
Abstract
Background
The bacteriocin nisin is naturally produced by Lactococcus lactis as an inactive prepeptide that is modified posttranslationally resulting in five (methyl-)lanthionine rings characteristic for class Ia bacteriocins. Export and proteolytic cleavage of the leader peptide results in release of active nisin. By targeting the universal peptidoglycan precursor lipid II, nisin has a broad target spectrum including important human pathogens such as Listeria monocytogenes and methicillin-resistant Staphylococcus aureus strains. Industrial nisin production is currently performed using natural producer strains resulting in rather low product purity and limiting its application to preservation of dairy food products.
Results
We established heterologous nisin production using the biotechnological workhorse organism Corynebacterium glutamicum in a two-step process. We demonstrate successful biosynthesis and export of fully modified prenisin and its activation to mature nisin by a purified, soluble variant of the nisin protease NisP (sNisP) produced in Escherichia coli. Active nisin was detected by a L. lactis sensor strain with strictly nisin-dependent expression of the fluorescent protein mCherry. Following activation by sNisP, supernatants of the recombinant C. glutamicum producer strain cultivated in standard batch fermentations contained at least 1.25 mg/l active nisin.
Conclusions
We demonstrate successful implementation of a two-step process for recombinant production of active nisin with C. glutamicum. This extends the spectrum of bioactive compounds that may be produced using C. glutamicum to a bacteriocin harboring complex posttranslational modifications. Our results provide a basis for further studies to optimize product yields, transfer production to sustainable substrates and purification of pharmaceutical grade nisin.
Similar content being viewed by others
Background
Massive over- and misuse of antibiotics in medicine and animal farming for food production has fueled the development of antibiotic resistance of a wide range of pathogens [1,2,3]. In 2014, the global increase in antibiotic resistant bacteria has been recognized by the World Health Organization (WHO) as one of the most urgent problems to human health. Consequently, the WHO has issued a warning of a post-antibiotic era in which infections with common bacteria become lethal are a realistic scenario [4]. Replacement of antibiotics with bacteriocins and other antimicrobial peptides may contribute to solve the problems of antibiotic resistance.
Bacteriocins are a group of ribosomally synthesized peptides produced by different bacteria that show high antimicrobial activity against various bacteria including antibiotic resistant human pathogens [5, 6]. Thus, bacteriocins may be an interesting alternative to classic antibiotics [5, 7]. One of the best characterized bacteriocins is the lanthipeptide nisin, which is naturally produced by different L. lactis strains [8]. There are several natural variants of nisin (e.g. nisin A, Z, and Q) with slightly different amino acid sequences but conserved secondary structure. The mature peptides consist of 34 amino acids with multiple posttranslational modifications including dehydrated amino acids and formation of so-called (methyl-)lanthionine rings [8].
The enzymes for nisin biosynthesis are encoded by a cluster of genes for the prepeptide (dependent on the nisin variant nisZ, nisA, etc.), modification (nisB and nisC), regulation (nisRK), transport (nisT) and immunity (nisI; nisFEG) [8,9,10]. The nisA- or nisZ-encoded precursor peptide (“prenisin”) consists of 57 amino acids and is subsequently processed and exported by the enzymes NisB, NisC, NisT and NisP [8]. NisB is a dehydratase that converts threonine and serine residues to the dehydroamino acids dehydroalanine and dehydrobutyrine, respectively [11, 12]. In a second modification step, the cyclase NisC couples the dehydroamino acids to specific cysteine residues by formation of thioether bonds resulting in five characteristic (methyl-)lanthionine rings [13, 14]. The fully modified but still inactive prenisin is then transported to the extracellular space via the ABC-transporter NisT [15,16,17]. Finally, proteolytic cleavage of the leader peptide between arginine and isoleucine residues in position 23–24 by the membrane anchored protease NisP leads to activation and release of mature and active nisin [18,19,20]. This final proteolytic activation by NisP requires a fully modified prenisin [21, 22].
To achieve growth inhibition and killing of bacteria, nisin targets the bacterial cell wall precursor lipid II [23, 24] located in the outer leaflet of the cell membrane by binding to the pyrophosphate unit of lipid II [25]. This leads to inhibition of growth by sequestering lipid II and preventing its incorporation into the nascent peptidoglycan chain at the cell septum. With increasing concentration of nisin at the membrane, the bactericidal activity of nisin is mediated by pore-forming complexes consisting of eight molecules of nisin and four molecules of lipid II [23, 26]. Nisin exhibits antimicrobial activity against a wide spectrum of Gram-positive bacteria including important human pathogens such as Listeria monocytogenes, Enterococcus sp., Staphylococcus aureus strains [27,28,29,30,31,32]. Due to its broad spectrum of target organisms and classification as a generally regarded as safe substance, nisin is authorized as food additive by the European Food Safety Authority (ESFA; E number: E234) and U.S. Food and Drug Authority (FDA) and widely used in food preservation [33]. The global market volume of nisin is steadily increasing and projected to exceed 500 Mio. USD in 2025 (https://www.marketsandmarkets.com/Market-Reports/nisin-market-29041412.html). Due to its activity against antibiotic resistant strains of Enterococcus sp. and S. aureus [29,30,31,32] it is also discussed as an alternative to treat infections with these organisms [5].
So far, industrial scale production of nisin is performed exclusively with natural L. lactis producer strains [34]. These production processes bear several disadvantages including expensive media and intensive downstream processing [34,35,36]. In consequence, nisin preparations are sold as partial purified product containing only 2.5% active nisin [34, 37]. Hence, recombinant production of nisin using a robust biotechnological workhorse may increase product yields and improve product purity by using defined media and well-established fermentation and down-stream processes. The Gram-positive bacterium Corynebacterium glutamicum is a well-established host for a wide range of compounds including high value active ingredients, therapeutic proteins and supplements for medial infusion solutions [38,39,40,41,42,43,44]. Recently, we successfully established C. glutamicum as a host to produce the bacteriocin pediocin PA-1, a class IIa bacteriocin that is not extensively modified [45]. With the present study, we extend the range of antimicrobial peptides that can be produced using C. glutamicum by implementing a two-step process for production of the completely modified class I bacteriocin nisin.
Results
C. glutamicum is not suitable for production of active nisin
Although previous studies reported C. glutamicum to be resistant to up to 40 µg/ml of nisin [46], our own results suggest that growth of C. glutamicum is completely inhibited by ~ 1 µg/ml of nisin [47, 48]. To corroborate these results and to determine the nisin concentration that is required for formation of pores in the membrane of C. glutamicum, we devised a biosensor expressing the pH-dependent fluorescent protein pHluorin2 [49] in a similar fashion as described recently for L. monocytogenes [50]. For this biosensor, a pHluorin2 gene sequence codon-optimized for C. glutamicum was fused to the strong, constitutive tuf promoter [51], cloned into the pPBEx2 plasmid [52] and introduced into C. glutamicum ATCC 13032 (Additional file 1: Fig. S1A, B).
Corynebacterium glutamicum ATCC 13032/pPB-pHinCg shows high fluorescence with two excitation maxima and the characteristic ratiometric, pH-dependent shift following cetyltrimethylammoniumbromid (CTAB)-induced disturbance of membrane integrity (Additional file 1: Fig. S1C, D). Exposure of this strain to a range of nisin concentrations revealed that as little as 195 ng/ml are sufficient to elicit signs of membrane damage and at concentrations above 781 ng/ml membrane integrity of all bacteria was completely disrupted (Fig. 1A). This indicates that recombinant production of active nisin is not possible using C. glutamicum as the product not only inhibits growth [47, 48] but kills producer cells at low concentrations.

Sensitivity of C. glutamicum to nisin. A Membrane damage of C. glutamicum CR099/pPB-pHin2Cg biosensor in the presence of different concentrations of nisin. Values are ratios of fluorescence intensity with emission at 510 nm after excitation at 400 and 470 nm (ratio RFU 400/470). B Optical density (OD600) of C. glutamicum CR099/pEKEx2 and CR099ΔporAΔporHΔporB/pEKEx-vraDE after 24 h of incubation in the presence of nisin at the indicated concentrations (195–12,500 ng/ml). For assays bacteria were harvested from fresh o/N cultures, resuspended in fresh medium at an OD600 of 0.05 in 2xTY medium. For CR099ΔporAΔporHΔporB/pEKEx-vraDE, medium was additionally supplemented with 2 g/l CaCl2. All values are mean ± standard deviation (SD) of triplicate measurements of one representative of three independent cultures per strain. The red and green lines indicate OD600 or ratio RFU 400/470 of the positive (i.e. complete inhibition of growth) or negative (i.e. in the absence of nisin) control, respectively
Recently, we tested several approaches to improve resistance of C. glutamicum to nisin by e.g. expression of nisin immunity genes, ABC-transporters of pathogenic bacteria known to confer protection against nisin and their homologs of C. glutamicum, deletion of porins, or modification of cell surface charge [47]. Some of these approaches yielded marginal improvements but, individually, none of them increased resistance more than two-fold. To test if they would synergize to create a more resistant strain suitable for production of active nisin, we combined several of these approaches creating C. glutamicum CR099ΔporAΔporHΔporB/pEKEx-vraDE. This strain carries clean deletions of the genes for porins PorA, PorH and PorB and harbours a plasmid for expression of the VraDE ABC-transporter of S. aureus, which is known to confer nisin resistance in this organism [53, 54]. When cultivated in the presence of 2 g/l CaCl2, i.e. a condition that also slightly increases resistance of C. glutamicum to nisin [47], this strain only showed about eightfold higher resistance to (Fig. 1B). Nevertheless, growth of C. glutamicum CR099ΔporAΔporHΔporB/pEKEx-vraDE under these conditions was also markedly reduced at concentrations around 5 µg/ml. Due to the rather low resistance to nisin and the observed pore formation at concentrations of ~ 200 ng/ml, we concluded that C. glutamicum is not suitable as a recombinant host for production of active nisin.
Construction of a nisin-specific biosensor
To be able to detect nisin in a specific manner, we first established a whole-cell biosensor based on nisin-inducible expression [55, 56] of a fluorescent protein in a L. lactis host. The promoter upstream of nisZ (Pnis) of L. lactis B1629 was amplified and fused to the gene for the fluorescent protein mCherry, which was optimized for codon usage of L. lactis (mcherryLl). This construct was cloned into the pNZ44 backbone and the obtained plasmid pNZ-Pnis-mcherryLl (Additional file 1: Fig. S2A) was introduced into L. lactis NZ9000 harbouring the genes coding for the two component nisin regulation system nisK (sensor kinase) and nisR (regulator) [56].
Following growth o/N in the presence of 10 ng/ml nisin, pellets of L. lactis NZ9000/ pNZ-Pnis-mcherryLl had a reddish colour indicating efficient expression of mCherry (Additional file 1: Fig. S2B). Further dose response experiments revealed a limit of detection of 0.1–0.2 ng/ml, maximum expression at 1.5–2 ng/ml (data not shown), and a linear dose response between 0 and 1 ng/ml (Additional file 1: Fig. S2C).
Production of prenisin using C. glutamicum and activation by trypsin
Based on the high sensitivity of C. glutamicum towards nisin and the unsuccessful attempts to create recombinant strains with significantly improved resistance we sought to establish a two-step process with production of (inactive) prenisin and downstream activation to (active) nisin by trypsin treatment as described previously [21, 22, 57]. To produce prenisin using C. glutamicum, the genes nisZ, nisB, nisT and nisC of the nisin Z biosynthesis operon of L. lactis B1629 (Additional file 1: Fig. S3A) were obtained as synthetic DNA fragments optimized for codon usage of C. glutamicum each equipped with a ribosomal binding site. The synthetic nisZBTCCg operon was cloned downstream of the Ptac promoter into pXMJ19 (Additional file 1: Fig. S3B) and the obtained plasmid pXMJ-nisZBTCCg was introduced into C. glutamicum CR099.
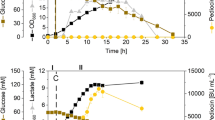
Growth of C. glutamicum CR099/pXMJ-nisZBTCCg on 2xTY complex medium containing 2% (w/v) glucose with addition of IPTG after 2 h was comparable to that of the empty vector control strain C. glutamicum CR099/pXMJ19 (Fig. 2A). Of note, supernatants of C. glutamicum CR099/pXMJ-nisZBTCCg were able to induce fluorescence in the nisin biosensor strain L. lactis NZ9000/pNZ-Pnis-mcherryLl following trypsin activation and fluorescence per OD increased with time (Fig. 2B). By contrast, supernatants of the empty vector control strain treated with trypsin did not induce fluorescence of the biosensor above background.

Production of prenisin using C. glutamicum and activation of prenisin to nisin culture supernatants. A Growth of C. glutamicum CR099/pXMJ-nisZBTCCg and CR099/pXMJ19 on 2xTY medium with 2% glucose and induction with IPTG (0.2 mM) after 2 h of growth. C Relative mCherry fluorescence normalized to OD600 (RFU/OD) of L. lactis NZ9000/pNZ-Pnis-mcherryLl grown o/N in the presence of supernatants of C. glutamicum CR099/pXMJ-nisZBTCg and CR099/pXMJ19 harvested at the indicated time points of the experiment shown in (B). Prior to assays, supernatants were activated by incubation with trypsin (0.5 mg/ml for 3.5 h) and diluted 1:100. Values are mean ± SD of n = 3 independent cultures
To further identify and characterize the compound produced by C. glutamicum CR099/pXMJ-nisZBTCCg, supernatant proteins were precipitated and analyzed by chromatography and mass spectrometry. Conditions of cultivation were slightly different with both precultures and main cultures supplemented with IPTG and supernatants harvested after o/N growth. Under these conditions, fluorescence of the biosensor incubated with supernatants diluted 1:1000 after trypsin activation was comparable to biosensors incubated in the presence of 0.5 ng/ml of nisin (Fig. 3A). This suggests the trypsin-treated supernatants of C. glutamicum CR099/pXMJ-nisZBTCCg contain at least 0.5 µg/ml of active nisin. Supernatants of these cultivations were harvested, and proteins were precipitated and purified by chromatography. In cation exchange (CIEX) chromatography a single peak in absorbance at 214 nm that coincided with a steep increase in conductivity was observed at the onset of elution (Fig. 3B). The peak fraction was collected and further analyzed by reverse phase (RP) chromatography with a two-step elution profile. This yielded a sharp peak in the second elution step at around 50% acetonitrile (Fig. 3B).

Purification and activation of prenisin produced by C. glutamicum. A Relative mCherry fluorescence normalized to OD (RFU/OD) of L. lactis NZ9000/pNZ-Pnis-mcherryLl grown o/N in the presence of supernatants (SN) of C. glutamicum CR099/pXMJ-nisZBTCCg. The producer was grown o/N in 2xTY with 2% Glc and 0.2 mM IPTG. B Purification of ammonium sulphate-precipitated SN proteins by cation exchange (CIEX) and subsequent reverse phase (RP) chromatography on the CIEX peak fraction. Indicated is absorbance at 214 nm (red) and conductance (mS/cm; black, in CIEX) or % of elution buffer (%B, black, in RP) over the elution volume. Boundaries of the peak fractions further analysed are marked with blue broken lines. C RFU/OD of L. lactis NZ9000/pNZ-Pnis-mcherryLl grown o/N in the presence of samples obtained at different steps during the purification of prenisin from SN shown in (A). prec: ammonium sulphate-precipitated SN proteins resuspended in pure H2O; CIEX and RP peak: peak fraction of the CIEX and RP chromatography. Prior to assays, samples were activated by incubation with trypsin (0.5 mg/ml for 3.5 h) and diluted 1:1000. As positive controls, the biosensor was grown in the presence of nisin Z at the indicated concentration. As negative controls, SN without trypsin treatment were included. D Mass spectrometry of the peak fraction obtained in RP chromatography in (B) with arbitrary peak intensity units (intensity [AU]) over mass/charge ratio (m/z). Values in (A) and (C) are mean ± SD of n = 3 independent cultures (A) or technical triplicates of one representative preparation (C)
Subsequent analysis of precipitated supernatant proteins, CIEX and RP peak fractions suggests that all samples contained a compound that induced fluorescence by the nisin-specific biosensor following activation by trypsin (Fig. 3C). Moreover, a signal with a mass/charge ratio (m/z) of 5666.7 was detected in MALDI-TOF/MS analyses (Fig. 3D). This is close to the predicted m/z of the nisin Z prepeptide harboring all required posttranslational modifications with a neutral net charge (Additional file 1: Fig. S3C). Collectively, these results suggest that recombinant production of fully modified prenisin using C. glutamicum and a protocol for purification and down-stream activation to nisin was established.
Improved activation of prenisin by recombinant NisP
In natural producers such as L. lactis B1629, prenisin is activated to nisin by the specific membrane anchored protease NisP [20, 22], which cleaves the prepeptide at the arginine residue in position 23 removing the leader peptide (Additional file 1: Fig. S3C). In line with previous studies [21, 22, 57, 58], we achieved activation of prenisin using the serine protease trypsin (Figs. 2 and 3), which cleaves proteins non-specifically at arginine and lysine residues [59]. However, nisin Z contains two internal lysin residues that also may serve as a substrate for trypsin (Fig. 4A). In fact, MS analyses revealed several of the predicted degradation products when commercial nisin Z was incubated with trypsin for 10 h and 24 h (Fig. 4B). Moreover, only a very weak signal corresponding to the m/z of a nisin standard (m/z = 3331.6) after 10 h of trypsinization and this signal was completely lost after 24 h of treatment.

MS analysis of trypsin-treated nisin Z samples. A Schematic representation of mature nisin Z. Potential trypsin cleavage sites are indicated by dashed lines and the possible nisin Z fragments that result from complete or partial cleavage and their corresponding molecular mass (in Da) are indicated by different letters (a–e). B Mass spectrometry of untreated nisin Z (−) or after treatment for 10 and 24 h with trypsin. Peaks corresponding to mature active nisin Z or fragments predicted to result from complete or partial cleavage by trypsin are highlighted with red boxes and arrows and their mass/charge ratios are indicated
To improve specificity of the activation step and increase product yield, we adopted a previously described approach employing a soluble NisP protease (sNisP) [21, 22]. An E. coli BL21 derivative harboring pEKEx-snisP-His6 for expression of a His-tagged version of sNisP was cloned and sNisP-His6 was purified form crude extract of this strain by immobilized metal affinity chromatography (IMAC; Fig. 5A). Analysis of the peak fraction by SDS-PAGE and following Western blot indicated the presence of a His6-tagged protein migrating around the size expected for sNisPHis6 (42 kDa; Fig. 5B).

Purification of a His6-tagged soluble variant of NisP. A Immobilized metal affinity chromatography (IMAC) of crude extracts of E. coli BL21/pEKEx-snisP-His6. Indicated is absorbance at 214 nm (red, left y-axis) and % of elution buffer (%B, black, right y-axis) over the elution volume. B Western blot analysis after SDS-PAGE using α-His6 antibody. Samples taken at different steps of the purification of sNisP-His6. CE: crude extract; FT: flowthrough of IMAC; E: eluate of IMAC peak fraction; M: molecular weight marker
This sNisP-His6 preparation was subsequently used to activate prenisin in supernatants of C. glutamicum CR099/pXMJ-nisZBTCCg. When activated with sNisP-His6, supernatants diluted 1:2500 induced fluorescence of the mCherry biosensor comparable to 0.5 ng/ml nisin (Fig. 6A) indicating presence of at least 1.25 µg/ml of active nisin. Additionally, the sNisP-His6 preparation of the IMAC peak fraction was concentrated by molecular weight cutoff filtration (cut-off 30 kDa) and used to activate prenisin in a concentrated RP peak fraction. As a control, the same concentrated RP peak fraction was activated with trypsin. Two-fold serial dilutions of these samples were investigated for activity using a growth-dependent assay with the nisin sensitive L. lactis strain IL1403 [60] as indicator strain (Fig. 6B). The results indicate that samples activated with sNisP-His6 contained about two-fold higher levels of active nisin than the standard (250 µg/ml), i.e. approx. 500 µg/ml of active nisin Z. By contrast, activation with trypsin yielded only half the activity of the standard solution, i.e. approx. 125 µg/ml. This suggests that the protocol for activation with sNisP-His6 yields four-fold higher levels of active nisin than trypsin activation. Additionally, MS-analyses of prenisin samples from RP purification activated by sNisP-His6 were performed (Fig. 6C). A signal with a mass/charge ratio (m/z) of 3331.1 was detected, which is in good agreement with the m/z (3330.4) of the mature nisin Z reported in a previous study [61]. Despite a number of additional peaks of unknown origin, the MS data indicated that sNisP-His6-dependent activation did not result in non-specific nisin degradation products as observed for trypsin treatment (Fig. 4B).

Detection of active nisin in SN of C. glutamicum CR099/pXMJ-nisZBTCCg or concentrated RP peak fractions activated with sNisP-His6. A RFU/OD of L. lactis NZ9000/pNZ-Pnis-mcherryLl grown o/N in the presence of supernatants (SN) of C. glutamicum CR099/pXMJ-nisZBTCCg or nisin (0.5 or 1 ng/ml). Prior to assays, supernatants were activated by incubation with the sNisP-His6 preparation shown in Fig. 5B for 3.5 h (sNisP) and diluted 1:2500. B Inhibition of growth (OD600) of L. lactis IL1403 by serial twofold dilutions concentrated RP peak fraction (RPC) or nisin Z (starting concentration of the standard solution 250 µg/ml). Where indicated, the RP peak fraction was activated with either trypsin (0.5 mg/ml) or sNisP-His6 for 3.5 h. All values are mean ± SD of n = 3 independent cultures. As negative controls untreated SN or RP were included. The broken red and green lines indicate OD600 of the positive (i.e. complete inhibition of growth) or negative (i.e. sterile medium) controls, respectively. C Mass spectrometry of sNisP-activated SN analysed in (A, B). The peak corresponding to mature, active nisin Z are highlighted with a red box
Discussion
At present, nisin is the only antimicrobial peptide approved as a food preservative by the FDA and EFSA [33, 62]. The global market for nisin reached 443 million USD in 2020 and is expected to increase to over 550 million USD by the year 2025 (marketsandmarkets.com). At the same time, the world is facing a dramatic increase in infections with antibiotic resistant bacteria and resistance to last resort antibiotics are rather common than an exception [4]. Hence, antimicrobial peptides are discussed as alternatives to classical antibiotics [5]. To allow use of bacteriocins for medical applications and ensure cost-efficient supply for other purposes, improved biotechnological production processes are needed. Production of nisin and other bacteriocins is currently performed with natural producer strains in batch fermentations on milk- or whey-based substrates [34, 36, 63]. These processes are well established and yield a product that is approved for use as food preservative and marketed world-wide. The complex media components of these substrates are, however, a major drawback for further downstream processing and purification steps [35] and limit the potential of nisin (and other bacteriocins) for clinical applications. Moreover, regulation of bacteriocin biosynthesis by feedback inhibition in natural producer strains results in low product yields [64]. An alternative to overcome these drawbacks may be transfer of nisin biosynthesis to a heterologous production host that can be cultivated to high cell densities on cheap, simple and well-defined media.
Heterologous production in E. coli was demonstrated for several lanthipeptides including nisin [65]. Similarly, biosynthesis of nisin and other lanthipeptides was transferred to Bacillus subtilis as expression host [66,67,68]. The present study was aimed at implementing a process for production of fully active nisin using C. glutamicum. This organism has several advantages: it has “generally regarded as safe” status, is widely used in biotechnology, can be cultivated to very high cell densities, is genetically well accessible, and several genome-reduced chassis strains are available [69,70,71]. Recently, we successfully demonstrated heterologous production of the class IIa bacteriocin pediocin PA-1 with C. glutamicum [45]. C. glutamicum lacks the receptor for pediocin, i.e. a mannose-PTS, and, consequently, is resistant to relevant concentrations of pediocin [45]. Nisin targets the universal cell wall precursor lipid II and hence a wide range of Gram-positive bacteria [26, 72]. It is thus not surprising that growth of C. glutamicum is inhibited by concentrations by far lower than the inhibitory concentrations of pediocin [45] making heterologous production of active nisin with C. glutamicum more difficult. We have thus tested various approaches to increase the resistance of C. glutamicum to nisin. These approaches included expression of (i) immunity proteins or (ii) ABC transporters known to confer nisin resistance in other organisms or their homologues in C. glutamicum, (iii) enzymes for alanylation or lysinylation of the cell envelope to introduce positive charges, and (iv) deletion of genes for porins of the outer membrane [47]. None of these approaches on their own yielded a substantial increase in resistance. Our results show that by combining, deletion of three porins, expression of an ABC transporter of S. aureus and addition of CaCl2, resistance of C. glutamicum to nisin was increased by a factor of 8. i.e. to around 5 µg/ml (Fig. 1B). Due to the low resistance of unmodified C. glutamicum to nisin and the rather marginal increase by various approaches, we decided to establish a two-step process with production of inactive prenisin using C. glutamicum and activation to nisin in a second downstream step.
Growth-dependent and pHluorin-dependent assays used and described here or elsewhere have a minimum detection limit in the low µg/ml range depending on the sensitivity of the sensor bacteria used [50, 73]. To establish a more sensitive and specific method to detect active nisin, we made use of the nisin autoregulation system of L. lactis [55]. An mcherry gene was fused to the promotor upstream of nisZ as reported previously [74], cloned into pNZ44 and introduced into L. lactis NZ9000 harboring the nisRK two-component system for sensing of mature nisin and regulation of the nisZ promoter. The obtained reporter stain showed strictly nisin-dependent mCherry florescence and had a limit of detection < 0.25 ng/ml of active nisin (Additional file 1: Fig. S2).
The minimal requirements for production of fully modified prenisin are the genes coding for the core peptide (nisZ) and for the modification and transport machinery (nisBCT), respectively [17]. In native producer strains, prenisin is cleaved by the membrane-anchored protease NisP [18] but presence of the arginine residue at position 23 allows cleavage and activation by trypsin [21, 22, 57]. For production of prenisin Z using C. glutamicum, a synthetic operon consisting of the genes nisZBTC codon-optimized for C. glutamicum each equipped with a strong ribosomal binding site was cloned under the IPTG-inducible Ptac promoter into pXMJ19. A recombinant C. glutamicum strain carrying this plasmid produced a compound that, after treatment with trypsin, was able to induce mCherry fluorescence in a highly specific nisin biosensor (Figs. 2 and 3). The purified compound had an m/z of 5666.7, which is in good agreement with the m/z of prenisin Z (5663). Of note, this compound was found in the supernatant of the recombinant producer. In a similar approach using expression of nisABC in E. coli active nisin was only obtained after purification of prenisin from crude extracts and activation by trypsin [65]. This clearly indicates functionality of the complete nisin biosynthesis, modification and export machinery in C. glutamicum, and successful production of completely modified prenisin Z that can be activated to mature nisin. Heterologous production of nisin (and other lantibiotics) in a two-step approach with downstream activation of the prepeptide has also been demonstrated with B. subtilis [66, 67]. However, in these studies successful production was demonstrated qualitatively and no quantification of the peptides or their bioactivity was performed. Thus, it is difficult to compare efficacy of (pre)nisin production with B. subtilis and C. glutamicum based on data available in literature.
Studies on heterologous production of (pre)nisin in B. subtilis made use of strains carrying an endogenous gene cluster for the lantibiotic subtilin [66, 67]. The recombinant producer strains were generated by replacing the gene for subtilin with a gene for prenisin plus other modification of the promoters. So, these strains produced prenisin with the biosynthetic machinery of subtilin. C. glutamicum lacks endogenous gene clusters for production of lanthipeptides and was modified to express the nisin biosynthetic machinery. This involved expression of membrane proteins for posttranslational modification and export. To our knowledge, this represents the first report on recombinant production of a posttranslationally modified peptide with heterologous expression of a complete set of enzymes for modification and transport.
It is hypothesized that trypsin only cleaves off the signal peptide and the presence of (methyl-)lanthionine rings prevents further proteolytic degradation at two lysin residues, i.e. potential trypsin cleavage sites, in the mature nisin molecule [75]. By contrast our MS data suggest unspecific cleavage of nisin at these lysin residues at least at longer incubation times (Fig. 4B). To increase specificity of prenisin activation, we adopted a strategy to produce a soluble version of the nisin-specific protease NisP as described previously [21, 22]. Following recombinant expression of sNisP-His6 in E. coli BL21 and purification by IMAC, prenisin was successfully activated in culture supernatants of C. glutamicum CR099/pXMJ-nisZBTCCg by adding sNisP-His6.
Based on semiquantitative determination using a standard solution of commercial nisin, at least 1.25 µg/ml were present in sNisP-activated culture supernatants. This is comparable to or slightly above levels reported for homologous production with L. lactis strains [58, 76]. Also, nisin activity was clearly improved compared to activation with trypsin (Fig. 6). Based on semiquantitative determination using a standard solution of commercial nisin, activity equivalent to approx. 500 µg/ml were obtained when RP-purified prenisin was activated with sNisP-His6. Moreover, this is about 20-fold higher than nisin levels obtained with E. coli [65].
Conclusions
In summary, our data demonstrates successful establishment of a two-step approach with recombinant production of prenisin using C. glutamicum and downstream activation using a soluble NisP protease. Thus, our results demonstrate that C. glutamicum may be used as heterologous production host not only for the non-modified class IIa bacteriocin pediocin [45] but also the fully modified lantibiotic nisin. Moreover, our approach may also be used to produce other lantibiotics by simply replacing the nisZ gene in the expression vector because the nisin modification and export machinery is promiscuous to other lanthipeptides as demonstrated in L. lactis [77]. Together with purification protocols e.g. by RP chromatography and downstream activation, this may be an interesting alternative for production of bacteriocins. As our experiments were carried out in rather simple batch cultivations in shake flasks, transfer to fed-batch or continuous fermentation processes may help to increase product yield and productivity. Moreover, C. glutamicum may be engineered to utilize a wide range of substrates [38] offering the possibility of production on sustainable substrates. Admittedly, the described two-step process yields a product derived from genetically modified organisms. Further studies are needed to establish production and purification processes that comply with good manufacturing practices required for clinical applications.
Methods
Strains and growth conditions
Strains and plasmids used in this study are listed in Additional file 1: Table S1. Bacteria were cultivated in 2xTY complex medium (C. glutamicum and E. coli) or GM17 medium (L. lactis) with constant agitation (130 rpm) at 30 °C (C. glutamicum and L. lactis) or 37 °C (E. coli). For heterologous production of prenisin with C. glutamicum, 5 ml reaction tubes were inoculated from a single colony and incubated overnight (o/N). The next morning, cultures were transferred to 50 ml 2xTY medium containing 0.05 mM isopropyl-β-d-thiogalactoside (IPTG) in baffled Erlenmeyer flasks for further 8 h of cultivation. Then, the complete 50 ml cell culture was transferred to 2 l baffled Erlenmeyer flask containing 450 ml 2xTY, glucose and IPTG were added to a final concentration of 2% (w/v) and 0.2 mM, respectively. Cells were then cultivated o/N at 30 °C with constant agitation (100 rpm).
For growth experiments in 50 ml 2xTY medium main cultures were inoculated from o/N precultivated cells to an optical density (OD600) of 1.2 and cultivated for 48 h in 500 ml baffled Erlenmeyer flasks. Two hours after inoculation, prenisin synthesis was induced by 2% (w/v) glucose and 0.2 mM IPTG.
For selection either 12.5 µg/ml chloramphenicol (Cm; for pXMJ-nisZBTCCg) or 50 µg/ml kanamycin (Kan; for pEKEx-nisZBTCCg or pEKEx-snisP-His6) was added where appropriate.
Cloning procedures
Molecular cloning procedures were performed using standard reagents according to the manufacturer’s instructions. The prenisin biosynthesis genes nisZ, nisB, nisC and nisT were codon-optimized for expression in C. glutamicum obtained as synthetic DNA fragments from a commercial service provider (Eurofins Genomics). PCR reactions were performed in a C100 thermocycler (Bio-Rad Laboratories, Munich, Germany), nucleotides were purchased from Bio-Budget (Krefeld, Germany). All primer and gene sequences are listed in Additional file 1: Table S2. Original gene sequences were extracted from the genome sequence of the nisin Z producer strain L. lactis B1629 isolated from fermented purple aubergine (collection of D.B. Diep, Laboratory of Microbial Gene Technology, Norwegian University of Life Sciences). For cloning of plasmids for production of prenisin, the nisZCg gene was excised from its cloning vector pEX-K168 by restrictions enzymes PstI and SalI and ligated into the shuttle vector pEKEx2 linearized by the same enzymes using T4-DNA ligase (Thermo Scientific). The resulting vector pEKEx-nisZCg was used as backbone for a Gibson Assembly [78] approach to introduce further genes. The nisBCg gene was amplified via PCR using Q5 high fidelity polymerase (New England Biolabs) and appropriate primers creating overlapping regions with additional ribosome binding sites (RBS: 5ʹ-AAGGAGTTTTC-3ʹ) and restriction sites. The amplified nisBCg fragment was fused via Gibson Assembly into the backbone pEKEx-nisZCg linearized by SalI. The resulting plasmid pEKEx-nisZBCg was then again linearized by SalI and fused with the PCR-amplified genes nisTCg and nisCCg in a second Gibson Assembly step yielding pEKEx-nisZBTCCg. Finally, pEKEx-nisZBTCCg and empty vector pXMJ19 were digested with SalI/SacI and the nisZBTCCg fragment was inserted into the pXMJ19 backbone by T4 DNA ligation yielding pXMJ-nisZBTCCg. Following cloning in E. coli DH5α, pXMJ-nisZBTCCg was transformed into C. glutamicum CR099 by electroporation as described previously [79].
For construction of a fluorescent nisin sensor plasmid the nisin-inducible promoter upstream of nisZ (Pnis) was PCR amplified from L. lactis spp. lactis B1629 genomic DNA. Additionally, the gene coding for the red-fluorescent protein mCherry was obtained as a synthetic DNA fragment codon-optimized for expression in L. lactis spp. cremoris (Eurofins Genomics; Additional file 1: Table S2) and also amplified via PCR. Both PCRs were performed using primers for generation of overlapping sequences for subsequent Gibson Assembly. The pNZ44 vector [80] was linearized by restriction enzymes BglII and NcoI thereby removing the p44 promotor. The Pnis and mcherryLl fragments were fused to the linearized vector via Gibson Assembly resulting in the sensor plasmid pNZ-Pnis-mcherryLl (Additional file 1: Fig. S2), which was transformed into L. lactis NZ9000 by electroporation [81] to obtain the nisin sensor strain L. lactis NZ9000/pNZ-Pnis-mcherryLl.
A soluble variant of the nisin protease nisP (sNisP) lacking the C-terminal LPXTG sortase motive was constructed as described previously [21]. The gene region coding for sNisP was amplified using L. lactis B1629 genomic DNA and primers snisP_fwd and snisP-6xH_rv (Additional file 1: Table S2) adding a 6× histidine tag (sNisP-His6). The PCR product was fused to the BamHI-linearized pEKEx2 vector by Gibson Assembly yielding pEKEx-snisP-His6. Following cloning in E. coli DH5α the construct was transformed into E. coli BL21 for efficient expression of sNisP-His6.
The plasmid pPB-pHin2Cg was constructed as follows. Plasmid pPBEx2 [52] was digested with PstI and PvuII to remove the Ptac promoter as well as the first 1044 bp of the lacI gene. A fragment containing 179 bp upstream of the tuf gene of C. glutamicum, i.e. the highly active constitutive tuf promoter [82] fused to a pHluorin2 gene [49] codon-optimized for C. glutamicum was synthesized by a commercial service provider (Eurofins Genomics), obtained in the pEX-K168 cloning vector. The insert was cut out and ligated to the pPBEx2 backbone to yield pPB-pHin2Cg. All plasmids were verified for correct cloning by restriction analysis and Sanger sequencing prior to transformation in their final hosts.
Assessment of membrane damage
For detection of membrane damage, the pHluorin assay described recently [50], which is based on the ratiometric pHluorin by Miesenböck et al. [83], was adapted for C. glutamicum. For this purpose, C. glutamicum ATCC 13032 was transformed with pPB-pHin2Cg. The sensor strain C. glutamicum ATCC 13032/pPB-pHin2Cg was grown o/N in 5 ml BHI containing 50 µg/ml Kan. The next day, cells were harvested by centrifugation and resuspended in Listeria minimal buffer (LMB, pH 6.2) at an optical density at 600 nm (OD600) of 3. Two-fold serial dilutions of samples for analysis were prepared in a black 96-well microtiter plates (Sarsted, Nümbrecht, DE) with a final volume of 100 µl in each well. Then, 100 µl of the sensor strain suspension was added and the plate was incubated at room temperature in the dark for 30 min. Readout was performed by measuring pHluorin2 fluorescence at 510 nm emission either across an excitation spectrum (350–490 nm) or with excitation at the distinct pHluorin2 maxima (400 and 470 nm) using an infinite M200 plate reader (Tecan, Männedorf, CH).
Detection of activated pre-nisin using mCherry-based sensor bacteria
For analysis of nisin/prenisin standards or samples, 5 ml of GM17 medium supplemented with 10 µg/ml Cm was inoculated with a single colony of L. lactis NZ9000/pNZ-Pnis-mCherryLl and grown for 8 h at 30 °C under continuous shaking (160 rpm). Afterwards, 100 µl of the preculture were used to inoculated 2.5 ml of fresh GM17 (+ 10 µg/ml Cm) medium containing the respective (pre-)nisin sample in the indicated dilution. As reference, different nisin standards and sole medium was used. The cultures were cultivated o/N at 30 °C under constant agitation (130 rpm). For mCherry fluorescence measurements, cells were centrifuged (3200g; 8 min; RT) and resuspended in 500 µl saline (0.9% (w/v)). Afterwards, 100 µl of each cell suspension was transferred to a black 96× well microtiter plate (Sarstedt) for measurements in technical triplicates. Fluorescence levels were determined in an Infinite M200 microplate reader (Tecan) with an excitation wavelength set to 570 nm and recorded emission at 610 nm. The optical density of the remaining cell suspension was determined spectroscopically and used to normalize the obtained fluorescence values.
Growth inhibition assay
If appropriate, antimicrobial activity of previously activated prenisin samples was assessed by a growth inhibition assay in a similar manner as described before [45] using the nisin-sensitive L. lactis IL1403 as a sensor strain. Bacteria were grown o/N at 30 °C in GM17 medium containing 10 µg/ml Cm and diluted 1:25 in fresh GM17 prior to the assay. 100 µl of serial twofold dilutions of samples (100 µl) or a nisin standard were mixed with 100 µl of indicator bacteria in sterile 96-well plates. The plates were incubated at 30 °C for 6 h and growth was monitored by measuring the OD600 in an Infinite M200 plate reader (Tecan). Based on nisin standards of known concentration the nisin levels in activated prenisin samples were determined in a semi-quantitative manner. Growth inhibition assays were also used to evaluate nisin resistance levels of different C. glutamicum strains as described previously [47].
Purification of prenisin
Purification of prenisin from cell-free supernatants was adopted from a previously described method [84]. In brief, supernatant proteins were precipitated o/N at 4 °C by (NH4)2SO4 (final concentration: 50% (w/v)). The precipitate was collected by centrifugation (45 min, 10,000g, 4 °C), resuspended in HPLC-grade H2O (1/10 of the initial volume), and pH was adjusted to 3.9 using 2 M HCl. After a further centrifugation step (45 min, 10,000g, 4 °C) to remove insoluble particles, the supernatant was applied to a HiPrep SP FF 16/10 column (GE Healthcare Life Sciences) for subsequent CIEX chromatography. Equilibration of column was done with 20 mM sodium phosphate buffer at pH 3.9 followed by 5 column volumes (CVs) washing step using 20 mM sodium phosphate buffer at pH 6.9. Elution was carried out with a single step using 5 CVs of 20 mM sodium phosphate buffer at pH 6.9 with 2 M NaCl. The eluate was collected to 5 ml fractions. Peak-fractions were pooled and applied to a Resource RP chromatography column (GE Healthcare Life Sciences). Elution of prenisin was done by a two-step protocol: (1) 5 CV of 10% buffer B (85% (v/v) acetonitrile with 0.1% (v/v) trifluoroacetic (TFA) acid), (2) linear increase of buffer B to 100% over 20 CV (flow rate for both steps: 1.0 ml/min). Prenisin eluted at approx. 50% of buffer B. All purification steps were performed with the Äkta-Pure system (GE Healthcare Life Sciences).
Purification a His-tagged, soluble NisP protease
A soluble variant of the nisin protease NisP carrying an 6×His-tag (sNisP-His6) for purification by immobilized metal affinity chromatography (IMAC) was produced using E. coli BL21/pEKEx2-snisP-His6. A single colony of this strain was used to inoculate 5 ml of 2xTY. This culture was incubated o/N at 37 °C and then transferred to 50 ml 2xTY containing 0.2 mM IPTG and incubated at 37 °C for 8 h. To produce sNisP-His6, 250 ml Terrific broth (TB) medium were inoculated with 10% (v/v) of the 50 ml preculture and incubated o/N at 30 °C in the presence of 1 mM IPTG. To prepare sNisP-His6, bacteria of 50 ml aliquots of the o/N culture were harvested by centrifugation and each resuspended in 30 ml IMAC binding buffer (20 mM sodium phosphate buffer, pH 7.4; 0.5 M NaCl; 20 mM imidazole). Bacteria were disrupted by four passages through French Press (SLM Instruments) at 1100 psi. After removal of cell debris (2 × 10,000 g; 30 min; 4 °C) the supernatant, i.e. crude cell extract, was applied to the HisTrap FF 1 ml column (GE Healthcare Life Sciences) using a 50 ml super-loop. Protein bound to the columns was eluted by step gradient with high imidazole buffer (20 mM sodium phosphate buffer, pH 7.4; 0.5 M NaCl; 300 mM imidazole). To remove imidazole, 500 µl of the eluate were applied to a 30 kDa cut-off filter (Carl Roth GmbH) and retained protein sample was resuspended in 100 µl activation buffer (50 mM MOPS, pH 6.8; 50 mM NaCl) and stored at − 20 °C until usage for prenisin activation.
SDS-PAGE and Western blot analysis
To confirm presence of sNisP-His6 in different fractions from previous purification, a Western blot was performed. First, a sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) on a tris-glycine, 10% polyacrylamide gel (Biorad) loaded with 10 µl of each sample fraction was performed. 5 µl of PageRuler™ prestained protein ladder 10–180 kDa (Thermo Scientific) were used as molecular weight marker. After electrophoresis (constant 30 mA; 250 V), gels were transferred for western blotting following a semi-dry blotting procedure in a Trans-blot turbo system (Biorad) according to the manufacturer’s protocol. For specific detection of sNisP-His6, a mouse mAb targeted against the 6×His-tag (Invitrogen, Thermo Scientific; Cat# MA1-21315, diluted 1:5000 in TBST buffer) was used. As secondary antibody, HRP-conjugated anti-Mouse goat IgG (Sigma-Aldrich; Cat# 12-349, diluted 1:5000 in TBST) was used. Detection was performed using the SuperSignal™ West Femto Maximum Sensitivity Substrate and the iBright Imaging System (ThermoFisher Scientific, Dreieich, Germany).
Prenisin activation
Inactive prenisin can be activated either by the natural nisin protease NisP or using trypsin [21, 84], which both cleave after the arginine residue at position 23 of the leader peptide. For trypsin activation, supernatants, precipitation or samples from different purification steps were incubated with 1/10 of a 5 mg/ml trypsin (Sigma Aldrich) solution for 3.5 h at 37 °C. Activation by sNisP-His6 was performed using concentrated (by 30 kDa cut-off filter) purified sNisP-His6 dissolved in activation buffer. The mix of prenisin and sNisP was incubated for the indicated time period in a prenisin:sNisP-His6 ratio of 10:1. Activated prenisin samples were directly used for further analyses or stored at − 20 °C. For activation of purified prenisin, RP fractions were concentrated by evaporation in a speed vacuum concentrator (Eppendorf) and resuspended in the same activation buffer as sNisP-His6.
Nisin trypsinization experiments
For trypsin digestion, nisin Z was purified from supernatants of the L. lactis producer strain by ammonium sulfate precipitation, followed by CIEX and RP chromatography. The purified nisin solution, containing 87% of nisin, was divided into three aliquots and dried at 55 °C in a SpeedVac concentrator (SPD2010 Integrated SpeedVac, ThermoFisher Scientific, USA). The pellet samples were resuspended in equal volumes of Tris-HCl 50 mM buffer, pH 8.0, containing 0.2 mg/ml of trypsin from porcine pancreas (Sigma Aldrich). The positive control sample was resuspended in Tris-HCl 50 mM buffer, pH 8.0 without trypsin. For partial digestion the nisin-trypsin mixture was incubated for 10 h at 37 °C. For total digestion the mixture was incubated for 24 h at 37 °C. Positive control sample was also incubated for 24 h at 37 °C.
To confirm the nisin digestion, activity was measured using a growth-dependent microtiter plate assay with L. lactis IL1403 as indicator as described above. Additionally, 5 µl of each sample was desalted with C18 loaded pipette tips (Millipore) and applied on MALDI-TOF spectrometer.
MALDI-TOF analysis
For verification and further analysis of (pre)-nisin MALDI-TOF was performed. RP elution samples were concentrated by evaporation in a speed vacuum concentrator (Eppendorf) and resuspended in activation buffer (50 mM MOPS, pH 6.8; 50 mM NaCl) to 1/10 of the initial volume. Concentrated prenisin RP samples were analysed either with or without activation by sNisP-His6 or trypsin MALDI-TOF spectra were recorded on an Ultraflex III MS (Bruker Daltonics) operated in reflection mode with delayed extraction. Ions of positive charge in the m/z range of 200 to 6000 were analysed using 25 kV acceleration voltage. The sample spectra were calibrated externally with a calibration standard, m/z range from 700 to 3100 (Bruker Daltonics, Bremen, Germany).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its additional information files.
Change history
17 February 2022
A Correction to this paper has been published: https://doi.org/10.1186/s12934-022-01744-1
Abbreviations
- A:
-
Ampere
- ABC-transporter:
-
ATP binding cassette transporter
- ATCC:
-
American Type Culture Collection
- BHI:
-
Brain heart infusion
- CTAB:
-
Cetyltrimethylammonium bromide
- Cm:
-
Chloramphenicol
- CIEX:
-
Cation ion exchange chromatography
- CV:
-
Column volume
- °C:
-
Degree celsius
- Da:
-
Dalton
- ESFA:
-
European Food Safety Authority
- et al.:
-
Et alii
- g:
-
Gram
- h:
-
Hour
- IPTG:
-
Isopropyl-β-d-thiogalactopyranoside
- IMAC:
-
Immobilized metal ion affinity chromatography
- k:
-
Kilo
- LAB:
-
Lactic acid bacteria
- l:
-
Liter
- MALDI/TOF:
-
Matrix-assisted laser desorption-ionization/time of flight
- MS:
-
Mass spectrometry
- MIC:
-
Minimal inhibitory concentration
- m/z:
-
Mass/charge
- M:
-
Molar
- m:
-
Milli
- min:
-
Minute
- µ:
-
Micro
- n:
-
Nano
- nm:
-
Nanometer
- OD600 :
-
Optical density at 600 nm
- o/N:
-
Over night
- %:
-
Percent
- RPC:
-
Reversed-phase chromatography
- RT:
-
Room temperature
- SN:
-
Supernatant
- SD:
-
Standard deviation
- SDS-PAGE:
-
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- V:
-
Volt
- WHO:
-
World Health Organization
- WT:
-
Wildtype
References
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol. 2010;8:251–9.
Aslam B, Wang W, Arshad MI, Khurshid M, Muzammil S, Rasool MH, et al. Antibiotic resistance: a rundown of a global crisis. Infect Drug Resist. 2018;11:1645–58.
Berendonk TU, Manaia CM, Merlin C, Fatta-Kassinos D, Cytryn E, Walsh F, et al. Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol. 2015;13:310–7.
World Health Organization. Antimicrobial resistance: global report on surveillance. 2014. https://www.who.int/publications/i/item/WHO-HSE-PED-AIP-2014.2. Accessed 14 June 2018.
Cotter PD, Ross RP, Hill C. Bacteriocins—a viable alternative to antibiotics? Nat Rev Microbiol. 2013;11:95–105.
Soltani S, Hammami R, Cotter PD, Rebuffat S, Said LB, Gaudreau H, et al. Bacteriocins as a new generation of antimicrobials: toxicity aspects and regulations. FEMS Microbiol Rev. 2021. https://doi.org/10.1093/femsre/fuaa039.
Meade E, Slattery MA, Garvey M. Bacteriocins, potent antimicrobial peptides and the fight against multi drug resistant species: resistance is futile? Antibiotics. 2020;9:32.
Lubelski J, Rink R, Khusainov R, Moll GN, Kuipers OP. Biosynthesis, immunity, regulation, mode of action and engineering of the model lantibiotic nisin. Cell Mol Life Sci. 2008;65:455–76.
Kuipers OP, Beerthuyzen MM, Siezen RJ, De Vos WM. Characterization of the nisin gene cluster nisABTCIPR of Lactococcus lactis. Requirement of expression of the nisA and nisI genes for development of immunity. Eur J Biochem. 1993;216:281–91.
Ra SR, Qiao M, Immonen T, Pujana I, Saris PEJ. Genes responsible for nisin synthesis, regulation and immunity form a regulon of two operons and are induced by nisin in Lactoccocus lactis N8. Microbiology. 1996;142:1281–8.
Koponen O, Tolonen M, Qiao M, Wahlström G, Helin J, Saris PEJ. NisB is required for the dehydration and NisC for the lanthionine formation in the post-translational modification of nisin. Microbiology. 2002;148:3561–8.
Sen AK, Narbad A, Horn N, Dodd HM, Parr AJ, Colquhoun I, et al. Post-translational modification of nisin. The involvement of NisB in the dehydration process. Eur J Biochem. 1999;261:524–32.
Li B, Van Der Donk WA. Identification of essential catalytic residues of the cyclase NisC involved in the biosynthesis of nisin. J Biol Chem. 2007;282:21169–75.
Okeley NM, Paul M, Stasser JP, Blackburn N, Van Der Donk WA. SpaC and NisC, the cyclases involved in subtilin and nisin biosynthesis, are zinc proteins. Biochemistry. 2003;42:13613–24.
Kuipers A, De Boef E, Rink R, Fekken S, Kluskens LD, Driessen AJM, et al. NisT, the transporter of the lantibiotic nisin, can transport fully modified, dehydrated, and unmodified prenisin and fusions of the leader peptide with non-lantibiotic peptides. J Biol Chem. 2004;279:22176–82.
Qiao M, Saris PEJ. Evidence for a role of NisT in transport of the lantibiotic nisin produced by Lactococcus lactis N8. FEMS Microbiol Lett. 1996;144:89–93.
Van Bart Den Berg Van Saparoea H, Bakkes PJ, Moll GN, Driessen AJM. Distinct contributions of the nisin biosynthesis enzymes NisB and NisC and transporter NisT to prenisin production by Lactococcus lactis. Appl Environ Microbiol. 2008;74:5541–8.
Siezen RJ, Rollema HS, Kuipers OP, De Vos WM. Homology modelling of the Lactococcus lactis leader peptidase nisp and its interaction with the precursor of the lantibiotic nisin. Protein Eng Des Sel. 1995;8:117–25.
Van der Meer JR, Polman J, Beerthuyzen MM, Siezen RJ, Kuipers OP, De Vos WM. Characterization of the Lactococcus lactis nisin A operon genes nisP, encoding a subtilisin-like serine protease involved in precursor processing, and nisR, encoding a regulatory protein involved in nisin biosynthesis. J Bacteriol. 1993;175:2578–88.
Van Der Meer JR, Rollema HS, Siezen RJ, Beerthuyzen MM, Kuipers OP, De Vos WM. Influence of amino acid substitutions in the nisin leader peptide on biosynthesis and secretion of nisin by Lactococcus lactis. J Biol Chem. 1994;269:3555–62.
Lagedroste M, Smits SHJ, Schmitt L. Substrate specificity of the secreted nisin leader peptidase NisP. Biochemistry. 2017;56:4005–14.
Montalbán-López M, Deng J, van Heel AJ, Kuipers OP. Specificity and application of the lantibiotic protease NisP. Front Microbiol. 2018;9:160.
Breukink E, Wiedemann I, Van Kraaij C, Kuipers OP, Sahl HG, De Kruijff B. Use of the cell wail precursor lipid II by a pore-forming peptide antibiotic. Science. 1999;286:2361–4.
Brötz H, Josten M, Wiedemann I, Schneider U, Götz F, Bierbaum G, et al. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol Microbiol. 1998;30:317–27.
Hsu STD, Breukink E, Tischenko E, Lutters MAG, De Kruijff B, Kaptein R, et al. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11:963–7.
Hasper HE, de Kruijff B, Breukink E. Assembly and stability of nisin−lipid II pores †. Biochemistry. 2004;43:11567–75.
Benkerroum N, Sandine WE. Inhibitory action of nisin against Listeria monocytogenes. J Dairy Sci. 1988;71:3237–45.
Harris LJ, Fleming HP, Klaenhammer TR. Sensitivity and resistance of Listeria monocytogenes ATCC 19115, Scott A, and UAL500 to nisin. J Food Prot. 1991;54:836–40.
Brumfitt W, Salton MRJ, Hamilton-Miller JMT. Nisin, alone and combined with peptidoglycan-modulating antibiotics: activity against methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci. J Antimicrob Chemother. 2002;50:731–4.
Jensen C, Li H, Vestergaard M, Dalsgaard A, Frees D, Leisner JJ. Nisin damages the septal membrane and triggers DNA condensation in methicillin-resistant Staphylococcus aureus. Front Microbiol. 2020;11:1–8.
Millette M, Dupont C, Shareck F, Ruiz MT, Archambault D, Lacroix M. Purification and identification of the pediocin produced by Pediococcus acidilactici MM33, a new human intestinal strain. J Appl Microbiol. 2008;104:269–75.
Reinseth IS, Ovchinnikov KV, Tønnesen HH, Carlsen H, Diep DB. The increasing issue of vancomycin-resistant enterococci and the bacteriocin solution. Probiotics Antimicrob Proteins. 2020;12:1203–17.
Delves-Broughton J. Applications of the bacteriocin, nisin. Antonie Van Leeuwenhoek Int J Gen Mol Microbiol. 1996;69:193–202.
de Arauz LJ, Jozala AF, Mazzola PG, Vessoni Penna TC. Nisin biotechnological production and application: a review. Trends Food Sci Technol. 2009;20:146–54.
Abbasiliasi S, Tan JSS, Tengku Ibrahim TAA, Bashokouh F, Ramakrishnan NRR, Mustafa S, et al. Fermentation factors influencing the production of bacteriocins by lactic acid bacteria: a review. RSC Adv. 2017;7:29395–420.
Juturu V, Wu JC. Microbial production of bacteriocins: latest research development and applications. Biotechnol Adv. 2018;36:2187–200.
Taylor TM, Davidson PM, Zhong Q. Extraction of nisin from a 2.5% commercial nisin product using methanol and ethanol solutions. J Food Prot. 2007;70:1272–6.
Becker J, Rohles CM, Wittmann C. Metabolically engineered Corynebacterium glutamicum for bio-based production of chemicals, fuels, materials, and healthcare products. Metab Eng. 2018;50:122–41.
Eggeling L, Bott M. A giant market and a powerful metabolism: l-lysine provided by Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2015;99:3387–94.
Wolf S, Becker J, Tsuge Y, Kawaguchi H, Kondo A, Marienhagen J, et al. Advances in metabolic engineering of Corynebacterium glutamicum to produce high-value active ingredients for food, feed, human health, and well-being. Essays Biochem. 2021. https://doi.org/10.1042/ebc20200134.
Bückle-Vallant V, Krause FS, Messerschmidt S, Eikmanns BJ. Metabolic engineering of Corynebacterium glutamicum for 2-ketoisocaproate production. Appl Microbiol Biotechnol. 2014;98:297–311.
Krause FS, Blombach B, Eikmanns BJ. Metabolic engineering of Corynebacterium glutamicum for 2-Ketoisovalerate production. Appl Environ Microbiol. 2010;76:8053–61.
Yim SS, Choi JW, Lee RJ, Lee YJ, Lee SH, Kim SY, et al. Development of a new platform for secretory production of recombinant proteins in Corynebacterium glutamicum. Biotechnol Bioeng. 2016;113:163–72.
Yim SS, An SJ, Choi JW, Ryu AJ, Jeong KJ. High-level secretory production of recombinant single-chain variable fragment (scFv) in Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2014;98:273–84.
Goldbeck O, Desef DN, Ovchinnikov KV, Perez-Garcia F, Christmann J, Sinner P, et al. Establishing recombinant production of pediocin PA-1 in Corynebacterium glutamicum. Metab Eng. 2021. https://doi.org/10.1016/j.ymben.2021.09.002.
Sieger B, Schubert K, Donovan C, Bramkamp M. The lipid II flippase RodA determines morphology and growth in Corynebacterium glutamicum. Mol Microbiol. 2013;90:966–82.
Weixler D, Goldbeck O, Seibold GM, Eikmanns BJ, Riedel CU. Towards improved resistance of Corynebacterium glutamicum against nisin. 2021. p. 2021.08.09.454123. https://www.biorxiv.org/content/10.1101/2021.08.09.454123v1
Goldbeck O, Weixler D, Eikmanns BJ, Riedel CU. In silico prediction and analysis of unusual lantibiotic resistance operons in the genus Corynebacterium. Microorganisms. 2021;9:646.
Mahon MJ. pHluorin2: an enhanced, ratiometric, pH-sensitive green florescent protein. Adv Biosci Biotechnol. 2011;02:132–7.
Crauwels P, Schäfer L, Weixler D, Bar NS, Diep DB, Riedel CU, et al. Intracellular pHluorin as sensor for easy assessment of bacteriocin-induced membrane-damage in Listeria monocytogenes. Front Microbiol. 2018;9:3038.
Becker J, Klopprogge C, Zelder O, Heinzle E, Wittmann C. Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl Environ Microbiol. 2005;71:8587–96.
Bakkes PJ, Ramp P, Bida A, Dohmen-Olma D, Bott M, Freudl R. Improved pEKEx2-derived expression vectors for tightly controlled production of recombinant proteins in Corynebacterium glutamicum. Plasmid. 2020;112:102540.
Arii K, Kawada-Matsuo M, Oogai Y, Noguchi K, Komatsuzawa H. Single mutations in BraRS confer high resistance against nisin A in Staphylococcus aureus. MicrobiologyOpen. 2019;8:1–14.
Hiron A, Falord M, Valle J, Débarbouillé M, Msadek T. Bacitracin and nisin resistance in Staphylococcus aureus: a novel pathway involving the BraS/BraR two-component system (SA2417/SA2418) and both the BraD/BraE and VraD/VraE ABC transporters. Mol Microbiol. 2011;81:602–22.
Kuipers OP, Beerthuyzen MM, De Ruyter PGGA, Luesink EJ, De Vos WM. Autoregulation of nisin biosynthesis in Lactococcus lactis by signal transduction. J Biol Chem. 1995;270:27299–304.
Kuipers OP, De Ruyter PGGA, Kleerebezem M, De Vos WM. Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol. 1998;64:15–21.
Plat A, Kluskens LD, Kuipers A, Rink R, Moll GN. Requirements of the engineered leader peptide of nisin for inducing modification, export, and cleavage. Appl Environ Microbiol. 2011;77:604–11.
Lagedroste M, Reiners J, Smits SHJ, Schmitt L. Systematic characterization of position one variants within the lantibiotic nisin. Sci Rep. 2019;9:1–11.
Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol Cell Proteomics. 2004;3:608–14.
Kramer NE, van Hijum SAFT, Knol J, Kok J, Kuipers OP. Transcriptome analysis reveals mechanisms by which Lactococcus lactis acquires nisin resistance. Antimicrob Agents Chemother. 2006;50:1753–61.
Schneider N, Werkmeister K, Pischetsrieder M. Analysis of nisin A, nisin Z and their degradation products by LCMS/MS. Food Chem. 2011;127:847–54.
Cotter PD, Hill C, Ross RP. Food microbiology: bacteriocins: developing innate immunity for food. Nat Rev Microbiol. 2005;3:777–88.
Li C, Bai J, Cai Z, Ouyang F. Optimization of a cultural medium for bacteriocin production by Lactococcus lactis using response surface methodology. J Biotechnol. 2002;93:27–34.
Özel B, Şimşek Ö, Akçelik M, Saris PEJ. Innovative approaches to nisin production. Appl Microbiol Biotechnol. 2018. https://doi.org/10.1007/s00253-018-9098-y.
Shi Y, Yang X, Garg N, Van Der Donk WA. Production of lantipeptides in Escherichia coli. J Am Chem Soc. 2011;133:2338–41.
Rintala H, Graeffe T, Paulin L, Kalkkinen N, Saris PEJ. Biosynthesis of nisin in the subtilin producer Bacillus subtilis ATCC6633. Biotechnol Lett. 1993;15:991–6.
van Tilburg AY, van Heel AJ, Stülke J, de Kok NAW, Rueff A-S, Kuipers OP. Mini bacillus PG10 as a convenient and effective production host for lantibiotics. ACS Synth Biol. 2020;9:1833–42.
Yuksel S, Hansen JN. Transfer of nisin gene cluster from Lactococcus lactis ATCC 11454 into the chromosome of Bacillus subtilis 168. Appl Microbiol Biotechnol. 2007;74:640–9.
Becker J, Wittmann C. Industrial microorganisms: Corynebacterium glutamicum. Ind Microorg. 2017. https://doi.org/10.1002/9783527807796.ch6.
Baumgart M, Unthan S, Rückert C, Sivalingam J, Grünberger A, Kalinowski J, et al. Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl Environ Microbiol. 2013;79:6006–15.
Unthan S, Baumgart M, Radek A, Herbst M, Siebert D, Brühl N, et al. Chassis organism from Corynebacterium glutamicum—a top-down approach to identify and delete irrelevant gene clusters. Biotechnol J. 2015;10:290–301.
Sahl HG, Jack RW, Bierbaum G. Biosynthesis and biological activities of lantibiotics with unique post-translational modifications. Eur J Biochem. 1995;230:827–53.
Papagianni M, Avramidis N, Filioussis G, Dasiou D, Ambrosiadis I. Determination of bacteriocin activity with bioassays carried out on solid and liquid substrates: assessing the factor “indicator microorganism.” Microb Cell Factories. 2006;5:30.
Landete JM, Langa S, Escudero C, Peirotén Á, Arqués JL. Fluorescent detection of nisin by genetically modified Lactococcus lactis strains in milk and a colonic model: application of whole-cell nisin biosensors. J Biosci Bioeng. 2020;129:435–40.
Chan WC, Leyland M, Clark J, Dodd HM, Lian LY, Gasson MJ, et al. Structure-activity relationships in the peptide antibiotic nisin: antibacterial activity of fragments of nisin. FEBS Lett. 1996;390:129–32.
Reiners J, Lagedroste M, Ehlen K, Leusch S, Zaschke-Kriesche J, Smits SHJ. The N-terminal region of nisin is important for the BceAB-type ABC transporter NsrFP from streptococcus agalactiae COH1. Front Microbiol. 2017;8:1–10.
Van Heel AJ, Kloosterman TG, Montalban-Lopez M, Deng J, Plat A, Baudu B, et al. Discovery, production and modification of five novel lantibiotics using the promiscuous nisin modification machinery. ACS Synth Biol. 2016;5:1146–54.
Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–5.
Tauch A, Kirchner O, Löffler B, Götker S, Pühler A, Kalinowski J. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol. 2002;45:362–7.
McGrath S, Fitzgerald GF, van Sinderen D. Improvement and optimization of two engineered phage resistance mechanisms in Lactococcus lactis. Appl Environ Microbiol. 2001;67:608–16.
Holo H, Nes IF. Transformation of lactococcus by electroporation. In: Nickoloff JA, editor. Electroporation protocols for microorganisms. New Jersey: Humana Press; 1995. p. 195–9.
Lange J, Müller F, Takors R, Blombach B. Harnessing novel chromosomal integration loci to utilize an organosolv-derived hemicellulose fraction for isobutanol production with engineered Corynebacterium glutamicum. Microb Biotechnol. 2018;11:257–63.
Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–5.
Ovchinnikov KV, Chi H, Mehmeti I, Holo H, Nes IF, Diep DB. Novel group of leaderless multipeptide bacteriocins from gram-positive bacteria. Appl Environ Microbiol. 2016;82:5216–24.
Funding
Open Access funding enabled and organized by Projekt DEAL. This project has received funding from the Bio Based Industries Joint Undertaking under the European Union’s Horizon 2020 research and innovation program (Grant agreement No 790507).
Author information
Authors and Affiliations
Contributions
DW performed molecular cloning and experimental work except for MALDI-TOF analysis and pHluorin studies and was involved in writing, review and editing of the manuscript. MB constructed initial pEKEx2-based plasmids for production of prenisin and established mCherry sensor assays. KVO performed mass spectrometry analyses. OG was involved in prenisin purification and writing, review and editing of the manuscript. SJR designed the C. glutamicum pHluorin2 biosensor, conducted the respective experiments, and was involved in writing, review and editing of the manuscript. GMS, CW, NSB, and BJE were involved in acquisition of funding, conceptualization of the study and writing, review and editing of the manuscript. DBD provided support performing and analyzing mass spectrometry data and was involved in in acquisition of funding, conceptualization of the study and writing, review and editing of the manuscript. CUR analyzed and visualized all data and was involved in acquisition of funding, conceptualization of the study and writing, review and editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
OG, DW, GMS, CW, NSB, DBD, and CUR are co-inventors on a patent application related to this research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the figure 3d is revised.
Supplementary Information
Additional file 1
: Figure S1. Generation and properties of a C. glutamicum biosensor for detection of membrane damage. Figure S2. Generation and properties of L. lactis NZ9000/pNZ-Pnis-mcherryLl biosensor for specific detection of nisin. Figure S3. Genetic organization of natural nisin Z operon of L. lactis B1629 and structure of prenisin Z. Table S1. Bacterial strains and plasmids used in this study. Table S2. Oligonucleotide primers and synthetic gene sequences used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Weixler, D., Berghoff, M., Ovchinnikov, K.V. et al. Recombinant production of the lantibiotic nisin using Corynebacterium glutamicum in a two-step process. Microb Cell Fact 21, 11 (2022). https://doi.org/10.1186/s12934-022-01739-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-022-01739-y