
Abstract
Background
Nicotinamide adenine dinucleotide phosphate (NADPH) is an important cofactor ensuring intracellular redox balance, anabolism and cell growth in all living systems. Our recent multi-omics analyses of glucoamylase (GlaA) biosynthesis in the filamentous fungal cell factory Aspergillus niger indicated that low availability of NADPH might be a limiting factor for GlaA overproduction.
Results
We thus employed the Design-Build-Test-Learn cycle for metabolic engineering to identify and prioritize effective cofactor engineering strategies for GlaA overproduction. Based on available metabolomics and 13C metabolic flux analysis data, we individually overexpressed seven predicted genes encoding NADPH generation enzymes under the control of the Tet-on gene switch in two A. niger recipient strains, one carrying a single and one carrying seven glaA gene copies, respectively, to test their individual effects on GlaA and total protein overproduction. Both strains were selected to understand if a strong pull towards glaA biosynthesis (seven gene copies) mandates a higher NADPH supply compared to the native condition (one gene copy). Detailed analysis of all 14 strains cultivated in shake flask cultures uncovered that overexpression of the gsdA gene (glucose 6-phosphate dehydrogenase), gndA gene (6-phosphogluconate dehydrogenase) and maeA gene (NADP-dependent malic enzyme) supported GlaA production on a subtle (10%) but significant level in the background strain carrying seven glaA gene copies. We thus performed maltose-limited chemostat cultures combining metabolome analysis for these three isolates to characterize metabolic-level fluctuations caused by cofactor engineering. In these cultures, overexpression of either the gndA or maeA gene increased the intracellular NADPH pool by 45% and 66%, and the yield of GlaA by 65% and 30%, respectively. In contrast, overexpression of the gsdA gene had a negative effect on both total protein and glucoamylase production.
Conclusions
This data suggests for the first time that increased NADPH availability can indeed underpin protein and especially GlaA production in strains where a strong pull towards GlaA biosynthesis exists. This data also indicates that the highest impact on GlaA production can be engineered on a genetic level by increasing the flux through the pentose phosphate pathway (gndA gene) followed by engineering the flux through the reverse TCA cycle (maeA gene). We thus propose that NADPH cofactor engineering is indeed a valid strategy for metabolic engineering of A. niger to improve GlaA production, a strategy which is certainly also applicable to the rational design of other microbial cell factories.

Similar content being viewed by others
Background
The filamentous fungus Aspergillus niger is one of the main cell factories used nowadays in the industry for homologous or heterologous protein production due to its extraordinary ability for protein expression and secretion [1,2,3]. The Design-Build-Test-Learn (DBTL) cycle is an increasingly adopted systematic metabolic engineering strategy to achieve the desired outcome through reconstructing heterologous metabolic pathways or rewiring native metabolic activities [4, 5]. Rational strain development of cell factories can be improved by the iterative application of the DBTL cycles, which not only contributes to the optimization of biomanufacturing processes, it is also advantageous to build a complete metabolic model of engineered cells to deepen our understanding of cellular metabolism. Noteworthy, the advance of genetic engineering has speeded up the DBTL cycle of metabolic engineering [6]. For the cell factory A. niger, several genetic approaches have proven their potency to improve its enzyme producing capability, including protein carrier approaches, tunable Tet-on driven gene expression, and morphology engineering, to name but a few [1, 7,8,9]. However, the impact of cofactor engineering, i.e., the rebalance of the intracellular redox status, on protein production has not been systematically studied in A. niger.
NADPH is a limiting factor for the biosynthesis of amino acids that are the building blocks of proteins. For instance, 3 mol and 4 mol of NADPH is required for producing 1 mol of arginine and lysine, respectively [10]. Thus, adequate cytosolic NADPH supply is indispensable to maintain the intracellular redox balance and serves as a driving force for efficient amino acid biosynthesis [11]. NADPH also provides the main anabolic reducing power for biomass growth, lipid formation, and also for natural product biosynthesis [12]. Indeed, cofactor engineering has been reported to improve productivities in the bacterial cell factories Escherichia coli [13, 14], and Corynebacterium glutamicum, as well as in the yeast cell factory Yarrowia lipolytica [15]. Two common strategies have mainly been employed to optimize the availability of NADPH. One is to activate the enzyme activities of NAD(H) kinases (EC 2.7.1.86, EC 2.7.1.23) which are used to obtain NADPH or NADP + through phosphorylation of NADH and NAD + , respectively. The other is to modulate the expression strength of typical NADPH generating enzymes of the glycolytic pathway, the pentose phosphate pathway or the citric acid cycle. These include glucose-6-phosphate dehydrogenase (G6PDH), 6-phospho-gluconate dehydrogenase (6PGDH), NADP-dependent isocitrate dehydrogenase (NADP-ICDH), and NADP-dependent malic enzyme (NADP-ME) [16, 17]. Notably, heterologous protein expression in Pichia pastoris and A. niger can be triggered through boosted carbon flux to the pentose phosphate pathway (PPP), a catabolic pathway also known to produce NADPH [18, 19]. This suggests that central carbon metabolism may have evolved to ensure the production of cellular components under the balance of energy production and consumption [4]. In agreement, the metabolic flux through the PPP increased by 15–26% compared to the parental strains when GlaA was overproduced in A. niger [20] or the enzyme amylase overproduced in A. oryzae [21].
In the past two decades, extensive studies have focused on engineering a high flux through the PPP in E. coli, C. glutamicum, A. nidulans, and A. niger [11, 22,23,24,25]. A block of the glycolytic pathway by down-regulating the pgi gene encoding a phosphoglucose isomerase was one successful strategy in C. glutamicum [11]. In order to elevate the NADPH pool originating from the PPP in A. niger, the gsdA gene (glucose 6-phosphate dehydrogenase), the gndA gene (6-phosphogluconate dehydrogenase) and the tktA gene (transketolase) were individually overexpressed in A. niger. Strong overexpression of gndA led to a nine-fold increase in intracellular NADPH concentration, while gsdA and tktA affected the NADPH level only weakly [25]. However, any correlation between the NADPH supply and enzyme overproduction remained unclear.
Irrespective of the importance of the PPP for NADPH regeneration, an efficient carbon economy is only guaranteed when the carbon flux enters the glycolytic pathway (Embden-Meyerhoff-Parnass pathway, EMP) instead of the PPP because the PPP releases one carbon as CO2 when oxidizing 1 mol of hexose. Takeno et al. [26] thus substituted the endogenous NAD-dependent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the EMP in C. glutamicum with a heterologous NADP-dependent GAPDH, leading to 2 mol of NADPH generation instead of 2 mol NADH from 1 mol of hexose. This genetic modification provoked a substantial improvement in the yield of L-lysine production by 70–120%. Similar strategies also have been followed to overproduce ethanol in the yeast Saccharomyces cerevisiae [27] or lycopene and ε-caprolactone in the bacterium Clostridium acetobutylicum [28]. Likewise, cytosolic NADP-ME has been shown to positively affect lipid accumulation in oleaginous fungi [29, 30].
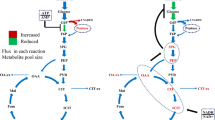
As summarized above, a wealth of metabolomic and fluxomic data in A. niger demonstrated that strains adapted to protein overproducing conditions channel a higher carbon flux through the PPP. However, cofactor engineering has not been considered yet or performed in A. niger to guide enzyme overproduction. We thus mined our recently published genome-scale metabolic network model (GSMM) developed for the A. niger protein producing reference strain CBS 513.88 [31]. This iHL1210 model identified the involvement of NADPH in 173 intracellular redox reactions in A. niger, including 49 NADPH generating reactions [31]. Notably, the GSMM did not predict any NADPH/NADP + shuttle in the mitochondrial membrane, and we thus concluded that any mitochondrial NADPH is unlikely to become directly consumed by cytosolic amino acid biosynthesis. Overall, the GSMM predicted that seven potential NADPH generating enzymes are of importance for GlaA production in A. niger (Table 1, Fig. 1): two enzymes of the cytosolic PPP (glucose-6-phosphate dehydrogenase, G6PDH; 6-phosphogluconate dehydrogenase 6PGDH), two cytosolic NADP-dependent enzymes (NADP-ICDH and NADP-ME) and three uncharacterized open reading frames (An12g04590, An14g00430, An16g02510). An12g04590, An14g00430 show high homology to NADP + oxidoreductases, and An16g02510 displays homology to alcohol dehydrogenases. In order to evaluate whether the model prediction is strain-dependent, we individually overexpressed all seven candidate genes in two A. niger host strains. Strain AB4.1 produces native levels of GlaA as it carries one glaA gene copy, and strain B36 is a derivative thereof, carrying seven glaA gene copies and is thus a high-yield GlaA producing strain [32]. All 14 strains were first investigated in shake flask-level cultivations. Based on the data gained, three engineered strains were selected for chemostat cultivations to decipher the association among genetic perturbation, NADPH availability, and GlaA production in A. niger.

Pathway map highlighting all seven genes modified during this study in red. The cytosolic glycolytic pathway, the pentose phosphate pathway and the mitochondrially located citric acid cycle are shown
Results
Strain generation using CRISPR/Cas9 technology and the synthetic Tet-on gene switch
In order to compare the effect of the seven selected genes on GlaA production in an A. niger strain carrying one glaA (AB4.1) or seven glaA (B36) gene copies, we first had to ensure that the introduced genetic modifications would allow us to directly compare the observed phenotypes. This required that the introduced genes would be under the same genetic control and furthermore introduced at the same genomic locus in both recipient strains. We thus decided to integrate an additional copy of all candidate genes under the control of the strong and tunable Tet-on gene switch into the pyrG locus of A. niger. This gene switch is inducible by the addition of doxycycline (DOX) to the culture medium, is tight in the absence of DOX and metabolism-independent in A. niger [33]. It has furthermore been shown to strongly induce gene expression up to levels above the glucoamylase gene, which is one of the highest expressed genes in A. niger [33,34,35].
Strain AB4.1 is a uridine-auxotroph due to a defective pyrG gene, a locus that is perfectly suited for gene targeting and screening purposes. The introduction of an intact pyrG copy at this locus occurs efficiently and confers uridine prototrophy in A. niger [36]. However, strain B36 does carry an intact pyrG gene [37]. We thus first mutated the pyrG locus in this strain in order to apply the same gene targeting strategy for all seven genes in both recipient strains. We edited the pyrG gene in B36 by following a CRISPR/Cas9 strategy that employed ribonucleoprotein particles. This approach was first published for the penicillin producer Penicillium chrysogenum [38] and has later been successfully established in other fungal cell factories [39]. As explained in detail in Additional file 1: Fig. S1, this approach enabled us to obtain a derivative of B36, strain YS20.2, which carries a 195 bp deletion within the pyrG ORF and is therefore unable to grow on medium lacking uridine or uracil. Both recipient strains, AB4.1 and YS20.2, were eventually used to integrate Tet-on driven candidate genes at the pyrG locus (for details see Materials and Methods and Additional file 1: Fig. S2). Respective genetic modifications were proven by PCR and Southern blot analyses (Additional file 1: Figs. S3, S5, S6, S7). All 14 strains obtained are summarized in Table 3. We finally also decided to delete the native ORFs of An14g00430 and An16g02510 in their respective Tet-on driven overexpression strains in order to analyze their deletion phenotypes.
The impact of NADPH engineering on GlaA production is strain-dependent
All 14 strains were subjected to batch cultivations in shake-flask format, whereby a medium containing maltose as GlaA-inducing carbon source was used. FW35.1 (a pyrG + derivative of AB4.1) and B36 were taken along as corresponding reference strains. DOX-induced gene expression in all seven AB4.1 derivatives was about 1.5–2.7 times higher compared to the reference strain FW35.1 as examined by qRT-PCR (Additional file 1: Fig. S8, Table S4). Although this led to an elevated NADPH pool of about 30% in the case of gndA, icdA or An16g02510, no significant increase in GlaA enzyme activity was observed for all of seven strains compared to the FW35.1 reference (Additional file 1: Fig. S8, Table S4). However, when all seven candidate genes were overexpressed in the YS20.2 background strain containing seven glaA gene copies, increased transcript levels were similar as in AB4.1, but for An16g02510 higher transcription levels (fourfold) were observed (Fig. 2b). Noteworthy, overexpression of gndA displayed the highest effect on the transcription of glaA (2.4-fold) compared to B36. A NADPH pool increase of about 30% was measured in three other strains overexpressing maeA, gndA, gsdA, respectively (Fig. 2e). With regard to Tet-on driven gene expression of maeA, gndA, gsdA, about 10% increase in secreted total protein, and about 10–18% higher GlaA activity was observed for these engineered strains (Fig. 2c, d, Additional file 1: Table S4). These observations encouraged our hypothesis that NADPH engineering might be a promising strategy to improve GlaA production.They furthermore implied that the success of such an approach is dependent on the glaA gene copy number as the impact on protein production is not linearly correlated with increased transcription of NADPH producing enzymes. To shed further light on these phenomena, we selected the three most promising YS20.2 background strains overexpressing maeA, gndA, gsdA, respectively, cultivated them under chemostat conditions, and compared their performance with the reference strain B36.

Data for shake flask-level cultivations of all engineered strains in the YS20.2 background in relation to the control strain B36. a Dry cell weight (DCW); b Relative expression level of glaA and engineered genes; c Total secreted protein per gram biomass at 72 h after inoculation; d Enzyme activity of GlaA per gram biomass at 72 h after inoculation; e Intracellular NADPH concentration in the exponential phase; f Comparison between engineered strains in the AB4.1 and YS20.2 background, respectively. All experiments were conducted in biological quadruplicates. Significance values were calculated with the two-tailed t-test with independent variables (*p < 0.05, **p < 0.01, ***p < 0.001)
Physiology and gene expression during maltose-limited chemostat cultivations
Strain B36 and the three strains overexpressing maeA, gndA, and gsdA, respectively, were run in duplicate maltose-limited chemostat cultures. To induce the expression of these three candidate genes, 10 µg/ml DOX was added during the early exponential growth phase, when the biomass reached 1–2 gDCW/kg. After about 22 h, the cultivation process was switched to the chemostat mode with a dilution rate D = 0.1 h−1, as described in Kwon et al. [32] and the Materials and Methods section. DOX was continuously added through the feed medium. During exponential and steady state conditions, culture samples were taken using an in-house developed fast-quenching sampling device (unpublished) for biomass determination, gene expression analyses (qRT-PCR), intracellular metabolite quantification (GC/LC–MS) and secreted protein determination. Carbon was accounted for in carbon balances of the feed medium, effluent culture broth, and exhaust gas. The carbon dioxide evolution rate (CER), the oxygen uptake rate (OUR), and biomass concentrations reached constant levels after about three-volume changes (Fig. 3a, c).

Physiological parameters for A. niger B36 and three overexpression strains during chemostat cultivation at a dilution rate of 0.1 h−1. a CO2 evolution rate (CER) in mmol/L/h; b Formation of the by-product oxalic acid in µmol/g cell dry weight; c Dry cell weight in gDCW/kg; d Yield of total secreted protein per gram biomass; e glaA relative gene expression level at the exponential growth phase and during steady state; f Relative gene expression level of the overexpressed genes during exponential growth and steady state; g Intracellular NADPH level at steady state. Data represent the mean ± SD from two independent cultures which were measured in technical triplicates. Significance values were calculated with the two-tailed t-test with independent variables (*p < 0.05, **p < 0.01, ***p < 0.001). Straight lines in a-d indicate the switch from batch to chemostat cultivation. OE gsdA: strain overexpressing gene gsdA; OE gndA: strain overexpressing gene gndA; OE maeA: strain overexpressing gene maeA
Interestingly, overexpression of gsdA or gndA increased biomass accumulation, whereas overexpression of maeA reduced it. This is also reflected by the final carbon-recoveries (Table 2). They were higher in both gsdA or gndA overexpressing strains (110%, 104%), but lower in maeA overexpressing strain (91%) compared to 99% of the B36 strain. Similar to shake flask-level cultivations, transcript levels of all three overexpressed genes were about 1.3 (gsdA, gndA) or 2.7 (maeA) times above their respective transcript levels in B36 during the exponential phase. They considerably increased 3.3-fold (gsdA, gndA) or 8.2-fold (maeA) during steady state conditions (Fig. 3e, f). This data might suggest that although all three genes are under the same Tet-on driven transcriptional control at the same locus (pyrG), other regulatory mechanisms, e.g., mRNA turnover or metabolic feedback regulation, might additionally control the activity of these three genes. Notably, overexpression of gsdA increased biomass accumulation (Table 2) but inhibited the yield of total secreted protein and GlaA by 40% (Fig. 3d), suggesting a competition between growth and protein production as previously proposed [11]. In contrast, overexpression of gndA and maeA positively impacted protein secretion by 60% and 30%, respectively (Table 2). This data was consistent with NADPH pool measurements during steady state conditions: Overexpression of gndA and maeA increased NADPH levels (46% and 66%, respectively), whereas only wild-type NADPH levels were observed for the strain overexpressing gsdA (Fig. 3g).
Metabolic differences revealed by multivariate statistical analysis
In total, 42 intracellular metabolites were identified and quantified for all eight chemostat runs. These included nine sugar phosphates, eight organic acids, 19 amino acids, and six currency metabolites (NAD, NADH, NADP, NADPH, ADP, ATP). Principal Component Analysis (PCA) uncovered that samples from all four strains separated into four distinct groups as shown in the score plot of Partial Least Squares Discrimination Analysis (PLS-DA) (Fig. 4a). Especially the strain overexpressing maeA displayed the strongest metabolic changes, whereas the strains overexpressing gsdA and gndA, respectively, showed only subtle differences when compared to the metabolic profile of strain B36. The loading map uncovered representative metabolites which mainly contributed to the separation of these four strains. Variations of relative abundances of pyruvate (PYR), succinate (SUC), histidine (HIS), maltose (MAL), 6-phosphogluconate (6PG) mainly contributed to distinguish both gsdA and gndA overexpressing strains, whereas the majority of variables contributed to the separation of OE maeA strain (Fig. 4a). A variable importance plot (VIP) displaying the relative contributions of these representative metabolites demonstrated that VIP values of 18 metabolites out of the 42 metabolites were above 1 (Fig. 4b), suggesting that these 18 metabolites could be considered as potential markers to discriminate all four strains. Pathway enrichment analyses highlighted that the PPP, the glyoxylate bypass, and dicarboxylate metabolism had a significant impact in the strains overexpressing gsdA and gndA, respectively. However, glycine, serine, and threonine metabolism was enriched in the strains overexpressing gsdA and maeA. The latter in general showed overrepresentation of amino acid metabolic pathways, including alanine, aspartate and glutamate metabolism, arginine and proline metabolism besides glycine, serine and threonine metabolism (Fig. 4c–e).

Bioplot (combining the loading plot and score plot). Three biological replicates from each strain were denoted with the same color (a), VIP score of 42 intracellular metabolites calculated using the PLS-DA (b), and respective pathway impact analysis for metabolic profiling at steady state (c–e)
Metabolic profiling of amino acid pools and central carbon metabolism
Figure 5 and Additional file 1: Table S5 summarize the amino acid pools in all three overexpression strains compared to B36. In general, Ala, Glu, Gly, Leu, and Lys are the top five amino acids in the biomass of A. niger, whereas Ser, Thr, Ala, Leu, and Gly account for about 50% of the total amino acids in GlaA [40]. Our metabolomics analyses uncovered that in all four strains, amino acids from the glutamate family are most and aromatic amino acids are least abundant, which is in general agreement to previously reported amino acid pools in A. niger [41]. Overall, the amino acid pool in the gndA overexpression strain was slightly reduced compared to B36 but was increased in strains overexpressing gsdA (22%) or maeA (30%), respectively (Additional file 1: Table S5). A general observation was also that the histidine pool dramatically increased when gsdA (increased by three folds) or gndA (60%) were overexpressed. Two out of the five amino acids dominating A. niger's biomass (Glu, Lys) accumulated in the strain overexpressing gsdA, while the pool sizes of three top GlaA composing amino acids (Ser, Thr, Gly) were less abundant compared to B36. This data might explain the reduced GlaA production in this strain. In the case of maeA overexpression, nearly all amino acids where higher abundant when compared to strain B36 (Additional file 1: Table S5), especially the pools for the GlaA dominating amino acids Thr, Ala, Leu, and Val, suggesting that increased amino acid pools might cause the extra driving force for GlaA formation in a maeA overexpressing strain. In contrast, the overall amino acid pools of the strain overexpressing gndA displayed only moderate reduction compared to B36 (Additional file 1: Table S5).

The pool sizes of amino acids for the A. niger reference strain B36 and three overexpression strains at steady state. Blocks in the heat map represent B36, OE gsdA (strain overexpressing gene gsdA), OE gndA (strain overexpressing gene gndA), and OE maeA (strain overexpressing gene maeA) from left to right. Amino acids labeled in yellow are the top five amino acids present in GlaA. Red arrows indicate the enzymes overexpressed. Quantified amino acid amounts and significance values are given in Additional file 1: Table S5
As summarized in Fig. 6 and Additional file 1: Table S6, overexpression of gsdA, gndA, and maeA, led to a significant flux redistribution of the central carbon metabolism, including PPP, EMP, and TCA. As expected, the glycolytic pathway intermediates G6P and F6P were significantly reduced in the two PPP engineered strains which overexpressed gsdA and gndA, respectively. Diverting the carbon flux towards the PPP at the branch node of G6P after overexpression of gsdA and gndA has in fact lowered the flux towards the EMP. The 6PG and PEP pools increased (due to gsdA overexpression), which in turn inhibited the upper glycolytic pathway (feedback inhibition via 6PG [42] and PEP [43]). Another consequence is a reduced carbon uptaken rate, which is indeed the case for the strain overexpressing gsdA (Table 2). On the lower glycolytic pathway, the accumulation of the intermediates PEP and PYR, respectively, in OE gsdA and OE gndA led to a reduced flux through 3PG (Additional file 1: Fig. S9), accompanied by a reduction of the serine family pool in these two strains. This data could be well explained by the close association between the abundance of metabolite precursors on the central metabolic network and the pool size of the correlated amino acid families as already reported elsewhere [44]. Importantly, the PPP is not the only key for NADPH regeneration but also provides two important precursors for amino acid biosynthesis, i.e., ribose 5-phosphate (R5P) and erythrose 4-phosphate (E4P), which in turn are required for His and aromatic amino acid biosynthesis. Overproduction of gndA channeled more carbon from 6PG to Ru5P, resulting in a lower abundance of 6PG as expected. Surprisingly, the R5P pools declined in both strains overexpressing gsdA and gndA, respectively. However, taking into account the increased biomass formation and higher His pools in these two strains, the increased carbon flux towards R5P was possibly channeled towards nucleotide biosynthesis. In agreement with previous reports studying high protein secretion in A. niger [18, 41] and our in silico metabolic flux simulations (Additional file 1: Fig. S9), the flux through the TCA cycle was also reduced during overexpression of the PPP genes gsdA and gndA, respectively. However, the picture for the TCA cycle intermediates was inhomogeneous. The TCA cycle intermediates AKG, FUM, and OAA were less abundant, whereas MAL accumulated when gsdA was overexpressed. In the case for gndA overexpression, the experimental data showed only weak differences for the pool sizes of TCA intermediates when compared to strain B36 (Fig. 6, Additional file 1: Fig. S9). Last but not least, overexpression of maeA facilitated not only amino acid biosynthesis but also improved the carbon flux towards the EMP and TCA cycle (Fig. 6) and elevated the pools of the key intermediates F6P, G3P, PEP, OAA, and FUM but not PYR. Worth mentioning in this context is oxalic acid, a major by-product during A. niger cultivation which stems from OAA [45]. Whereas the oxalic acid pool did not differ in the strains overexpressing gsdA or gndA, respectively, it was substantially increased by about 30% in OE maeA, suggesting that the elevated pool size of OAA could be responsible for the accumulation of this by-product (Fig. 3b).

The pool sizes of a part of the metabolites on central carbon metabolism pathways for the A. niger reference strain B36 and three engineered strains at steady state. Blocks in the heat map represent B36, OE gsdA (strain overexpressing gene gsdA), OE gndA (strain overexpressing gene gndA), and OE maeA (strain overexpressing gene maeA) from left to right. Red arrows indicate the enzymes overexpressed. Quantified sugar and organic acid amounts including significance values are given in Additional file 1: Table S6
Discussion
NADPH regeneration is a limiting step for amino acid biosynthesis; hence NADPH availability and allocation are essential for efficient protein production. This study demonstrates that the NADPH pool can be increased by 30% in A. niger through genetic engineering by the gndA, gsdA, and maeA genes, respectively. However, the physiological consequences of this increased NADPH pool are strain-dependent. For the native GlaA producing strain AB4.1, which carries only one glaA copy, we assume that the excess of NADPH might not be allocated to increased amino acid biosynthesis but was channeled to other NADPH consumption pathways, e.g., for the production of steroids, lipids, and nucleotides and thus biomass formation. In contrast, due to the higher glaA gene number in B36, it is tempting to speculate that the excess in NADPH was indeed used for GlaA production because of the higher intracellular pull towards GlaA production and its amino acid precursors. Future 13C metabolic flux analyses targeting sugars, amino acids, steroids, lipids, and nucleotides in these strains will prove or disprove this hypothesis. These analyses will also clarify whether NADPH supply is a bottleneck for protein biosynthesis, as previously proposed for the yeast Y. lipolytica [46].
Metabolic flux analysis is the most authoritative method for measuring in vivo fluxes [47]. In this study, we used our previously updated A. niger GSMM iHL1210 [31] to predict the flux distribution of the central carbon metabolic network in silico during steady-state conditions of three strains overexpressing gndA, gsdA, and maeA, respectively. These simulation results indicated that the relative flux of the EMP and TCA cycle decreased after redirecting a higher flux through the PPP when gndA or gsdA were overexpressed, a situation which was reverse predicted when maeA was overexpressed. This predicted computational flux distribution was congruent with the in vivo metabolite pools measured by mass spectrometry: The upper glycolytic pathway intermediates G6P and F6P were significantly lower (p < 0.05) in the two strains overexpressing gndA or gsdA, which both encode enzymes of the PPP. When compared with other autonomously evolved metabolic networks from forced protein overproduction [18, 41, 48], it illustrates a comparable flux pattern through the PPP and TCA cycle as observed here, suggesting similarities of core carbon metabolism in the central metabolic network in diverse high-yield protein overproducing A. niger cell factories.
Notably, overexpression of the gsdA gene improved biomass accumulation and only slightly increased NADPH production, which were paralleled by reduced protein production capacity by about 40%. This observation is generally consistent with previous reports for A. niger and other filamentous fungal cell factories. For example, a high specific protein production rate has been shown to correlate with relatively low growth rates in Trichoderma reesei [49], due to a reduced proteome allocated for central metabolism [12]. Also, a recent multi-omics analysis from our group that integrated transcriptomics, metabolomics and GSMM simulations proposed that an increased GlaA production is probably achieved through reduced growth, which likely is regulated at several metabolic mechanisms: (i) an increased carbon catabolism that generates more amino acid precursors for protein production, (ii) a reduced fatty acid and ribosome biogenesis and, thus, reduced growth, and (iii) an increased flux through the glyoxylate bypass to reduce NADH formation from the citric acid cycle and to maintain the cellular redox balance [50]. In the current study, at least for the two strains overexpressing gndA and gsdA, respectively, we observed that both the PPP and the glyoxylate pathway changed considerably (Fig. 4). The general view might be that once growth is limited, more reducing equivalents (NADH and NADPH), and precursors can be channeled into protein and glucoamylase production. We have shown previously that A. niger benefits from a very flexible transcriptional machinery that ensures adaptation to the burden of high protein loads [32]. When forced to overexpress glaA, A. niger increases the transcription of secretory pathway genes involved in translation, protein folding, and protein secretion. Under this circumstance, the expression of genes less important for growth and survival becomes decreased loads [32]. This phenomenon is called 'Repression under secretion stress' (RESS) and was first discovered in T. reesei [51]. A. niger can, thus, fall back on a very efficient gene regulatory and metabolic machinery that balances cellular capacities with the intracellular burden. However, it is currently impossible to predict nor to understand all regulatory mechanisms and their correlations behind these phenomena. Additionally, 13C-flux analysis detected that the two NADPH generating reactions, i.e., NADP-ICDH and G6PDH, contributed only negligibly (below 5%) to the total NADPH production [52]. This could be an alternative reason for the minor changes in NADPH when comparing the strain overexpressing gsdA to the reference strain B36. Moreover, more NADPH might have been consumed for growth in this strain. Yet, this observation is opposite to the critical role of G6PDH in the production of NADPH in Saccharomyces cerevisiae [53]. Our study clearly uncovered that overexpression of the gndA or maeA gene, respectively, enhanced total protein production and specifically GlaA production in A. niger. Whereas the gndA gene encodes one enzyme of the PPP, the maeA encoded enzyme is part of the cytosolic reverse TCA cycle. Overexpression of both genes gave rise to both the NADPH pool (46%, 66%, respectively) and protein production rates (65%, 30%, respectively) compared to B36, thus implying that our in silico predicted cofactor engineering approach is a biological valid and thus successful approach to improve protein production capacities of A. niger. Notably, overexpression of maeA provoked the most complex metabolic changes (Fig. 5 and 6). NADP-ME is critical not only for pyruvate supply but also enhanced the flux through glycolysis and TCA cycle to provide sufficient precursors for amino acid production. Based on our previous multi-omics analysis and exogenous amino acid addition experiments [50], Ala, Glu, Gly, and Asp were confirmed as four amino acid limiting GlaA production in A. niger. This was further corroborated by our recent study reporting that removing the amino acid limitation is supporting GlaA production [54]. Hence, enriched pools of amino acids, carbon precursors and NADPH due to increased maeA transcription could altogether boost GlaA production. However, increased maeA transcription was paralleled by the accumulation of the by-product oxalic acid, which could in turn be targeted in the next DBTL cycle to even further increase GlaA production.
Conclusions
In this study, we followed a DBTL approach to systematically understand and optimize GlaA production in A. niger. Our previous multi-omics integration analyses predicted that NADPH regeneration could be one so far unstudied bottleneck for enzyme production in A. niger. To test this hypothesis, we genetically engineered strains with improved NADPH biosynthetic capacities. Metabolic profiling and multivariate statistical analysis revealed that three NADPH regeneration enzymes encoded by the gsdA, gndA, and maeA genes play the most significant decisive role during batch and continuous cultivations, whereby overexpression of the gndA and maeA genes provoked the highest flux redistributions towards protein biosynthesis. Future studies will examine whether combined overexpression of these candidate genes, e.g., by polycistronic gene expression [55], will further improve protein production capacities of A. niger and thus evaluate whether combinatorial cofactor engineering is a further successful approach for strain engineering.
Materials and methods
Plasmids construction
Integrative plasmids used for overexpressing NADPH generating genes in the A. niger strains were cloned by Gibson assembly or AQUA cloning [56]. The backbone vector was obtained by digesting pFW22.1 with PmeI, and then the 8.1 kb fragment was purified by gel extraction. Plasmid pFW22.1 carries the Tet-on inducible gene expression system, and an unfunctional pyrG, which was used as a selective marker for transformant screening [35]. Open reading frames (ORF) of NADPH generating enzymes (6PGDH (gsdA), G6PDG (gndA), NADP-ICDH (icdA), NADP-ME (maeA), An14g00430, An15g04590, and An16g02510) were amplified by PCR, using genomic DNA from the A. niger B36 strain as a template. PCR products were integrated into pFW22.1 at the PmeI site (Additional file 1: Fig. S2). Primers carrying overlapping regions to the backbone vector and plasmids constructed in this study are listed in Additional file 1: Tables S7 and S8, respectively.
Strains
A. niger strains used in this study are listed in Table 3. AB4.1 is a lab strain with only one glaA copy and uridine auxotrophic [36]. B36 is an engineered strain with multiple copies of glaA and derived from N402 [37]. YS20.2 is a uridine auxotrophic derivative of B36, which was engineered using CRISPR/Cas9 (see below). Overexpression strains of NADPH generating genes were constructed in the background of AB4.1 or YS20.2, and transformants were screened based on uridine prototrophy. Gene deletions were obtained using the split marker method [57]. PCR products containing either ~ 1.5 kb 5′ or 3′ flanking regions of the corresponding gene and a part of the selective marker hygromycin were used for transformation (for details please refer to Additional file 1: Fig. S4 and Table S7). To avoid a lethal phenotype, all deletion strains were built in their corresponding overexpression strains. A. niger transformation, genomic DNA extraction, and Southern hybridization were performed as previously described in Arentshorstet et al. [58] with the following exceptions: strain YS20.2 was cultivated at 80 rpm to obtain young mycelium for protoplastation. PEG mediated transformation was done using 60% PEG 4000 as described in Pohl et al. [38]. This improved the transformation rate from 1 CFU/µgDNA to 4–5 CFU/µgDNA. All overexpression and deletion strains were generated without CRISPR. Positive transformants were confirmed by diagnostic PCR and Southern analysis (Fig. S3, S5, S6, and S7).
Medium
Strains were grown at 30 °C using a complete or minimal medium supplemented with 1 mM uridine when necessary [58]. Shake flask medium (g/L): 3% Maltose·H2O, 10 g tryptone, 5 g Yeast extract, 1 g KH2PO4, 0.5 g MgSO4·7H2O, 0.03 g ZnCl2, 0.02 g CaCl2, 0.0076 g MnSO4·H2O, 0.3 g FeSO4·7H2O, 3 ml Tween 80, pH was adjusted to 5.5 by 1 M HCl. Maltose-limited chemostat medium was as described in Kwon et al. [32] with the following slight modification: the medium contained 1% (w/v) of maltose in batch cultures and 0.8% (w/v) during chemostat cultivations. Germination was induced by the addition of 0.003% (w/w) yeast extract. Media used for CRISPR/Cas9 gene editing: 200 µg/ml hygromycin and 500 µg/ml caffeine were supplied into MM transformation media. Filtered-sterilized 0.75 g/L 5-FOA, 10 mM proline and 10 mM uridine were added to MM medium when subcultivating uridine auxotrophic transformants.
CRISPR/Cas9 genetic modification
CRISPR/Cas9 modifications were done using the ribonucleoprotein (RNP) approach [38]. The Cas9 protein was expressed from the pET28aCas9cys vector (Addgene: 53,261) and purified by AKTA FPLC (Fast protein liquid chromatography, GE, USA). The sgRNA was selected online using the Cas-Designer website (https://www.rgenome.net/cas-designer/). Two sgRNAs were designed to test the gene targeting efficiency, which located at 205 bp and 393 bp after the start codon of pyrG. DNA templates for in vitro sgRNA synthesis (MegaScript T7 Transcription Kit, Thermo Fisher Scientific, USA) were constructed as DNA oligos incorporating a T7-promoter sequence, 20 bp protospacer and a 77 bp sgRNA tail (Primers are listed in Additional file 1: Table S9). During transformation, 5 µl purified Cas9 protein, 1 µl or 2 µl sgRNA, and 2 µg pMA171.1 plasmid carrying hygromycin as a selective marker [59] were used. Transformants were subcultivated on MM plates containing uridine, proline, and 5-FOA (details on concentrations are above). Consequently, the ORF region of pyrG from sporulating single colonies on 5-FOA medium was amplified to confirm mutations by sequencing (Additional file 1: Fig. S1).
Shake flask-level cultivation
To evaluate the performance of engineered strains as GlaA producers, 106 spores/ml of reference strains FW35.1 [35] or B36 [32] and engineered strains were inoculated into 50 ml shake flask liquid medium and cultivated at 30 °C and 250 rpm. After 18 h of cultivation, 20 µg/mL doxycycline (DOX) was used to induce gene expression. In order to ensure gene overexpression, the same amount of DOX was added every 12 h after the initial induction. Samples were taken at 24, 48, and 72 h after inoculation. Physiological parameters (dry weight, total secreted protein, residual glucose, and enzyme activity of GlaA in the broth) were measured. Samples for NADPH measurement and qRT-PCR were taken in the exponential phase. Experiments were performed in biological quadruplicates.
Chemostat cultivation
Submerged cultivations were carried out in 5 L bioreactors (NCBIO, Shanghai, China). Batch cultivations were adapted from Kwon et al. [32]. Chemostat cultivations were initiated in the late exponential growth phase when OUR (Oxygen Uptake Rate) or CER (Carbon-dioxide Evolution Rate) started to decrease and DO (Dissolved Oxygen) started to increase. The dilution rate (D) was set at 0.1 h−1. The steady-state was reached after approximately three residence times (≈ 30 h) and indicated by constant CO2, O2, and biomass concentrations. Samples were taken regularly (6–8 h) to monitor growth and to determine if a steady-state had been reached. All samples were quickly frozen in liquid nitrogen. 10 µg/ml DOX was added when the biomass reached 1–2 g/kg. To ensure induction, 10 µg/mL DOX was also applied to the feed medium. Samples used for extracting intracellular metabolites were taken during steady-state and intracellular metabolites were quantified by GC/LC–MS. Samples for NADPH measurement and qRT-PCR were taken both in the mid-exponential phase (4 h after DOX induction) and at steady-state.
Determination of dry weight, total secreted protein, residual glucose and enzyme activity of glucoamylase
4 mL of samples were taken at the indicated time points from shake flask or bioreactor cultures. Biomass was harvested through vacuum filtration, washed 3 times with deionized water, then frozen at − 80 °C and finally freeze-dried overnight to determine dry weight. Total extracellular protein in the culture supernatant was determined via the Bradford assay (BioRad, California, USA) according to the manufacturer's protocols, and absorbance (600 nm) was measured using a GloMax®-Multi Detection System (Promega, Madison, USA). Quantification of residual glucose in the cultivation medium was performed with the Glucose GOD/PAP Liqui-color kit (Human, Wiesbaden, Germany) according to the manufacturer's manual. The enzyme activity of GlaA was measured as described in Lu et al. [50].
Quantification of extracellular organic acid
By-products such as extracellular acetic acid, citric acid, oxalic acid, pyruvic acid, and succinic acid were determined by HPLC (Shimadzu, Kyoto, Japan) using a VARIAN Metacarb H plus column and 5 mM H2SO4 as a mobile phase at a flow rate of 0.4 ml/min and 50 °C. Acids were detected at a wavelength of 210 nm.
Quantification of intracellular metabolites
Quantification of intracellular metabolites was based on Lu et al. [41], and slight modifications were made. This study adopted the Isotope Dilution Mass Spectrometry (IDMS) method to accurately quantify intracellular metabolites [60]. 1–2 ml broth was rapidly taken from bioreactors by fast-sampling equipment to tubes with 10 ml precooled quench solution (− 27.6 °C 60% v/v methanol solution) at steady-state. To precisely determine the amount of broth taken, the tubes were weighed before and after sampling. In order to remove the extracellular metabolites promptly, the mixture was filtered with a vacuum pump. Then, 20 ml precooled quench solution was used to rinse the filter cake. The washed filter cake and 100 µl 13C internal standard solution were added to prewarmed 25 ml 75% (v/v) ethanol solution and extracted 3 min at 95 °C. The tubes were cooled down on the ice to room temperature. The filtrate was collected after vacuum filtration and concentrated to 600 µl by rotary evaporation. Subsequently, the metabolite pools were quantified with the LC–MS/MS (Thermo Fisher Scientific Corporation, USA) and GC–MS (Agilent, Santa Clara, CA, USA).
Quantification of intracellular NADPH
1 ml broth was quickly taken from the shake flask or bioreactor and frozen immediately in liquid nitrogen. Samples were thawed before measurement, diluted 2 or 5 times with 1 × PBS, and centrifuged at 12,000 rpm for 5 min to remove the supernatant. Intracellular NADPH was quantified by the EnzyFluo™ Assay Kit (BioAssay Systems Corporation, USA) according to the manufacturer's manual.
Quantitative real-time PCR
Mycelium harvested for RNA extraction was ground in liquid nitrogen and then extracted by the Fungal Total RNA Isolation Kit (Sangon, Shanghai, China). About 1 μg of total RNA was used for cDNA synthesis using the PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, Shiga, Japan) according to the manufacturer's instructions. The real-time PCR reaction system was prepared with TB Green™ Premix Ex Taq™ II (Takara, Shiga, Japan) in a volume of 25 μl with diluted cDNA (about 1 µg) as a template. Diluted cDNA was used to keep mean Ct (threshold cycles) values between 20 and 30. Each reaction was carried out in triplicates. Oligonucleotide primers used for qPCR are listed in Additional file 1: Table S7. The 28S rRNA and 18S rRNA were used as the internal standard. PCR conditions were as follows: 95 °C for 3 min, followed by subsequent 40 cycles of the three-steps: 95 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s.
Multivariate statistical analysis of intracellular metabolites
Hierarchy clustering analysis of metabolomics data at steady state was plotted using the R package pheatmap. Principal component analysis and partial least squares discriminant analysis (PLS-DA) were then performed by the R package ggbiplot. Pathway enrichment analysis was performed by the online metabolomics analysis website MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/MetaboAnalyst/faces/home.xhtml).
Simulation of relative metabolic flux by A. niger GSMM
Relative metabolic fluxes were predicted as described in Lu et al. [50] through A. niger GSMM iHL1210, which was updated recently by our lab [31]. Maximization of cell growth was set as the objective function, and maltose was the sole carbon source. qS, qP, qO2, qCO2 from the chemostat fermentations were set as constraints during the simulation.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- PPP:
-
Pentose phosphate pathway
- EMP:
-
Embden-Meyerhof-Parnas pathway
- TCA:
-
Tricarboxylic acid cycle
- CER:
-
Carbon dioxide evolution rate
- OUR:
-
Oxygen uptake rate
- DO:
-
Dissolved oxygen
- PLS-DA:
-
Partial least squares discrimination analysis
- G6PDH:
-
Glucose 6-phosphate dehydrogenase
- 6PGDH:
-
Phosphogluconate dehydrogenase
- NADP-ICDH:
-
NADP + dependent isocitrate dehydrogenase
- NADP-ME:
-
NADP + dependent malic enzyme
- 3PG:
-
3-Phosphoglycerate
- 3HBCoA:
-
(S)-3-Hydroxybutanoyl-CoA
- AACCoA:
-
Acetoacetyl-CoA
- AC:
-
Acetate
- ACCoA:
-
Acetyl-CoA
- ACAL:
-
Acetaldehyde
- AKG:
-
α-Ketoglutaric acid
- Ala:
-
Alanine
- Arg:
-
Arginine
- Asn:
-
Asparagine
- CIT:
-
Citrate
- DHAP:
-
Dihydroxyacetone-phosphate
- DHF:
-
7:8-Dihydrofolate
- E4P:
-
Erythrose-4-phosphate
- ETH:
-
Ethanol
- F6P:
-
Fructose-6-phosphate
- FBP:
-
1,6 Fructose diphosphate
- FUM:
-
Fumarate
- GLC:
-
Glucose
- G1P:
-
Glucose-1-phosphate
- G6P:
-
Glucose-6-phosphate
- GlaA:
-
Glucoamylase
- GLCNT:
-
Gluconate
- G3P:
-
Glyceraldehyde-3-phosphate
- Gln:
-
Glutamine
- Glu:
-
Glutamate
- His:
-
Histidine
- ICT:
-
Isocitrate
- Ile:
-
Isoleucine
- Leu:
-
Leucine
- Lys:
-
Lysine
- Mal:
-
Malate
- Met:
-
Methionine
- OA:
-
Oxalate
- OAA:
-
Cytosolic oxaloacetate
- Orn:
-
Ornithine
- PEP:
-
Phosphoenolpyruvate
- Phe:
-
Phenylalanine
- Pro:
-
Proline
- PYR:
-
Pyruvate
- R5P:
-
Ribose-5-phosphate
- Ru5P:
-
Ribulose-5-phosphate
- S7P:
-
Sedoheptulose-7-phosphate
- Ser:
-
Serine
- SUC:
-
Succinate
- Thr:
-
Threonine
- Trp:
-
Tryptophan
- Tyr:
-
Tyrosine
- Val:
-
Valine
- X5P:
-
D-xylulose-5-phosphate
References
Cairns TC, Nai C, Meyer V. How a fungus shapes biotechnology: 100 years of Aspergillus niger research. Fungal Biol Biotechnol. 2018;5:13–27.
Meyer V, Andersen MR, Brakhage AA, Braus GH, Caddick MX, Cairns TC, de Vries RP, Haarmann T, Hansen K, Hertz-Fowler C, et al. Current challenges of research on filamentous fungi in relation to human welfare and a sustainable bio-economy: a white paper. Fungal Biol Biotechnol. 2016;3:6.
Meyer V, Basenko EY, Benz JP, Braus GH, Caddick MX, Csukai M, de Vries RP, Endy D, Frisvad JC, Gunde-Cimerman N, et al. Growing a circular economy with fungal biotechnology: a white paper. Fungal Biol Biotechnol. 2020;7:5.
Nielsen J, Keasling JD. Engineering Cellular Metabolism. Cell. 2016;164:1185–97.
Ando D, Garcia MH. Two-Scale 13C metabolic flux analysis for metabolic engineering. Methods Mol Biol. 2018;1671:333–52.
Liu Y, Nielsen J. Recent trends in metabolic engineering of microbial chemical factories. Curr Opin Biotechnol. 2019;60:188–97.
Cairns TC, Zheng X, Zheng P, Sun J, Meyer V. Moulding the mould: understanding and reprogramming filamentous fungal growth and morphogenesis for next generation cell factories. Biotechnol Biofuels. 2019;12:77–96.
Fiedler MRM, Barthel L, Kubisch C, Nai C, Meyer V. Construction of an improved Aspergillus niger platform for enhanced glucoamylase secretion. Microb Cell Fact. 2018;17:95–106.
Fiedler MRM, Cairns TC, Koch O, Kubisch C, Meyer V. Conditional expression of the small GTPase ArfA impacts secretion, morphology, growth, and actin ring position in Aspergillus niger. Front Microbiol. 2018;9:878–94.
Moritz B, Striegel K, de Graaf AA, Sahm H. Changes of pentose phosphate pathway flux in vivo in Corynebacterium glutamicum during leucine-limited batch cultivation as determined from intracellular metabolite concentration measurements. Metab Eng. 2002;4:295–305.
Park SH, Kim HU, Kim TY, Park JS, Kim SS, Lee SY. Metabolic engineering of Corynebacterium glutamicum for L-arginine production. Nat Commun. 2014;5:4618.
Nielsen J. Cell factory engineering for improved production of natural products. Nat Prod Rep. 2019;36:1233–6.
Li W, Wu H, Li M, San KY. Effect of NADPH availability on free fatty acid production in E. coli. Biotechnol Bioeng. 2018;115:444–52.
Yao R, Li J, Feng L, Zhang X, Hu H. 13C metabolic flux analysis-guided metabolic engineering of Escherichia coli for improved acetol production from glycerol. Biotechnol Biofuels. 2019;12:29.
Wasylenko TM, Ahn WS, Stephanopoulos G. The oxidative pentose phosphate pathway is the primary source of NADPH for lipid overproduction from glucose in Yarrowia lipolytica. Metab Eng. 2015;30:27–39.
Marx A, de Graaf AA, Wiechert W, Eggeling L, Sahm H. Determination of the fluxes in the central metabolism of Corynebacterium glutamicum by nuclear magnetic resonance spectroscopy combined with metabolite balancing. Biotechnol Bioeng. 2015;49:111–29.
Marx A, Eikmanns BJ, Sahm H, de Graaf AA, Eggeling L. Response of the central metabolism in Corynebacterium glutamicumto the use of an NADH-dependent glutamate dehydrogenase. Metab Eng. 1999;1:35–48.
Driouch H, Melzer G, Wittmann C. Integration of in vivo and in silico metabolic fluxes for improvement of recombinant protein production. Metab Eng. 2012;14:47–58.
Nie Y, Huang M, Lu J, Qian J, Lin W, Chu J, Zhuang Y, Zhang S. Impacts of high beta-galactosidase expression on central metabolism of recombinant Pichia pastoris GS115 using glucose as sole carbon source via (13)C metabolic flux analysis. J Biotechnol. 2014;187:124–34.
Pedersen H, Christensen B, Hjort C, Nielsen J. Construction and characterization of an oxalic acid nonproducing strain of Aspergillus niger. Metab Eng. 2000;2:34–41.
Pedersen H, Carlsen M, Nielsen J. Identification of enzymes and quantification of metabolic fluxes in a wild type and a recombinant Aspergillus oryzae strain. Appl Environ Microbiol. 1999;65:11–9.
Canonaco F, Hess TA, Heri S, Wang T, Szyperski T, Sauer U. Metabolic flux response to phosphoglucose isomerase knock-out in Escherichia coli and impact of overexpression of the soluble transhydrogenase UdhA. FEMS Microbiol Lett. 2001;204:247–52.
Lim SJ, Jung YM, Shin HD, Lee YH. Amplification of the NADPH-related genes zwf and gnd for the oddball biosynthesis of PHB in an E. coli transformant harboring a cloned phbCAB operon. J Biosci Bioeng. 2002;93:543–9.
Panagiotou G, Grotkjær T, Hofmann G, Bapat PM, Olsson L. Overexpression of a novel endogenous NADH kinase in Aspergillus nidulans enhances growth. Metab Eng. 2009;11:31–9.
Poulsen BR, Nohr J, Douthwaite S, Hansen LV, Iversen JJL, Visser J, Ruijter GJG. Increased NADPH concentration obtained by metabolic engineering of the pentose phosphate pathway in Aspergillus niger. FEBS J. 2010;272:1313–25.
Takeno S, Murata R, Kobayashi R, Mitsuhashi S, Ikeda M. Engineering of Corynebacterium glutamicum with an NADPH-generating glycolytic pathway for l-lysine production. Appl Environ Microbiol. 2010;76:7154–60.
Verho R, Londesborough J, Penttila M, Richard P. Engineering redox cofactor regeneration for improved pentose fermentation in Saccharomyces cerevisiae. Appl Environ Microbiol. 2003;69:5892–7.
Martínez I, Zhu J, Lin H, Bennett GN, San KY. Replacing Escherichia coli NAD-dependent glyceraldehyde 3-phosphate dehydrogenase (GAPDH) with a NADP-dependent enzyme from Clostridium acetobutylicum facilitates NADPH dependent pathways. Metab Eng. 2008;10:352–9.
Zhang Y, Adams IP, Ratledge C. Malic enzyme: the controlling activity for lipid production? Overexpression of malic enzyme in Mucor circinelloides leads to a 2.5-fold increase in lipid accumulation. Microbiology. 2007;153:2013–25.
Hao G, Chen H, Wang L, Gu Z, Song Y, Zhang H, Chen W, Chen YQ. Role of malic enzyme during fatty acid synthesis in the oleaginous fungus Mortierella alpina. Appl Environ Microbiol. 2014;80:2672–8.
Lu H, Cao W, Ouyang L, Xia J, Huang M, Chu J, Zhuang Y, Zhang S, Noorman H. Comprehensive reconstruction and in silico analysis of Aspergillus niger genome-scale metabolic network model that accounts for 1210 ORFs. Biotechnol Bioeng. 2016;114:685–95.
Kwon MJ, Jørgensen TR, Nitsche BM, Arentshorst M, Park J, Ram AF, Meyer V. The transcriptomic fingerprint of glucoamylase overexpression in Aspergillus niger. BMC Genomics. 2012;13:701–18.
Meyer V, Wanka F, van Gent J, Arentshorst M, van den Hondel CA, Ram AF. Fungal gene expression on demand: an inducible, tunable, and metabolism-independent expression system for Aspergillus niger. Appl Environ Microbiol. 2011;77:2975–83.
Schäpe P, Kwon MJ, Baumann B, Gutschmann B, Jung S, Lenz S, Nitsche B, Paege N, Schutze T, Cairns TC, Meyer V. Updating genome annotation for the microbial cell factory Aspergillus niger using gene co-expression networks. Nucleic Acids Res. 2019;47:559–69.
Wanka F, Cairns T, Boecker S, Berens C, Happel A, Zheng X, Sun J, Krappmann S, Meyer V. Tet-on, or Tet-off, that is the question: advanced conditional gene expression in Aspergillus. Fungal Genet Biol. 2016;89:72–83.
van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwels PH, van den Hondel CA. Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet. 1987;206:71–5.
Verdoes JC, Punt PJ, Schrickx JM, van Verseveld HW, Stouthamer AH, van den Hondel CA. Glucoamylase overexpression in Aspergillus niger : molecular genetic analysis of strains containing multiple copies of the glaA gene. Transgenic Res. 1993;2:84–92.
Pohl C, Kiel JA, Driessen AJ, Bovenberg RA, Nygard Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synth Biol. 2016;5:754–64.
Kwon MJ, Schutze T, Spohner S, Haefner S, Meyer V. Practical guidance for the implementation of the CRISPR genome editing tool in filamentous fungi. Fungal Biol Biotechnol. 2019;6:15.
Sui YF, Ouyang LM, Chu J, Cao WQ, Liang LF, Zhuang YP, Cheng S, Norrman H, Zhang S-L, Zhang G-Y. Global transcriptional response of Aspergillus niger in the process of glucoamylase fermentation. Bioresour Bioprocess. 2017;4:44–56.
Lu H, Liu X, Huang M, Xia J, Chu J, Zhuang Y, Zhang S, Noorman H. Integrated isotope-assisted metabolomics and (13)C metabolic flux analysis reveals metabolic flux redistribution for high glucoamylase production by Aspergillus niger. Microb Cell Fact. 2015;14:147–61.
Sachla AJ, Helmann JD. A bacterial checkpoint protein for ribosome assembly moonlights as an essential metabolite-proofreading enzyme. Nat Commun. 2019;10:1526.
Ogawa T, Mori H, Tomita M, Yoshino M. Inhibitory effect of phosphoenolpyruvate on glycolytic enzymes in Escherichia coli. Res Microbiol. 2007;158:159–63.
Jordà J, Rojas HC, Carnicer M, Wahl A, Ferrer P, Albiol J. Quantitative metabolomics and instationary 13C-metabolic flux analysis reveals impact of recombinant protein production on trehalose and energy metabolism in Pichia pastoris. Metabolites. 2014;4:281–99.
Kubicek CP, Schreferl-Kunar G, Wöhrer W, RöHr M. Evidence for a cytoplasmic pathway of oxalate biosynthesis in Aspergillus niger. Appl Environ Microbiol. 1988;54:633.
Zhang H, Zhang L, Chen H, Chen YQ, Wei C, Song Y, Ratledge C. Enhanced lipid accumulation in the yeast Yarrowia lipolytica by over-expression of ATP: citrate lyase from Mus musculus. J Biotechnol. 2014;192:78–84.
Lawson CE, Harcombe WR, Hatzenpichler R, Lindemann SR, Loffler FE, O’Malley MA, Garcia Martin H, Pfleger BF, Raskin L, Venturelli OS, et al. Common principles and best practices for engineering microbiomes. Nat Rev Microbiol. 2019;17:725–41.
Ohnishi J, Katahira R, Mitsuhashi S, Kakita S, Ikeda M. A novel gnd mutation leading to increased l-lysine production in Corynebacterium glutamicum. FEMS Microbiol Lett. 2005;242:265–74.
Castillo S, Barth D, Arvas M, Pakula TM, Pitkanen E, Blomberg P, Seppanen-Laakso T, Nygren H, Sivasiddarthan D, Penttila M, Oja M. Whole-genome metabolic model of Trichoderma reesei built by comparative reconstruction. Biotechnol Biofuels. 2016;9:252.
Lu H, Cao W, Liu X, Sui Y, Ouyang L, Xia J, Huang M, Zhuang Y, Zhang S, Noorman H, Chu J. Multi-omics integrative analysis with genome-scale metabolic model simulation reveals global cellular adaptation of Aspergillus niger under industrial enzyme production condition. Sci Rep. 2018;8:14404.
Pakula TM, Laxell M, Huuskonen A, Uusitalo J, Saloheimo M, Penttila M. The effects of drugs inhibiting protein secretion in the filamentous fungus Trichoderma reesei—Evidence for down-regulation of genes that encode secreted proteins in the stressed cells. J Biol Chem. 2003;278:45011–20.
Peyraud R, Schneider K, Kiefer P, Massou S, Vorholt JA, Portais JC. Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst Biol. 2011;5:189.
Kwolek-Mirek M, Maslanka R, Molon M. Disorders in NADPH generation via pentose phosphate pathway influence the reproductive potential of the Saccharomyces cerevisiae yeast due to changes in redox status. J Cell Biochem. 2018;26:8521–33.
Cao W, Wang G, Lu H, Ouyang L, Chu J, Sui Y, Zhuang Y. Improving cytosolic aspartate biosynthesis increases glucoamylase production in Aspergillus niger under oxygen limitation. Microb Cell Fact. 2020;19:81.
Schütze T, Meyer V. Polycistronic gene expression in Aspergillus niger. Microb Cell Fact. 2017;16:162.
Beyer HM, Gonschorek P, Samodelov SL, Meier M, Weber W, Zurbriggen MD. AQUA Cloning: a versatile and simple enzyme-free cloning approach. PLoS ONE. 2015;10:e0137652.
Nielsen ML, Albertsen L, Lettier GL, Nielsen JB, Mortensen UH. Efficient PCR-based gene targeting with a recyclable marker for Aspergillus nidulans. Fungal Genet Biol. 2006;43:54–64.
Arentshorst M, Ram AFJ, Meyer V. Using non-homologous end-joining-deficient strains for functional gene analyses in filamentous fungi. Methods Mol Biol. 2012;835:133–50.
Carvalho ND, Arentshorst M, Kwon MJ, Meyer V, Ram AF. Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol. 2010;87:1463–73.
Wu L, Mashego MR, van Dam JC, Proell AM, Vinke JL, Ras C, van Winden WA, van Gulik WM, Heijnen JJ. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13 C-labeled cell extracts as internal standards. Anal Biochem. 2005;336:164–71.
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. The work of YFS has been funded by the Chinese scholarship council.
Author information
Authors and Affiliations
Contributions
YFS constructed all strains, characterized them, and performed shake flask cultivations under the supervision of TS. YFS, XZX, and JQ carried out the chemostat runs and analyzed the metabolome data. YFS and PL performed metabolite quantification by GC/LC–MS detection and GSMM simulations. YPZ and VM initiated this study and coordinated the project. YFS, LMQ, TS, HZL, and VM co-wrote the final text. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sui, Yf., Schütze, T., Ouyang, Lm. et al. Engineering cofactor metabolism for improved protein and glucoamylase production in Aspergillus niger. Microb Cell Fact 19, 198 (2020). https://doi.org/10.1186/s12934-020-01450-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-020-01450-w