29]. In this system, the discovery of an “LA-polymerizing enzyme (LPE),” which is a mutant of PHA synthase PhaC1S325T/Q481K(PhaC1STQK), originally from Pseudomonas sp. 61-3, enabled us to construct a one-step biosynthetic system for LA-based polyesters under mild conditions. P(LA-co-3HB) is intracellularly synthesized by the following successive enzymatic reaction steps: (i) generation of lactyl-coenzyme A (LA-CoA) by propionyl-CoA transferase (PCT) derived from Megasphaera elsdenii, (ii) supply of 3-hydroxybutyryl-CoA (3HB-CoA) via the dimerization pathway (PhaA and PhaB) derived from Cupriavidus necator, and (iii) copolymerization of the CoA esters by the LPE. Currently, these sets of genes for the P(LA-co-3HB) production are cloned into a plasmid called ‘pTV118NpctphaC1ps(ST/QK)AB’, and E. coli transformants are used. With this plasmid, the phaC1STQK-phaA-phaB operon is driven by the phaC promoter of C. necator, and pct is driven by the lac promoter of E. coli.
As described above, P(LA-co-3HB) is a copolymer that can have a wide range of properties owing to the LA fraction, which must be produced by an enzymatic reaction for the polymerization of both LA and 3HB, and there is a need to improve not only the production volume but also alteration of the LA fraction. The following various efforts have been made regarding P(LA-co-3HB) production in E. coli as a platform; the nature of the carbon source of the medium [5, 14, 16, 17, 21, 27], enzymatic modification of enzymes involved in P(LA-co-3HB) synthesis [30], deletion or overexpression of carbon source metabolism genes and sugar transporter genes [15, 31], modification of the transcriptional regulatory network by deletion of transcriptional regulators [9, 10, 12], and cell membrane flexibility [11].
In addition, we have accumulated extensive fundamental findings related to the transcription network governing global gene expression in E. coli [7]. In particular, we have developed a Genomic SELEX system to elucidate the genomic regulatory networks of transcription factors based on the identification of the binding sites in vitro of the test transcription factor on the E. coli genome [20]. These long-term studies are logically applicable to the microbial production of value-added products. In general, cell growth phase-separated regulation is appropriate for efficient production of the target product of interest. In particular, caution should be exercised with intracellular biosynthesis of polymeric materials, such as hydrophobic PHA polymers, to avoid cell growth inhibition caused by artificial compound production [3]. Recently, it was reported that membrane vesicles are produced by envelope stress caused by the accumulation of PHB [13]. However, the selection of appropriate promoters for efficient PHA production has not been reported to date.
In the present study, we investigated the effectiveness of promoter selection for increased productivity and altered LA fraction of P(LA-co-3HB), considering the cell growth phase of the recombinant E. coli strain used. In general, the intercellular synthesis rate of a target compound is affected not only by the activity of the enzyme itself but also by its expression level. Although genetic modifications of E. coli as a host strain, carbon sources, and culture conditions have been studied, it might be argued that the plasmid pTV118NpctphaC1ps(ST/QK)AB has been conventionally used for P(LA-co-3HB) production. First, we investigated the expression status of the P(LA-co-3HB)-synthesizing genes cloned into the plasmid, and we then attempted to alter P(LA-co-3HB) production and the LA fraction by optimizing the expression system through promoter replacement of a set of genes involved in P(LA-co-3HB) synthesis in the plasmid. As expected, improved production of P(LA-co-3HB) and altered LA fraction were achieved using a promoter replacement-based approach in combination with the selection of sugar-based carbon sources.
Results and discussion
Correlation of P(LA-co-3HB) production pattern with phaC1STQK-AB expression pattern by use of the conventional plasmid pTV118NpctphaC1ps(ST/QK)AB
P(LA-co-3HB) was produced by culturing E. coli harboring the conventional pTV118NpctphaC1ps(ST/QK)AB plasmid in glucose-supplemented LB medium. To determine the adequate amount of glucose in the medium for P(LA-co-3HB) production, the production volume and LA fraction of P(LA-co-3HB) by pTV118NpctphaC1ps(ST/QK)AB transformants in LB medium with various glucose concentrations were measured. It was found that a glucose concentration of 3% (w/v) was most suitable for P(LA-co-3HB) production (Additional file 1: Fig. S1), and a sugar concentration of 3% was fixed in the subsequent experiments.
The production and LA fraction of P(LA-co-3HB) in E. coli harboring pTV118NpctphaC1ps(ST/QK)AB under these conditions were measured over 48 h. P(LA-co-3HB) was measured by two methods: GC-FID was used to quantify the P(LA-co-3HB) copolymer in terms of the amount of 3HB and LA in dried cell weight %, and Nile-red staining was used as a semi-quantative measure of the total amount of P(LA-co-3HB). From the GC-FID results, accumulation of P(LA-co-3HB) was observed after 9 h of cultivation, followed by a gradual increase, reaching 2.7 g/L and an LA fraction of approximately 7.4 mol% after 48 h (Fig. 2A). This result was consistent with the quantification by Nile-red staining. Accumulation of P(LA-co-3HB) did not change with further incubation (data not shown). Next, the cell density under culture conditions was measured, and cell growth started to slow at 9 h and transitioned to the stationary phase by 12 h after inoculation (Fig. 2B). These results suggest that, under these conditions, the production of P(LA-co-3HB) begins with the transition of cells into the stationary phase.

P(LA-co-3HB) production and expression levels of phaC1STQK-AB operon over the course of cultivation time by E. coli harboring pTV118NpctphaC1ps(ST/QK)AB in glucose-supplemented LB medium. A The bars indicate the amount of 3HB units in the polymer (black), the amount of LA units in the polymer (white), and the fluorescent intensities of Nile-red-stained cells (gray). B Diagonal stripe bars indicate the mRNA levels of phaC. The line graph shows cell growth
In contrast, the mRNA expression of the phaC1STQK-AB operon responsible for P(LA-co-3HB) production was measured under the same conditions and was found to increase from the exponential growth phase to the early stationary phase, followed by a sharp decrease (Fig. 2B). This result indicates that the phaC promoter of C. necator functions as an exponential phase-dependent promoter in E. coli that is active during the exponential phase and inactive during the stationary phase. These results suggest that the enzymes involved in P(LA-co-3HB) synthesis expressed in the exponential phase contribute little to P(LA-co-3HB) production during the exponential phase. It is also suggested that stationary phase P(LA-co-3HB) production, which is responsible for the main production of P(LA-co-3HB) accumulation, may be carried out by enzymes expressed in the exponential phase by an exponential phase-dependent promoter. This imbalanced correlation between the promoter-mediated gene expression responsible for polymer synthesis and the P(LA-co-3HB) production phase was clearly identified as an issue in need of further improvement.
Effect of promoter replacement of the phaC1STQK-AB operon for 3HB-CoA synthesis and copolymerization
Since the production of P(LA-co-3HB) occurs mostly after the transition to the stationary phase, it was thought that the expression of genes involved in the production of P(LA-co-3HB) should also transition into the stationary phase. Previously, we comprehensively measured the activities and properties of E. coli stationary-phase promoter by cloning it into a multi-copy plasmid vector and fusing it to a gfp reporter gene [18]. Based on these results, we selected 18 stationary-phase-inducible promoters, including those with weak to strong activities, and three constitutive promoters for comparison, and we constructed 21 promoter replacement plasmids instead of the existing phaC promoter in pTV118NpctphaC1ps(ST/QK)AB. Each of these plasmids was transformed into the E. coli BW25113 strain, and the effect of the promoter replacement on P(LA-co-3HB) accumulation was assessed by Nile-red staining of cells 48 h after inoculation (Fig. 3, Table 1). Different amounts of P(LA-co-3HB) accumulation were observed for each promoter, and a 1.5- to 2.3-fold increase in production was observed for the stationary-phase-inducible promoters dps, sodC, yliH, gadB, and treA relative to the original phaC promoter. All these promoters have been reported to be dependent on the stationary-phase sigma factor RpoS, which recognizes stationary-phase promoters [18]. In contrast, for the constitutive promoters glpD, modA, and uxuA, which have been shown to exhibit strong constitutive activity in previous reports, the production of P(LA-co-3HB) decreased below that of the original phaC promoter.

P(LA-co-3HB) production by replacement of the promoter fused to the phaC1STQK-AB operon. Each P(LA-co-3HB) production was measured by the fluorescent intensities of Nile-red staining. pTV indicates the conventional plasmid pTV118NpctphaC1ps(ST/QK)AB. The gene name indicates the promoter replaced with the phaC promoter
To confirm the correlation between the activity of these 18 stationary-phase-inducible promoters and the P(LA-co-3HB) accumulation amount, the mRNA expression levels of phaC expressed by each promoter at different incubation times (9, 12, 16, and 24 h) and P(LA-co-3HB) accumulation were compared. The results show that the expression level at 16 h post-incubation correlated best with the P(LA-co-3HB) accumulation (correlation coefficient = 0.700) and was lower at 9 h (0.454) (Fig. 4 and Additional file 1: Fig. S2). To determine the exact amount and LA fraction of P(LA-co-3HB) products, copolymers accumulated by promoter replacement plasmids with significantly increased production were measured. We obtained results showing > twofold increase in production relative to the original phaC promoter, with the dps promoter showing > threefold increase to 8.8 g/L (Table 1, Additional file 1: Fig. S3). In accordance with the effect of this promoter activity optimization, a suitable glucose concentration was re-examined using the yliH promoter as a representative example, and 3% glucose was found to be optimal (Additional file 1: Fig. S4). Replacing the phaC promoter upstream of the phaC1STQK-AB operon with a stationary-phase-inducible promoter successfully increased P(LA-co-3HB) production but had little effect on the LA fraction, which was approximately 12 mol% as opposed to 8 mol% for the original promoter (Table 1, Additional file 1: Fig. S3).

Correlation between P(LA-co-3HB) production and the mRNA levels of phaC. The x-axis shows the expression ratio of phaC mRNA to the ribosomal RNA, rrsA, quantified by RT-qPCR after 16 h of incubation. The y-axis shows the production of P(LA-co-3HB) as measured by Nile-red staining of cells after 48 h of incubation. The gene name of the promoter used for expression of the phaC1STQK-AB operon is indicated next to each dot
Effect of promoter replacement of pct for LA-CoA synthesis and ldhA for l-lactate synthesis
To increase the LA fraction of P(LA-co-3HB), the degree of LA-CoA polymerization in the copolymer needs to be increased. LA is converted from pyruvate by lactate dehydrogenase encoded by ldhA located in the E. coli genome, which is then converted to LA-CoA by propionyl-CoA transferase (PCT) encoded by pct driven by the lac promoter in the pTV118NpctphaC1ps(ST/QK)AB plasmid (Fig. 1) [29]. Plasmids were constructed in which the lac promoter upstream of pct in the plasmid was replaced with a stationary-phase-inducible promoter dps, treA, or yliH, which had a critical effect on the expression of phaC1STQK-AB operon. However, the results indicated that there was no significant effect on the amount of P(LA-co-3HB) or the LA fraction measured for the transformants harboring these plasmids (Fig. 5). This suggests that PCT activity was not the rate-limiting reaction for LA-CoA production under these experimental conditions.

P(LA-co-3HB) production by replacement of the promoter fused to pct. The bars indicate the amount of 3HB units in the polymer (black), the amount of LA units in the polymer (white), and the LA fraction (vertical stripe). pTV indicates the conventional plasmid pTV118NpctphaC1ps(ST/QK)AB. The gene name indicates the promoter replaced with the lac promoter located upstream of pct
Next, we cloned E. coli ldhA encoding lactate dehydrogenase into a plasmid to control the expression level of LdhA. During this process, we were able to clone the ORF of ldhA without a promoter. However, when we attempted to fuse certain promoters to the ldhA ORF, we noted nonsense mutations, frameshifts in the ldhA ORF sequence, or mutations in the promoter sequence in dozens of the clones obtained (data not shown). Similar results were obtained by supplementing the medium with alanine to alleviate the decrease in the amount of intracellular pyruvate due to lactate production by LdhA [1] as well as by using a buffer solution as the medium to alleviate the effect of lactate production on pH (data not shown). As a result, plasmids for LdhA expression were not obtained. Recently, it was reported that expression of ldhA induces persister formation in E. coli [32], which may explain why ldhA cannot be expressed on a multi-copy plasmid in E. coli.
Improvement of LA fraction in xylose medium using promoter replacement plasmids
Previously, Nduko et al. [14] demonstrated that using xylose as a sugar source in the medium led to an increase in the LA fraction of P(LA-co-3HB) by suppressing the production of NADPH, thereby lowering the supply of NADPH required for 3HB-CoA synthesis compared to the use of glucose. Next, we measured P(LA-co-3HB) production and the LA fraction in xylose-supplemented LB medium using both the conventional plasmid pTV118NpctphaC1ps(ST/QK)AB and stationary-phase-inducible promoter replacement plasmids instead of the phaC promoter, in which P(LA-co-3HB) production was enhanced in the glucose-supplemented LB medium. In the case of the conventional transformants harboring the pTV118NpctphaC1ps(ST/QK)AB, the P(LA-co-3HB) production was 2.7 g/L and the LA fraction was 7.4 mol% in the glucose-supplemented LB medium (Table 1, Additional file 1: Fig. S3), but in the xylose-supplemented LB medium, the production amount was increased slightly to 3.6 g/L and the LA fraction was increased to 22.6 mol% (Fig. 6A, Table 1). In contrast, in transformants harboring the yliH promoter replacement plasmid, P(LA-co-3HB) production increased to 5.6 g/L and the LA fraction increased markedly to 40.2 mol% (Fig. 6A, Table 1). Transformants harboring the gadB promoter replacement plasmid resulted in a slight increase in production to 4.4 g/L, but the LA fraction was 23.4 mol%, the same fraction as with the original plasmid, and the same level or lower for dps and gadB promoter replacement plasmids. Since these results suggest that these promoter activities differed between glucose- and xylose-supplemented LB media, we quantified the mRNA level of the replaced promoter-dependent phaC in this xylose-supplemented LB medium and compared it with the amount and LA fraction of P(LA-co-3HB) produced. The results indicate that the P(LA-co-3HB) accumulation and LA fraction were correlated with the intracellular phaC mRNA level in the stationary phase 24 h after the start of incubation (Fig. 6B, C). At present, there is no knowledge of direct regulation of the yliH or dps promoters by transcription factors responsive to glucose or xylose in E. coli, so the obvious cause for the altered activity of these promoters depending on the sugar type is unknown. It has been reported that LdhA is induced in the stationary phase regardless of the sugar type in E. coli [8], suggesting that LA-CoA is synthesized in the stationary phase under this condition. These results indicate that in xylose-supplemented LB medium, both the accumulation of P(LA-co-3HB) and the LA fraction depend on the expression of the phaC1STQK-AB operon in the stationary phase.

P(LA-co-3HB) production and expression level of the phaC1STQK-AB operon by replacement of the promoter fused to the phaC1STQK-AB operon in xylose-supplemented LB medium. A The bars indicate the amount of 3HB units in the polymer (black), the amount of LA units in the polymer (white), and the LA fraction (gray). B, C The x-axis shows the expression ratio of phaC mRNA to the ribosomal RNA, rrsA, quantified by RT-qPCR after 24 h of incubation. The y-axis shows the production of P(LA-co-3HB) after 48 h of incubation (B), and the LA fraction (C). pTV indicates the conventional plasmid pTV118NpctphaC1ps(ST/QK)AB. The gene name indicates the promoter replaced with the phaC promoter
In this study, we aimed to improve P(LA-co-3HB) production and alter the LA fraction. We demonstrated the importance of optimizing the expression levels of a set of P(LA-co-3HB) synthesis genes in the stationary phase by testing the effects of promoter replacement. In glucose-supplemented LB medium, replacing the phaC1STQK-AB promoter of pTV118NpctphaC1ps(ST/QK)AB with the dps promoter that showed successfully increased P(LA-co-3HB) production threefold to 8.8 g/L. In xylose medium, replacing the yliH promoter successfully increased its production by 1.6-fold to 5.6 g/L and increased the LA fraction from 23 to 40 mol%. Thus, it is interesting to note that the same promoter in glucose- and xylose-supplemented LB medium resulted in different effects on P(LA-co-3HB) production and the LA fraction, owing to changes in the promoter activity in the stationary phase (Figs. 4 and 6). E. coli has approximately 300 different transcriptional regulators, which form a complex hierarchical structure in the cell. As facing the environment changes, each promoter activity changes under the influence of its transcriptional regulatory network [6, 7]. To efficiently produce useful materials using microorganisms, such as P(LA-co-3HB), it is important to adapt a suitable gene expression system to fit each condition of the production system used.
Conclusions
Using a universal model microbial factory of E. coli, we have provided proof-of-concept that the replacement of cell growth phase-dependent promoters represents a promising approach for improving the productivity and functional alteration of value-added target products such as P(LA-co-3HB) in a microbial factory other than E. coli. Notably, this approach synergistically exhibited the beneficial effects in combination with the selection of sugar-based carbon sources. The optimized strain will be subjected to the established high-cell density cultivation [5] for over-production of P(LA-co-3HB) to gain a multiple information on polymer properties. In the near future, the polymeric material properties of microbially synthesized P(LA-co-3HB) could be systematically altered using advanced synthetic biology based on this approach.
Materials and methods
Bacterial strains and medium
Escherichia coli K-12 BW25113 [2] was used as a host for polymer production [obtained from the E. coli Stock Center (National Bio-Resource Center, Chiba, Japan)]. E. coli JM109 cells were used for plasmid amplification and construction.
For polymer production, recombinant E. coli harboring pTV118NpctphaC1ps(ST/QK)AB or its derivative plasmids were grown in 20 mL of LB medium containing several different concentrations of glucose or xylose at 30 °C with reciprocal shaking at 180 rpm. Ampicillin (Amp; 100 μg/mL) and kanamycin (25 μg/mL) were added as required. Cell growth, which also reflects P(LA-co-3HB) production, was monitored by measuring the turbidity at 600 nm.
Plasmids construction
The expression vector pTV118NpctphaC1ps(ST/QK)AB [derivative of pTV118N (Takara, Japan), Ampr], which harbors genes encoding propionyl-CoA transferase from Megasphaera elsdenii (pct), engineered polyhydroxyalkanoate (PHA) synthase, termed LPE, with LA-polymerizing activity [phaC1ps(ST/QK)] from Pseudomonas sp. 61-3, and 3HB-CoA supplying enzymes β-ketothiolase and acetoacetyl-CoA reductase (phaA and phaB) from Cupriavidus necator (formerly Ralstonia eutoropha), was used for P(LA-co-3HB) production [29, 30].
Twenty-one phaC promoter replacement plasmids were constructed in this study from the original pTV118NpctphaC1ps(ST/QK)AB, with the Cupriavidus necator-derived phaC promoter replaced with various promoters in E. coli. Using the original pTV118NpctphaC1ps(ST/QK)AB as a template, a DNA fragment without the promoter upstream of phaC was amplified by PCR using primers 5′-ttattttttcagtcccatgggaccg-3′ and 5′-atgagtaacaagaatagcgatgacttg-3′. The primer sequences with sequences homologous to the vector added at the 5′ end in Additional file 2: Table S1B were then used to amplify the promoter sequences from the E. coli genome using the E. coli BW25113 genome as the template. The DNA fragments were joined by homologous recombination using the Gibson assembly method to obtain each phaC promoter replacement plasmid.
The same procedure was used for the three pct promoter replacement plasmids, with the lac promoter replaced upstream of pct. The plasmid vector fragment was amplified with 5′-atgagaaaagtagaaatcattacagctgaacaag′-3 and 5′-attgcgttgcgctcactg-3′ primers and joined by homologous recombination with the E. coli promoter fragment amplified using primers listed in Additional file 2: Table S1C.
ldhA from E. coli was cloned into pTV118NpctphaC1ps(ST/QK)AB and linked to the E. coli promoter using the Gibson assembly method. Vector DNA fragments were amplified using 5′-ccggcatgcaagcttgg-3′ and 5′-atcctatgcccaacaaggcac-3′ primers, and the ldhA coding region from the E. coli genome was amplified using 5′-atgaaactcgccgtttatagcac-3′ and 5′-ttaaaccagttcgttcgggcag-3′ primers with sequences homologous to the respective promoters added at the 5′ end. The primers listed in Additional file 2: Table S1D were used to amplify the E. coli promoter sequence upstream of ldhA.
The sequences of the cloned promoters and genes of the constructed plasmids and the sequences of the homologous recombined linkage regions were confirmed by DNA sequencing.
RT-qPCR analysis
RT-qPCR was performed according to standard procedures [19]. E. coli cells were inoculated into M9 minimal medium supplemented with CAA (0.2%) at 37 °C with aeration by constant shaking at 150 rpm. Total RNA was extracted from exponential phase E. coli cells (OD600 = 0.4) using ISOGEN solution (Nippon Gene, Tokyo, Japan). Total RNA (1 μg) was transcribed into cDNA with random primers using the THUNDERBIRD™ SYBR® qPCR/RT Set (TOYOBO, Osaka, Japan). Quantitative P CR (qPCR) was performed using the THUNDERBIRD™ SYBR® qPCR Mix (TOYOBO) and a LightCycler® 96 system (Roche, Basel, Switzerland). The primer pairs are listed in Additional file 2: Table S1A. The cDNA templates were serially diluted four-fold and used for qPCR analysis. The qPCR mixtures, containing 10 μL of THUNDERBIRD™ SYBR® qPCR Mix (TOYOBO), 1 μL of each primer (5 μM stock), 7 μL of water, and 1 μL of cDNA, were amplified under the following thermal cycling conditions: 2 min at 95 °C, 45 cycles of 10 s at 95 °C and 20 s at 55 °C, and then 20 s at 72 °C. The 16S rRNA expression level was used to normalize the phaC mRNA levels of the test samples, and the relative expression levels were quantified using Relative Quantification Software provided by Roche. The results are presented as the averages of three independent experiments.
Polymer extraction and analyses
Samples from the E. coli shake-flask cultures were taken periodically during cultivation and centrifuged at 7500 rpm for 3 min to separate the cells from the medium. The cells were then lyophilized and used for cell growth and polymer analyses. The polymers were analyzed by gas chromatography, as described previously [21]. To determine the cellular polyester content and polymer fraction, approximately 15 mg of dry cells was subjected to methanolysis with a solution consisting of 1.7 mL methanol, 0.3 mL 98% sulfuric acid, and 2.0 mL chloroform at 100 °C for 140 min to convert the constituents to their methyl esters. Addition of 1 mL water to the reaction mixture induced phase separation The lower chloroform layer was used for gas chromatography (GC) on a GL Sciences GC353B FID system equipped with an InertCap-1 capillary column (30 m × 0.25 mm) and a flame ionization detector. Each experiment was repeated at least three times, and the average values are shown.
Measurement of polymers by Nile-red staining
The Nile-red staining method was performed according to Spiekermann et al. [22]. One milliliter of the cell culture medium was mixed with 10 μL of Nile-red (Sigma-Aldrich Co., St. Louis, MO, USA) dissolved in DMSO (0.4 μg/μL). After vigorous vortexing for 20 s, the mixture was incubated for 5 min at room temperature in the dark. The cells were centrifuged (3 min, 5000×g), washed twice with PBS(−) buffer, and the absorbance was measured at 430 nm using a spectrophotometer (V-630BIO, Nihon Bunko, Japan). Each experiment was repeated at least three times, and the average values are shown.
Abbreviations
- LA:
-
Lactate
- 3HB:
-
3-Hydroxybutyrate
- P(LA-co-3HB):
-
Poly(LA-co-3-hydroxybutyrate)
- RT-qPCR:
-
Reverse transcription-quantitative real-time PCR
- LB:
-
Luria–Bertani
- ORF:
-
Open reading frame
- OD:
-
Optical density
- PHAs:
-
Polyhydroxyalkanoate
References
Bunch PK, Mat-Jan F, Lee N, Clark DP. The ldhA gene encoding the fermentative lactate dehydrogenase of Escherichia coli. Microbiology. 1997;143:187–95.
Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–5.
de Almeida A, Catone MV, Rhodius VA, Gross CA, Pettinari MJ. Unexpected stress-reducing effect of PhaP, a poly(3-hydroxybutyrate) granule-associated protein, in Escherichia coli. Appl Environ Microbiol. 2011;77:6622–9.
Hori C, Sugiyama T, Watanabe K, Sun J, Kamada Y, Ooi T, Isono T, Satoh T, Sato S, Taguchi S, Matsumoto K. Isolation of poly[d-lactate (LA)-co-3-hydroxybutyrate]-degrading bacteria from soil and characterization of D-LA homo-oligomer degradation by the isolated strains. Polym Degrad Stab. 2020;179: 109231.
Hori C, Yamazaki T, Ribordy G, Takisawa K, Matsumoto K, Ooi T, Zinn M, Taguchi S. High-cell density culture of poly(lactate-co-3-hydroxybutyrate)-producing Escherichia coli by using glucose/xylose-switching fed-batch jar fermentation. J Biosci Bioeng. 2019;127:721–5.
Ishihama A, Shimada T. Hierarchy of transcription factor network in Escherichia coli K-12: H-NS-mediated silencing and anti-silencing by global regulators. FEMS Microbiol Rev. 2021;45: fuab032.
Ishihama A, Shimada T, Yamazaki Y. Transcription profile of Escherichia coli: genomic SELEX search for regulatory targets of transcription factors. Nucleic Acids Res. 2016;44:2058–74.
Jiang GR, Nikolova S, Clark DP. Regulation of the ldhA gene, encoding the fermentative lactate dehydrogenase of Escherichia coli. Microbiology. 2001;147:2437–46.
Kadoya R, Kodama Y, Matsumoto K, Ooi T, Taguchi S. Genome-wide screening of transcription factor deletion targets in Escherichia coli for enhanced production of lactate-based polyesters. J Biosci Bioeng. 2017;123:535–9.
Kadoya R, Kodama Y, Matsumoto K, Taguchi S. Indirect positive effects of a sigma factor RpoN deletion on the lactate-based polymer production in Escherichia coli. Bioengineered. 2015;6:307–11.
Kadoya R, Matsumoto K, Ooi T, Taguchi S. MtgA deletion-triggered cell enlargement of Escherichia coli for enhanced intracellular polyester accumulation. PLoS ONE. 2015;10: e0125163.
Kadoya R, Matsumoto K, Takisawa K, Ooi T, Taguchi S. Enhanced production of lactate-based polyesters in Escherichia coli from a mixture of glucose and xylose by Mlc-mediated catabolite derepression. J Biosci Bioeng. 2018;125:365–70.
Koh S, Sato M, Yamashina K, Usukura Y, Toyohuku M, Nomura N, Taguchi S. Controllable secretion of multilayer vesicles driven by microbial polymer accumulation. Sci Rep. 2022;12:3393.
Nduko JM, Matsumoto K, Ooi T, Taguchi S. Effectiveness of xylose utilization for high yield production of lactate-enriched P(lactate-co-3-hydroxybutyrate) using a lactate-overproducing strain of Escherichia coli and an evolved lactate-polymerizing enzyme. Metab Eng. 2013;15:159–66.
Nduko JM, Matsumoto K, Ooi T, Taguchi S. Enhanced production of poly(lactate-co-3-hydroxybutyrate) from xylose in engineered Escherichia coli overexpressing a galactitol transporter. Appl Microbiol Biotechnol. 2014;98:2453–60.
Salamanca-Cardona L, Scheel RA, Bergey NS, Stipanovic AJ, Matsumoto K, Taguchi S, Nomura CT. Consolidated bioprocessing of poly(lactate-co-3-hydroxybutyrate) from xylan as a sole feedstock by genetically-engineered Escherichia coli. J Biosci Bioeng. 2016;122:406–14.
Salamanca-Cardona L, Scheel RA, Mizuno K, Bergey NS, Stipanovic AJ, Matsumoto K, Taguchi S, Nomura CT. Effect of acetate as a co-feedstock on the production of poly(lactate-co-3-hydroxyalkanoate) by pflA-deficient Escherichia coli RSC10. J Biosci Bioeng. 2017;123:547–54.
Shimada T, Makinoshima H, Ogawa Y, Miki T, Maeda M, Ishihama A. Classification and strength measurement of stationary-phase promoters by use of a newly developed promoter cloning vector. J Bacteriol. 2004;186:7112–22.
Shimada T, Furuhata S, Ishihama A. The whole set of constitutive promoters for RpoN sigma factor and the regulatory role of its enhancer protein NtrC in Escherichia coli K-12. Microb Genom. 2021. https://doi.org/10.1099/mgen.0.000653.
Shimada T, Ogasawara H, Ishihama A. Genomic SELEX screening of regulatory targets of Escherichia coli transcription factors. Methods Mol Biol. 2018;1837:49–69.
Shozui F, Matsumoto K, Nakai T, Yamada M, Taguchi S. Biosynthesis of novel terpolymers poly(lactate-co-3-hydroxybutyrate-co-3-hydroxyvalerate)s in lactate-overproducing mutant Escherichia coli JW0885 by feeding propionate as a precursor of 3-hydroxyvalerate. Appl Microbiol Biotechnol. 2010;85:949–54.
Spiekermann P, Rehm BH, Kalscheuer R, Baumeister D, Steinbüchel A. A sensitive, viable-colony staining method using Nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds. Arch Microbiol. 1999;171:73–80.
Steinbüchel A, Hein S. Biochemical and molecular basis of microbial synthesis of polyhydroxyalkanoates in microorganisms. Adv Biochem Eng Biotechnol. 2001;71:81–123.
Sudesh K, Abe H, Doi Y. Synthesis, structure and properties of polyhydroxyalkanoates: biological polyesters. Prog Polym Sci. 2000;25:1503–55.
Sun J, Matsumoto K, Nduko JM, Ooi T, Taguchi S. Enzymatic characterization of a depolymerase from the isolated bacterium Variovorax sp. C34 that degrades poly(enriched lactate-co-3-hydroxybutyrate). Polym Degrad Stab. 2014;110:44–9.
Sun J, Matsumoto K, Tabata Y, Kadoya R, Ooi T, Abe H, Taguchi S. Molecular weight-dependent degradation of d-lactate-containing polyesters by polyhydroxyalkanoate depolymerases from Variovorax sp. C34 and Alcaligenes faecalis T1. Appl Microbiol Biotechnol. 2015;99:9555–63.
Sun J, Utsunomia C, Sasaki S, Matsumoto K, Yamada T, Ooi T, Taguchi S. Microbial production of poly(lactate-co-3-hydroxybutyrate) from hybrid Miscanthus-derived sugars. Biosci Biotechnol Biochem. 2016;80:818–20.
Taguchi S, Tsuge T. Natural polyester-related proteins: structure, function, evolution and engineering. In: Protein engineering handbook. Weinheim: Wiley-VCH; 2008.
Taguchi S, Yamada M, Matsumoto K, Tajima K, Satoh Y, Munekata M, Ohno K, Kohda K, Shimamura T, Kambe H, Obata S. A microbial factory for lactate-based polyesters using a lactate-polymerizing enzyme. Proc Natl Acad Sci USA. 2008;105:17323–7.
Yamada M, Matsumoto K, Shimizu K, Uramoto S, Nakai T, Shozui F, Taguchi S. Adjustable mutations in lactate (LA)-polymerizing enzyme for the microbial production of LA-based polyesters with tailor-made monomer composition. Biomacromolecules. 2010;11:815–9.
Yamada M, Matsumoto K, Uramoto S, Motohashi R, Abe H, Taguchi S. Lactate fraction dependent mechanical properties of semitransparent poly(lactate-co-3-hydroxybutyrate)s produced by control of lactyl-CoA monomer fluxes in recombinant Escherichia coli. J Biotechnol. 2011;154:255–60.
Yamamoto N, Isshiki R, Kawai Y, Tanaka D, Sekiguchi T, Matsumoto S, Tsuneda S. Stochastic expression of lactate dehydrogenase A induces Escherichia coli persister formation. J Biosci Bioeng. 2018;126:30–7.
Acknowledgements
We thank Kadoya Ryosuke (Sugiyama Jogakuen University) and Michihisa Maeda (Meiji University) for helpful discussions.
Funding
This research was funded by MEXT Grants-in-Aid for Scientific Research (C) (19K06618) and (C) (22K06184).
Author information
Authors and Affiliations
Contributions
Conceptualization, TS and ST; methodology, YN, ST, SK, and TS; formal analysis, YN and TS; investigation, YN and SK; resources, TS and ST; writing—original draft preparation, TS and SK; writing—review and editing, TS, SK, YN, and SK; funding acquisition, TS. All authors have read and agreed to the current version of the manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
P(LA-co-3HB) production and the LA fraction at various glucose concentrations by E. coli BW25113 harboring pTV118NpctphaC1ps(ST/QK)AB plasmid in glucose-supplemented LB medium. P(LA-co-3HB) production after 48 h of incubation. Figure S2. Correlation between P(LA-co-3HB) production and the mRNA levels of phaC with cultivation time. The x-axis shows the expression ratio of phaC mRNA to the ribosomal RNA, rrsA, quantified by RT-qPCR at 9 (A), 12 (B), 16 (C), and 24 (D) hours of incubation. The y-axis shows the production of P(LA-co-3HB) as measured by Nile-red staining of cells after 48 h of incubation. Correlation coefficients between phaC mRNA levels and P(LA-co-3HB) productions were obtained by the Pearson correlation coefficient and are shown in the lower right corner of each panel. Figure S3. P(LA-co-3HB) production by replacement of the promoter fused to the phaC1STQK-AB operon in glucose-supplemented LB medium. The bars indicate the amount of 3HB units in the polymer (black), the amount of LA units in the polymer (white), and the LA fraction (vertical stripe). pTV indicates the conventional plasmid pTV118NpctphaC1ps(ST/QK)AB. The gene name indicates the promoter replaced with the phaC promoter located upstream of the phaC1STQK-AB operon. Figure S4. P(LA-co-3HB) production and the LA fraction at various glucose concentrations by E. coli BW25113 harboring pTV118NpctyliHps(ST/QK)AB plasmid in glucose-supplemented LB medium. P(LA-co-3HB) production after 48 h of incubation.
Additional file 2: Table S1A.
Primer sequences used in RT-qPCR. Table S1B. Primer sequences used for construction of phaC promoter replacement plasmid. Table S1C. Primer sequences used for construction of pct promoter replacement plasmid. Table S1D. Primer sequences used for construction of ldhA promoter replacement plasmid.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nagao, Y., Koh, S., Taguchi, S. et al. Cell-growth phase-dependent promoter replacement approach for improved poly(lactate-co-3-hydroxybutyrate) production in Escherichia coli. Microb Cell Fact 22, 131 (2023). https://doi.org/10.1186/s12934-023-02143-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-023-02143-w