
Abstract
Background
Biological nitrogen fixation converting atmospheric dinitrogen to ammonia is an important way to provide nitrogen for plants. Pseudomonas stutzeri DSM4166 is a diazotrophic Gram-negative bacterium isolated from the rhizosphere of cereal Sorghum nutans. Endogenous constitutive promoters are important for engineering of the nitrogen fixation pathway, however, they have not been systematically characterized in DSM4166.
Results
Twenty-six candidate promoters were identified from DSM4166 by RNA-seq analysis. These 26 promoters were cloned and characterized using the firefly luciferase gene. The strengths of nineteen promoters varied from 100 to 959% of the strength of the gentamicin resistance gene promoter. The strongest P12445 promoter was used to overexpress the biological nitrogen fixation pathway-specific positive regulator gene nifA. The transcription level of nitrogen fixation genes in DSM4166 were significantly increased and the nitrogenase activity was enhanced by 4.1 folds determined by the acetylene reduction method. The nifA overexpressed strain produced 359.1 µM of extracellular ammonium which was 25.6 times higher than that produced by the wild-type strain.
Conclusions
The endogenous strong constitutive promoters identified in this study will facilitate development of DSM4166 as a microbial cell factory for nitrogen fixation and production of other useful compounds.
Similar content being viewed by others
Background
Biological nitrogen fixation (nif) plays an important role in the global nitrogen cycle [1, 2]. It converts atmospheric dinitrogen to ammonia under microaerobic and nitrogen-free condition [3, 4] which can be used by plants for growth and development. Naturally organisms capable of fixing nitrogen are all prokaryotes including bacteria and archaea [5]. Legumes obtain most of their nitrogen from rhizobia bacteria residing in root nodules, however, many crop plants including cereals are nonleguminous and cannot form symbiotic associations [6].To meet the demand of improving crop yield, chemically synthesized nitrogen fertilizers have been widely applied in the world. Unfortunately, overuse of these chemical fertilizers has raised serious environmental problems [7,8,9]. Engineering of biological nitrogen fixation capacity in bacteria associating with non-legume crops will help to improve agricultural sustainability by reducing the dependence on chemically fertilizers [10,11,12,13].
Nif genes including the nitrogenase genes, the cofactor biosynthesis genes and electron transporter genes are organized in clusters on the chromosome of bacteria [14, 15]. The biological reduction of dinitrogen to ammonia are catalyzed by nitrogenases which are complex metalloenzymes [16, 17]. There are three classes of nitrogenases: the Mo-Nitrogenase, the V-Nitrogenase and the Fe-Nitrogenase, among which the Mo-containing nitrogenase is the most prevalent and the best characterized [18]. Mo-Nitrogenase is composed of two oxygen-sensitive components, the MoFe protein and the Fe protein. MoFe protein encoded by nifD and nifK is a heterotetramer which provides the active site for substrate reduction. Fe protein encoded by nifH is a homodimer which serves as an electron donor to the MoFe protein during catalysis [19,20,21,22]. Expression of nif genes is tightly regulated in diazotrophic bacteria by the pathway specific positive regulatory protein NifA [23] which belongs to bacterial enhancer-binding proteins (bEBPs) containing the AAA+ (ATPases associated with various cellular activities) domain [24, 25]. NifA protein binds to the specific sequence upstream of promoters of nif genes to activate their transcription. Transcription of nifA is regulated by the NtrB-NtrC two-component system in response to nitrogen source in diazotrophic proteobacteria such as Klebsiella pneumoniae, the FixL-FixJ two-component system in response to oxygen in symbiotic diazotrophs such as Sinorhizobium meliloti, or the RegS-RegR two-component system in response to redox in symbiotic diazotrophs such as Bradyrhizobium japonicum [6]. The activity of the NifA protein is modulated by the anti-activator protein NifL which is in response to oxygen and fixed nitrogen through interaction with GlnK in γ-Proteobacteria [12, 26,27,28,29]. In symbiotic diazotrophs that lack NifL, the domain structure of NifA proteins is different to those subject to NifL inhibition, and the activity of these NifA proteins is directly modulated by the oxidation state of the cell [30].
Pseudomonas stutzeri DSM4166 is a diazotrophic isolate from the rhizosphere of a “high fixing” Sorghum nutans cultivar [31]. Its nif gene cluster is 49 kb and contains 58 genes [32]. DSM4166 has the potential to be used as nitrogen fertilizer, therefore it is vital to improve its nitrogen fixation capacity. The nitrogenase activity of Rhodobacter capsulatus was enhanced by two folds through overexpressing the rnf electron transporter genes [33]. Overexpression of the NifH nitrogenase structural protein increased the nitrogen-fixing efficiency of Azotobacter vinelandii by two folds [34]. Increasing the copy number of the nif gene cluster in the cyanobacterium Synechocystis enhanced its nitrogen fixation activity by three folds [35]. The ammonia excretion capacity of Azotobacter vinelandii were enhanced through genetic manipulation by deleting the ammonium transporter AmtB [36], or introducing mutations in the NifA protein [37], the NifL protein [12, 38], or the glutamine synthetase [39]. The nitrogen fixation and the ammonia excretion capacities of Pseudomonas stutzeri A1501 were increased by overexpressing NifA and deleting the ammonium transporter AmtB [10, 13]. Novel noncoding RNAs involved in post-transcriptional regulation of nitrogenase expression were identified and characterized in Pseudomonas stutzeri A1501 [40,41,42]. Engineering of DSM4166 to increase its nitrogen fixation capacity needs promoters with different strengths, however, both exogenous and endogenous promoters have not been systematically characterized in DSM4166.
In this study, we characterized 26 candidate promoters identified by RNA-seq analysis of DSM4166 cultured in the LB rich medium and the PMM minimum medium. Ten promoters were stronger than the promoter of gentamicin resistance gene [43]. When we used the endogenous strong constitutive promoters to overexpress the nitrogen fixation pathway specific positive regulator gene nifA, the nitrogen fixation efficiency of DSM4166 were increased by 25.6 folds determined by the production of extracellular ammonium.
Results and discussion
Screening of strong constitutive promoters in the P. stutzeri DSM4166 transcriptome by RNA-seq
The LB rich medium and the PMM minimal medium were used for transcription analysis of all 4427 genes in DSM4166 (GenBank accession number: NC_017532.1). The cells in the mid-exponential phase and the early stationary phase were used for RNA-seq analysis (Fig. S1). Expression levels of genes from each sample were ranked from the highest to the lowest based on their values of read counts (Table S1). In the four samples (two time points in two media), there were 39 genes in the 3% cutoff, which means that 39 genes were in the top 3% of the most highly expressed genes under all culturing conditions (Fig. S2). Among these 39 genes, 13 genes with very short promoter regions (shorter than 100 bp) were excluded. Therefore, we chose 26 highly expressed genes with their corresponding promoter regions (Table 1) for further characterization.
Characterization of chosen promoters using the firefly luciferase reporter gene
Each promoter region that we selected to clone is the intergenic region between the highly expressed gene and its upstream gene. Sequences of these promoters were listed in Table S3. Each promoter and the firefly luciferase gene were assembled into a pBBR1 vector (Fig. 1A) by recombineering [44]. The promoter of the gentamicin resistance gene (Pgenta) was used as the control because the gentamicin resistance gene is a good selection marker in DSM4166. The verified plasmids were electroporated into DSM4166 for the luciferase activity assay in the LB medium under the aerobic (air) condition. As shown in Fig. 1B, activities of these promoters varied from 17 to 959% of that of Pgenta. Thirteen promoters had the same strength as Pgenta. Ten promoters were stronger than Pgenta. P12445 was the strongest among them.

Luciferase assay of constitutive promoters in P. stutzeri DSM4166 in the LB medium under the aerobic condition. (A) Schematic of the plasmid containing different promoters. pBBR1: replicon; kan: kanamycin resistance gene; firefly: firefly luciferase reporter gene. (B) Luciferase activity of plasmids containing different promoters in DSM4166. Vector: the promoterless plasmid. Pgenta: the promoter of the gentamicin resistance gene. PnifA: the promoter of the nitrogen fixation pathway specific positive regulator gene nifA. Error bars indicate the standard deviations of three replicates (n = 3). The P-value cutoff for all the plots is 0.05. * P < 0.05, ** P < 0.01, and *** P < 0.001. One-way ANOVA test with Tukey Pairwise comparisons was used to compute statistical significance
DSM4166 strains harboring the pBBR1-kan-firefly plasmids carrying promoters weaker than Pgenta (P22180 and P17270), promoters having the same strength as Pgenta (P20930, P12415), and promoters stronger than Pgenta (P16030, P10410, P02720, P18595, P11670, and P12445) in the LB medium under the aerobic condition were then cultivated under the nitrogen fixation condition in which the nitrogen-free medium K [41] and the microaerobic (1% oxygen) condition was used for the luciferase activity assay. As shown in Fig. 2, activities of these promoters under the nitrogen fixation condition showed almost the same pattern as that in the LB medium under the aerobic condition. P22180 was the weakest and P12445 was the strongest. P02720, P18595, P11670, and P12445 were significantly stronger than Pgenta. P12445 was significantly stronger than PnifA.

Luciferase activity of plasmids containing different promoters in DSM4166 in the nitrogen free medium K under the under the microaerobic (1% oxygen) condition. Vector: the promoterless plasmid. Pgenta: the promoter of the gentamicin resistance gene. PnifA: the promoter of the nitrogen fixation pathway specific positive regulator gene nifA. Error bars indicate the standard deviations of three replicates (n = 3). The P-value cutoff for all the plots is 0.05. * P < 0.05, ** P < 0.01, and *** P < 0.001. One-way ANOVA test with Tukey Pairwise comparisons was used to compute statistical significance
Enhancing the nitrogen fixation activity of DSM4166 using endogenous strong constitutive promoters
As nitrogenases are highly sensitive to oxygen, the nitrogenase activity of the wild-type DSM4166 strain cultivated in the nitrogen-free medium K was detected at a range of oxygen concentrations (0.5%, 1%, 2%, and 4%) by the acetylene reduction method [41]. As shown in Fig. S3, the oxygen concentration for optimal nitrogenase activity of DSM4166 is 0.5% or 1%. The nitrogenase activity of DSM4166 at 0.5% oxygen (278 nmol ethylene h − 1 (mg protein) −1) is almost the same as that at 1% oxygen (287 nmol ethylene h − 1 (mg protein) −1). The nitrogenase activity at 2% oxygen is very low and it cannot be detected at 4% oxygen. Therefore, we used 1% oxygen for the nitrogenase activity assay in the following experiments.
Because PnifA is significantly weaker than P12445 under the nitrogen fixation condition (Fig. 2), the nitrogenase activity of DSM4166 could be improved if the nitrogen fixation pathway specific positive regulator NifA is overexpressed by the stronger P12445 promoter. The P11670 promoter was also selected to overexpress the NifA protein because of its high transcriptional activity under the nitrogen fixation condition. To facilitate evaluating expression levels of different DNA constructs at an identical locus in DSM4166, the phiC31 attB site was inserted at the PSTAA_RS19520 site (GenBank accession number: NC_017532.1) in the chromosome by single crossover to generate the DSM4166-attB recombinant strain (Fig. S4A). The PSTAA_RS19520 gene encodes a hypothetical protein and insertion of the phiC31 attB site did not affect the nitrogenase activity of DSM4166 (Fig. S4B). All DNA constructs can be integrated at the same attB site in the DSM4166 chromosome via site-specific recombination mediated by the PhiC31 integrase. The nifA gene was cloned downstream of P11670 and P12445 in the p15A-oriT-phiC31 plasmid (Fig. S4C) and then the P11670-nifA and P12445-nifA cassettes were respectively integrated at the phiC31 attB site on the chromosome of the DSM4166-attB strain to obtain recombinant strains DSM4166-P11670-nifA and DSM4166-P12445-nifA.

Enhanced nitrogenase activity by overexpressing nifA using endogenous strong constitutive promoters in DSM4166. (A) The nitrogenase activity of DSM4166 and nifA overexpression strains cultivated in the nitrogen-free medium K at 1% oxygen determined by the acetylene reduction method. (B) The concentration of extracellular ammonium produced by DSM4166 and nifA overexpression strains. (C) The transcriptional level of selected 25 nif genes in the P12445-nifA overexpression strain relative to DSM4166 cultivated in the same condition as A. Error bars indicate the standard deviations of three replicates (n = 3). The P-value cutoff for all the plots is 0.05. * P < 0.05, ** P < 0.01, and *** P < 0.001. ns, not significant. One-way ANOVA test with Tukey Pairwise comparisons was used to compute statistical significance
The nitrogenase activities of DSM4166-P11670-nifA and DSM4166-P12445-nifA under the nitrogen fixation condition determined by the acetylene reduction method were increased by 1.8 and 4.1 folds compared with the wild-type DSM4166 respectively (Fig. 3A). Quantitative real-time PCR (qRT-PCR) analysis results suggested that transcription levels of nif genes in the DSM4166-P12445-nifA strain were 25 ~ 127 times higher than that in the wild-type DSM4166 (Fig. 3C).
The ammonium secretion of the wild-type DSM4166, DSM4166-P11670-nifA, and DSM4166-P12445-nifA strains were determined by measuring the concentration of ammonium in the supernatant of cultures (Fig. 3B). The highest ammonium concentration (359.1 µM) was obtained in the supernatant of the DSM4166-P12445-nifA strain. The ammonium concentration of the DSM4166-P11670-nifA strain was 1.6 times higher than that of the wild-type DSM4166 strain and 16.3 times lower than that of the DSM4166-P12445-nifA strain. This result suggested that the surplus ammonium was secreted by DSM4166 strains. Secretion of the excess nitrogen fixed by DSM4166 strains will favor fertilizing crops or producing ammonium in the bioreactor. However, we did not test if the engineered DSM4166 strains would work in the rhizosphere or during plant-bacteria interaction.
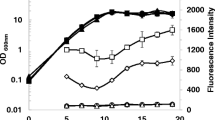
An ammonium-excreting P. stutzeri A1501 engineered strain 1568/pVA3 constructed by overexpressing NifA and deleting the ammonium transporter AmtB was recently reported to produce 20.3 µM of extracellular ammonium after 72 h incubation [13]. In this study, the DSM4166-P12445-nifA strain produced 18 times more extracellular ammonium than the 1568/pVA3 strain with a shorter incubation (24 h). Furthermore, nifA overexpression under the P12445 promoter had little effect on the growth of DSM4166 in the medium K at 99% N2 and 1% O2 (Fig. 4).

The growth curve of DSM4166 and nifA overexpression strains cultivated in the medium K at 99% N2 and 1% O2
Conclusion
With the development of synthetic biology, promoters have been widely used for modulating gene expression to optimize biosynthetic pathways for the synthesis of important compounds. Twenty-three genes for biosynthesis of the insecticide spinosad and twenty-five genes for biosynthesis of the anticancer drug salinomycin had been refactored using multiple endogenous strong constitutive promoters to obtain artificial gene clusters with higher transcription level and higher compound productions [45, 46]. Gene cluster refactoring with strong constitutive promoters was also used to activate silent gene clusters for discovery and characterization of new natural products [47,48,49]. The nitrogen fixation gene cluster from Klebsiella oxytoca was refactored into artificial operons under the control of synthetic promoters, ribosome binding sites, and terminators to removes all native regulations [50]. The nif gene cluster refactoring also facilitated controlling the nitrogen fixation activity of bacteria under different growth conditions [51]. In this study, we identified a panel of endogenous strong constitutive promoters in Pseudomonas stutzeri strain DSM4166. The nitrogen fixation activity of DSM4166 was enhanced by overexpressing the nitrogen fixation pathway specific positive regulator using these promoters.
The Gram-negative soil bacterium P. stutzeri DSM4166 has the potential to be developed as a good microbial cell factory. Large DNA constructs can be easily transformed into this strain and integrated into its chromosome through transposition with high efficiency [52]. The phiC31 site-specific recombination system was also established in this study. Its whole genome sequence had been published in 2011 and rhizosphere competence genes required in root colonization were identified such as denitrification and chemotaxis besides nitrogen fixation [32]. The endogenous constitutive promoters characterized in this study will facilitate engineering of P. stutzeri DSM4166 to further improve its nitrogen fixation capacity or produce useful chemicals.
Methods
Bacteria strains and culture conditions
Escherichia coli strains were cultured at 37 °C in LB medium. The auxotrophic conjugation donor strain E. coli WM3064 was maintained in the LB medium containing 1 mM of DL-α,ε-diaminopimelic acid [53]. Pseudomonas stutzeri DSM4166 strains were cultured at 30 °C in LB medium or PMM medium (8.0 g L− 1 K2HPO4·3H2O, 5.0 g L− 1 KH2PO4, 1.0 g L− 1 (NH4)2SO4, 6.6 g L− 1 sodium succinate, pH 7.0. Add 1.2 ml 1 M MgSO4 after autoclaving) under the aerobic condition or in the nitrogen-free medium K (0.4 g L− 1 KH2PO4, 0.1 g L− 1 K2HPO4, 0.1 g L− 1 NaCl, 0.2 g L− 1 MgSO4·7H2O, 0.01 g L− 1 MnSO4·H2O, 0.01 g L− 1 Fe2(SO4)3·H2O, 0.01 g L− 1 Na2MoO4·H2O, 10 mL L− 1 Sodium DL-lactate, pH 6.8) under microaerobic (nitrogen fixation) condition. Concentrations of antibiotics used in this study were: kanamycin, 15 µg mL− 1; apramycin, 20 µg mL− 1; gentamicin 2 µg mL− 1.
Pseudomonas stutzeri cultivation and RNA-seq analysis
P. stutzeri DSM4166 culture was grown in the LB rich medium and the PMM minimal medium at 30 °C overnight with shaking at 950 rpm in an Aoheng thermomixer. Each 500 µL of overnight culture was inoculated into 50 mL of fresh LB medium or PMM medium in a 250-mL flask, and cultivated at 30 °C. The growth curve was determined by measuring OD600 of cell cultures every 2 h (Fig. S1). Cells were collected at 10 h (mid-exponential phase) and 14 h (early stationary phase) in LB medium, and 28 h (mid-exponential phase) and 38 h (early stationary phase) in PMM medium. Three replicates were set for each culturing condition. Twenty milliliters of culture from each replicate were mixed and centrifuged. The cell pellets frozen in dry ice were sent to Novogene Corporation (Beijing, China) for total RNA extraction and sequencing. The RNA integrity number (RIN) of samples was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). RIN of all samples were larger than 8.9 (Table S4) and recognized as good quality for library preparation. Samples were sequenced on an Illumina Hiseq 2500 platform and paired-end reads were generated. The P. stutzeri DSM4166 reference genome (GenBank accession number: NC_017532.1) was used for alignment. HTSeq v0.6.1 was used to count the reads numbers mapped to each gene (Table S1). Genes in each sample were ranked from the highest to the lowest expression using the values of read counts.
DNA manipulation
The pBBR1-kan vector, the firefly reporter gene and promoters were amplified by PCR using the PrimeSTAR Max DNA Polymerase (Takara, cat. no. R045A) and oligonucleotides listed in Table S5. These three fragments were assembled by linear plus linear homologous recombination (LLHR) mediated by RecET in E. coli GB05-dir [44] to form the plasmid pBBR1-kan-promoter-firefly. After restriction analysis and sequencing, the correct plasmids were transformed into P. stutzeri DSM4166 by electroporation for luciferase assay.
For construction of the DSM4166-attB strain, the pK18mob vector [54] and the PSTAA_19520 segment were amplified by PCR using oligonucleotides listed in Table S6. These two fragments were assembled by LLHR in E. coli GB05-dir to form the plasmid pK18mob-19520-attB. After restriction analysis and sequencing, the correct pK18mob-19520-attB plasmid was transformed into E. coli WM3064 for conjugation. P. stutzeri DSM4166-attB exconjugants were screened by colony PCR using 19520-check-F/lac-seq and 19520-check-R/pK18-seq listed in Table S6.
For construction of DSM4166-P12445-nifA and DSM4166-P11670-nifA strains, the p15A vector, P12445, P11670, and the nifA gene were amplified by PCR using oligonucleotides listed in Table S7. Corresponding PCR products were assembled by LLHR in E. coli GB05-dir. After restriction analysis and sequencing, the correct plasmids were transfer into E. coli WM3064 for conjugation. P. stutzeri DSM4166 exconjugants were screened by colony PCR using 19520-check-F/nifA-5out and lac-seq/nifA-3out listed in Table S7.
Luciferase assay
The Single-Luciferase (Firefly) Reporter Assay Kit (TransDetect, cat. no. FR101-01) was used to check the strength of the promoters. P. stutzeri DSM4166 cells harboring pBBR1-kan-promoter-firefly were inoculated into 1.0 mL of LB supplemented with kanamycin and incubated at 30 °C overnight with shaking at 950 rpm in an Eppendorf thermomixer. Cells were collected by centrifugation and washed once with phosphate buffer saline. Cells suspended in phosphate buffer saline with the OD600 = 1.0 were used to detect the fluorescence according to the manufacturer’s protocol.
For detecting the strength of the promoters in microaerobic (nitrogen fixation) condition, the overnight cultures of P. stutzeri DSM4166 cells harboring pBBR1-kan-promoter-firefly were collected and washed by nitrogen-free medium K. Then 1 mL of bacterial suspension (OD600 = 1.0) was added to a 100 mL anaerobic serum bottle which contains 9 mL of nitrogen-free medium K. The gas mixture (99% Ar + 1% O2) was blown into anaerobic serum bottle for 3 min to replace the headspace air, and the culture was incubated at 30 °C for 16 h with shaking at 200 rpm. Cells were collected by centrifugation and washed once with phosphate buffer saline. Cells suspended in phosphate buffer saline with the OD600 = 1.0 were used to detect the fluorescence according to the manufacturer’s protocol.
Quantitative real-time PCR (qRT-PCR) analysis
The overnight cultures of the engineered P. stutzeri DSM4166 strains incubated in LB liquid medium were collected by centrifugation and washed with nitrogen-free medium K. Then 1 mL of diluted bacterial suspension (OD600 = 1.0) were added to a 100 mL anaerobic serum bottle which contains 9 mL of nitrogen-free medium K. The gas mixture (99% Ar + 1% O2) was blown into anaerobic serum bottle for 3 min to replace the headspace air, and the culture was incubated at 30 °C for 4 h with shaking at 200 rpm. After incubation, cells were collected by centrifugation. Total RNA was extracted using the RNAprep pure Kit (Tiangen, cat. no. DP430). DNA elimination and reverse transcription was performed with the PrimeScript RT reagent Kit with gDNA Eraser (Takara, cat. no. RR047A). The qRT-PCR was performed on StepOnePlus Real-Time PCR System (Applied Biosystems) using TB Green Premix Ex Taq (Takara, cat. no. RR420A) according to the manufacturer’s protocol. Oligonucleotides were listed in Table S8. The endogenous gene gapdh, encoding glyceraldehyde phosphate dehydrogenase, was used as the internal control.
Nitrogenase activity assay
Nitrogenase activity was determined using the acetylene reduction assay [55]. P. stutzeri DSM4166 strains were incubated in LB liquid medium supplemented with appropriate antibiotics at 30 °C overnight with shaking. Cells were then collected by centrifugation and washed with nitrogen-free medium K, and resuspended to achieve an OD600 of 1.0 in medium K. Then 1 mL of diluted bacterial suspension were added to a 100 mL anaerobic serum bottle which contains 9 mL of nitrogen-free medium K. The gas mixture (99% Ar + 1% O2) was blown into anaerobic serum bottle for 3 min to replace the headspace air, 10% of acetylene gas (10 mL) was injected and the culture was incubated at 30 °C for 4 h with shaking at 200 rpm. Ethylene production was detected by Shimadzu GC2014 gas chromatograph equipped with a flame ionization detector (FID). We used a capillary column (KB-Al2O3/Na2SO4, 30-m length, 0.53-mm inner diameter, 20-µm film thickness; Shimadzu) with the following detection conditions: the injection port temperature was 100 °C, the transfer line temperature was 100 °C, the FID temperature was 180 °C. The carrier gas, ultra-high-purity nitrogen, flowed at a constant rate of 3 mL min− 1. One mL of gas samples was injected and the injections were split at a ratio of 1:40. The data was collected for 5 min. The ethylene gas was used as standard. After that, the cells were collected by centrifugation to determine the concentration of total proteins by the Bradford method [56] using bovine-serum albumin as standard. The nitrogenase activity was calculated by the following formula:
Quantification of ammonia concentration
To detect the concentration of ammonia excreted from engineered strains, cells from an overnight culture in LB medium supplemented with appropriate antibiotics were centrifuged and resuspended in 100 mL anaerobic serum bottle containing 10 ml nitrogen-free medium K at OD600 of 0.1. The gas mixture (99% N2 + 1% O2) was blown into anaerobic serum bottle for 3 min to replace the headspace air. After inoculation at 30 °C with shaking at 200 rpm for 24 h, the concentration of ammonium in the supernatant was measured using the Nessler’s reagent method [57]. Ammonium chloride was used as the standard.
Data Availability
All data for this study are included in this published article and its additional file.
References
Capone DG. Marine nitrogen fixation: what’s the fuss? Curr Opin Microbiol. 2001;4(3):341–8.
Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai ZC, Freney JR, et al. Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science. 2008;320(5878):889–92.
Yan YL, Ping SZ, Peng JP, Han YL, Li L, Yang J et al. Global transcriptional analysis of nitrogen fixation and ammonium repression in root-associated Pseudomonas stutzeri A1501.BMC Genomics. 2010;11.
Threatt SD, Rees DC. Biological nitrogen fixation in theory, practice, and reality: a perspective on the molybdenum nitrogenase system.FEBS Lett. 2022.
Raymond J, Siefert JL, Staples CR, Blankenship RE. The natural history of nitrogen fixation. Mol Biol Evol. 2004;21(3):541–54.
Dixon R, Kahn D. Genetic regulation of biological nitrogen fixation. Nat Rev Microbiol. 2004;2(8):621–31.
Zhang X, Davidson EA, Mauzerall DL, Searchinger TD, Dumas P, Shen Y. Managing nitrogen for sustainable development. Nature. 2015;528(7580):51–9.
Liu EK, Yan CR, Mei XR, He WQ, Bing SH, Ding LP, et al. Long-term effect of chemical fertilizer, straw, and manure on soil chemical and biological properties in northwest China. Geoderma. 2010;158(3–4):173–80.
Yan L, Wang YY, Tumbalam P, Zhang TL, Gao Q, Zhang WJ, et al. Spatiotemporal distribution of chemical fertilizer application and manure application potential in China. Environ Eng Sci. 2019;36(10):1337–48.
Zhang T, Yan Y, He S, Ping S, Alam KM, Han Y, et al. Involvement of the ammonium transporter AmtB in nitrogenase regulation and ammonium excretion in Pseudomonas stutzeri A1501. Res Microbiol. 2012;163(5):332–9.
Oldroyd GE, Dixon R. Biotechnological solutions to the nitrogen problem. Curr Opin Biotechnol. 2014;26:19–24.
Plunkett MH, Knutson CM, Barney BM. Key factors affecting ammonium production by an Azotobacter vinelandii strain deregulated for biological nitrogen fixation. Microb Cell Fact. 2020;19(1):107.
Jiang S, Li J, Wang Q, Yin C, Zhan Y, Yan Y, et al. Maize Growth Promotion by Inoculation with an Engineered ammonium-excreting strain of Nitrogen-Fixing Pseudomonas stutzeri. Microorganisms. 2022;10(10):1986.
Yan YL, Yang J, Dou YT, Chen M, Ping SZ, Peng JP, et al. Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501. Proc Natl Acad Sci USA. 2008;105(21):7564–9.
Yang JG, Xie XQ, Xiang N, Tian ZX, Dixon R, Wang P. Polyprotein strategy for stoichiometric assembly of nitrogen fixation components for synthetic biology. Proc Natl Acad Sci USA. 2018;115(36):E8509–E17.
Rees DC, Howard JB. Nitrogenase: standing at the crossroads. Curr Opin Chem Biol. 2000;4(5):559–66.
Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem Rev. 2014;114(8):4041–62.
Yang JG, Xie XQ, Wang X, Dixon R, Wang YP. Reconstruction and minimal gene requirements for the alternative iron-only nitrogenase in Escherichia coli. Proc Natl Acad Sci USA. 2014;111(35):E3718–E25.
Burgess BK, Lowe DJ. Mechanism of molybdenum nitrogenase. Chem Rev. 1996;96(7):2983–3011.
Howard JB, Rees DC. Structural basis of biological nitrogen fixation. Chem Rev. 1996;96(7):2965–82.
Christiansen J, Dean DR, Seefeldt LC. Mechanistic features of the Mo-containing nitrogenase. Annu Rev Plant Physiol Plant Mol Biol. 2001;52:269–95.
Rees DC, Tezcan FA, Haynes CA, Walton MY, Andrade S, Einsle O, et al. Structural basis of biological nitrogen fixation. Philos Trans A Math Phys Eng Sci. 2005;363(1829):971–84.
Passaglia LMP, Van Soom C, Schrank A, Schrank IS. Purification and binding analysis of the nitrogen fixation regulatory NifA protein from Azospirillum brasilense. Braz J Med Biol Res. 1998;31(11):1363–74.
Bush M, Dixon R. The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription. Microbiol Mol Biol Rev. 2012;76(3):497–529.
Studholme DJ, Dixon R. Domain architectures of sigma54-dependent transcriptional activators. J Bacteriol. 2003;185(6):1757–67.
Little R, Reyes-Ramirez F, Zhang Y, van Heeswijk WC, Dixon R. Signal transduction to the Azotobacter vinelandii NIFL-NIFA regulatory system is influenced directly by interaction with 2-oxoglutarate and the PII regulatory protein. EMBO J. 2000;19(22):6041–50.
Little R, Colombo V, Leech A, Dixon R. Direct interaction of the NifL regulatory protein with the GlnK signal transducer enables the Azotobacter vinelandii NifL-NifA regulatory system to respond to conditions replete for nitrogen. J Biol Chem. 2002;277(18):15472–81.
Martinez-Argudo I, Little R, Shearer N, Johnson P, Dixon R. The NifL-NifA system: a multidomain transcriptional regulatory complex that integrates environmental signals. J Bacteriol. 2004;186(3):601–10.
Barrett J, Ray P, Sobczyk A, Little R, Dixon R. Concerted inhibition of the transcriptional activation functions of the enhancer-binding protein NIFA by the anti-activator NIFL. Mol Microbiol. 2001;39(2):480–93.
Fischer HM. Genetic regulation of nitrogen fixation in rhizobia. Microbiol Rev. 1994;58(3):352–86.
Krotzky A, Werner D. Nitrogen fixation in Pseudomonas stutzeri. Arch Microbiol. 1987;147(1):48–57.
Yu HY, Yuan ML, Lu W, Yang J, Dai SX, Li Q, et al. Complete genome sequence of the nitrogen-fixing and rhizosphere-associated bacterium Pseudomonas stutzeri strain DSM4166. J Bacteriol. 2011;193(13):3422–3.
Jeong HS, Jouanneau Y. Enhanced nitrogenase activity in strains of Rhodobacter capsulatus that overexpress the rnf genes. J Bacteriol. 2000;182(5):1208–14.
Nag P, Pal S. Fe protein over-expression can enhance the nitrogenase activity of Azotobacter vinelandii. J Basic Microbiol. 2013;53(2):156–62.
Liu D, Liberton M, Yu JJ, Pakrasi HB, Bhattacharyya-Pakrasi M. Engineering nitrogen fixation activity in an oxygenic phototroph.mBio. 2018;9(3).
Barney BM, Eberhart LJ, Ohlert JM, Knutson CM, Plunkett MH. Gene deletions resulting in increased Nitrogen Release by Azotobacter vinelandii: application of a Novel Nitrogen Biosensor. Appl Environ Microbiol. 2015;81(13):4316–28.
Bueno Batista M, Brett P, Appia-Ayme C, Wang YP, Dixon R. Disrupting hierarchical control of nitrogen fixation enables carbon-dependent regulation of ammonia excretion in soil diazotrophs. PLoS Genet. 2021;17(6):e1009617.
Mus F, Khokhani D, MacIntyre AM, Rugoli E, Dixon R, Ane JM, et al. Genetic determinants of ammonium excretion in nifL mutants of Azotobacter vinelandii. Appl Environ Microbiol. 2022;88(6):e0187621.
Ortiz-Marquez JC, Do Nascimento M, Curatti L. Metabolic engineering of ammonium release for nitrogen-fixing multispecies microbial cell-factories. Metab Eng. 2014;23:154–64.
Zhan Y, Deng Z, Yan Y, Zhang H, Lu C, Yang Z et al. NfiR, a New Regulatory Noncoding RNA (ncRNA), Is Required in Concert with the NfiS ncRNA for Optimal Expression of Nitrogenase Genes in Pseudomonas stutzeri A1501.Appl Environ Microbiol. 2019;85(14).
Zhan Y, Yan Y, Deng Z, Chen M, Lu W, Lu C, et al. The novel regulatory ncRNA, NfiS, optimizes nitrogen fixation via base pairing with the nitrogenase gene nifK mRNA in Pseudomonas stutzeri A1501. Proc Natl Acad Sci USA. 2016;113(30):E4348–56.
Zhang H, Zhan Y, Yan Y, Liu Y, Hu G, Wang S et al. The Pseudomonas stutzeri-Specific Regulatory Noncoding RNA NfiS Targets katB mRNA Encoding a Catalase Essential for Optimal Oxidative Resistance and Nitrogenase Activity.J Bacteriol. 2019;201(19).
Yin J, Zheng WT, Gao YS, Jiang CJ, Shi HB, Diao XT, et al. Single-stranded DNA-binding protein and exogenous RecBCD inhibitors enhance phage-derived homologous recombination in Pseudomonas. iScience. 2019;14:1–14.
Wang HL, Li Z, Jia RN, Hou Y, Yin J, Bian XY, et al. RecET direct cloning and Redαβ recombineering of biosynthetic gene clusters, large operons or single genes for heterologous expression. Nat Protoc. 2016;11(7):1175–90.
Song CY, Luan J, Cui QW, Duan QY, Li Z, Gao YS, et al. Enhanced heterologous spinosad production from a 79-kb synthetic multi-operon assembly. ACS Synth Biol. 2019;8(1):137–47.
Jiang CJ, Zhou HB, Sun HL, He RT, Song CY, Cui TQ, et al. Establishing an efficient salinomycin biosynthetic pathway in three heterologous Streptomyces hosts by constructing a 106-kb multioperon artificial gene cluster. Biotechnol Bioeng. 2021;118(12):4668–77.
Luo YZ, Huang H, Liang J, Wang M, Lu L, Shao ZY, et al. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat Commun. 2013;4:2894.
Shao ZY, Rao GD, Li C, Abil Z, Luo YZ, Zhao HM. Refactoring the silent spectinabilin gene cluster using a plug-and-play scaffold. ACS Synth Biol. 2013;2(11):662–9.
Luo YZ, Zhang L, Barton KW, Zhao HM. Systematic identification of a panel of strong constitutive promoters from Streptomyces albus. ACS Synth Biol. 2015;4(9):1001–10.
Temme K, Zhao DH, Voigt CA. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc Natl Acad Sci USA. 2012;109(18):7085–90.
Ryu MH, Zhang J, Toth T, Khokhani D, Geddes BA, Mus F, et al. Control of nitrogen fixation in bacteria that associate with cereals. Nat Microbiol. 2020;5(2):314–30.
Jing X, Cui Q, Li X, Yin J, Ravichandran V, Pan D, et al. Engineering Pseudomonas protegens Pf-5 to improve its antifungal activity and nitrogen fixation. Microb Biotechnol. 2020;13(1):118–33.
Ali M, Sun Y, Xie L, Yu H, Bashir A, Li L. The pathogenicity of Pseudomonas syringae MB03 against Caenorhabditis elegans and the Transcriptional response of nematicidal genes upon different nutritional conditions. Front Microbiol. 2016;7:805.
Schafer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145(1):69–73.
Yu FN, Jing XS, Li XC, Wang HL, Chen HN, Zhong L, et al. Recombineering Pseudomonas protegens CHA0: an innovative approach that improves nitrogen fixation with impressive bactericidal potency. Microbiol Res. 2019;218:58–65.
Bradford MM. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem. 1976;72(1–2):248–54.
Gao X, Wen YJ, Qu D, An L, Luan SL, Jiang WS, et al. Interference effect of Alcohol on Nessler’s reagent in Photocatalytic Nitrogen fixation. ACS Sustainable Chem Eng. 2018;6(4):5342–8.
Acknowledgements
The authors thank Jingyao Qu, Jing Zhu and Zhifeng Li from State Key Laboratory of Microbial Technology of Shandong University for help and guidance in GC-MS.
Funding
This work was supported by the National Key Research & Development Program of China 2021YFC2101000, National Natural Science Foundation of China 32122049, the 111 Project B16030, the Fund for Distinguished Young Scholars of SDU, and the Taishan Scholar Program of Shandong Province tsqn201812008.
Author information
Authors and Affiliations
Contributions
JL, HW, JF and YZ designed the experiments. GY, XL and QD conducted the experiments. JL and GY wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
I hereby give Microbial Cell Factories the right and permission to publish this article.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12934_2023_2085_MOESM1_ESM.pdf
Additional file 1: Figure S1. The growth curve of P. stutzeri DSM4166 in the LB medium and the PMM medium; Figure S2. Venn diagram of genes which were selected in 3% cut off according to gene expression level in RNA-seq; Figure S3. Schematic of construction of DSM4166-attB strain and plasmids of overexpressing nifA gene; Table S3. The sequence of screened endogenous promoters from P. stutzeri DSM4166. Table S4. Quality evaluation of samples for RNA-seq; Table S5. Oligonucleotides for cloning of screened endogenous promoter regions from P. stutzeri DSM4166; Table S6. Oligonucleotides used for construction of DSM4166-attB strain; Table S7. Oligonucleotides used for construction of DSM4166-P12445-nifA and DSM4166-P11670-nifA; Table S8. Oligonucleotides for quantitative real-time PCR; Table S9. Nitrogenase activity of DSM4166, DSM4166-P11670-nifA and DSM4166-P12445-nifA; Table S10. Quantitative real-time PCR analysis of nif genes in DSM4166 and DSM4166-P12445-nifA strains; Table S11. The concentration of extracellular ammonium produced by DSM4166 and nifA overexpression strains.
12934_2023_2085_MOESM2_ESM.xlsx
Additional file 2: Table S1. Read counts of RNA-seq samples.
12934_2023_2085_MOESM3_ESM.xlsx
Additional file 3: Table S2. The top 3% of the most highly expressed genes in the four samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, G., Li, X., Duan, Q. et al. Systematic identification of endogenous strong constitutive promoters from the diazotrophic rhizosphere bacterium Pseudomonas stutzeri DSM4166 to improve its nitrogenase activity. Microb Cell Fact 22, 91 (2023). https://doi.org/10.1186/s12934-023-02085-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-023-02085-3