
Abstract
Vitamin B12 is an essential vitamin that is widely used in medical and food industries. Vitamin B12 biosynthesis is confined to few bacteria and archaea, and as such its production relies on microbial fermentation. Rational strain engineering is dependent on efficient genetic tools and a detailed knowledge of metabolic pathways, regulation of which can be applied to improve product yield. Recent advances in synthetic biology and metabolic engineering have been used to efficiently construct many microbial chemical factories. Many published reviews have probed the vitamin B12 biosynthetic pathway. To maximize the potential of microbes for vitamin B12 production, new strategies and tools are required. In this review, we provide a comprehensive understanding of advances in the microbial production of vitamin B12, with a particular focus on establishing a heterologous host for the vitamin B12 production, as well as on strategies and tools that have been applied to increase microbial cobalamin production. Several worthy strategies employed for other products are also included.
Similar content being viewed by others


Background
Vitamin B12, also known as cyanocobalamin, belongs to the cobalamin family of compounds, which are composed of a corrinoid ring and an upper and lower ligand. The upper ligand can be adenosine, methyl, hydroxy, or a cyano group [1]. Vitamin B12 is synthesized by prokaryotes and inhibits the development of pernicious anemia in animals.
Microbial de novo biosynthesis of vitamin B12 occurs through two alternative routes: the aerobic or anaerobic pathway, in bacteria and archaea, respectively. Some strains can also synthesize cobalamin by absorbing corrinoids via a salvage pathway, as shown in Table 1. Tetrapyrrole compounds including cobalamin, heme, and bacteriochlorophyll, are derived from δ-aminolevulinate (ALA) and a complex interdependent and interactional relationship exists among these tetrapyrrole compounds in numerous bacterial species [2]. To maintain vitamin B12 at stable levels, its biosynthesis and transportation is regulated by a cobalamin riboswitch in the 5′ untranslated regions (UTR) of the corresponding genes.
Large scale industrial production of vitamin B12 occurs via microbial fermentation, predominantly utilizing Pseudomonas denitrificans, Propionibacterium shermanii, or Sinorhizobium meliloti [7]. However, these strains have several shortcomings, such as long fermentation cycles, complex and expensive media requirements, and a lack of suitable genetic systems for strain engineering. To date, most of the research on these producers has focused on traditional strategies, such as random mutagenesis and fermentation process optimization, with only limited research on metabolic engineering. Recently, engineers have shifted their attention to Escherichia coli as a platform for vitamin B12 production. E. coli has become a well-studied cell factory that has been extensively used for the production of various chemicals, such as terpenoids, non-natural alcohols, and poly-(lactate-co-glycolate) [8–10]. Furthermore, metabolic engineering and synthetic biology strategies have been extensively applied to improve the production of these compounds [11, 12]. Escherichia coli synthesizes ALA via the C5 pathway and has been used as a microbial cell factory to produce ALA via C4 and C5 pathways [13, 14] and E. coli can also synthesize vitamin B12 via the salvage pathway. The closely related Salmonella typhimurium is able to synthesize vitamin B12 de novo. Many genes involved in vitamin B12 biosynthesis in S. typhimurium have been shown to be functional in E. coli [15–17]. Transfer of 20 genes from the S. typhimurium cob locus allowed the production of vitamin B12 in E. coli [18]. These advantages facilitate the de novo production of vitamin B12 in E. coli.
Due to the complexity of the pathway and its metabolic regulation, numerous studies have been performed by various groups on the vitamin B12 biosynthesis. This review provides an overview of the vitamin B12 biosynthesis and its metabolic regulation, along with an investigation of several strategies for the development of microbial cell factories, including synthetic biology and metabolic engineering, among others. Since research on the engineering of vitamin B12 producing strains remains limited, related strategies for other chemicals are also outlined.
Cobalamin biosynthetic pathway
De novo pathway
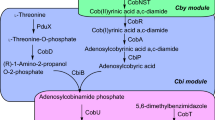
As mentioned above, cobalamin can be synthesized de novo in prokaryotes through two alternative routes according to the timing of cobalt insertion and the molecular oxygen requirement. These pathways are the aerobic pathway, which has been best studied in P. denitrificans, and the anaerobic pathway, which has been best studied in S. typhimurium, Bacillus megaterium, and P. shermanii, [19]. Both routes differ in terms of cobalt chelation (via hydrogenobyrinic a, c-diamide with the CobNST complex in the aerobic pathway, and via precorrin-2 with CbiK in S. typhimurium) and oxygen requirements (the aerobic pathway requires oxygen to promote ring-contraction, while the anaerobic pathway does not require oxygen in this step) (Fig. 1).

Biosynthetic pathways of tetrapyrrole compounds. ALA is synthesized by either the C4 or the C5 pathway. Adenosylcobalamin is synthesized via the de novo or via salvage pathways. The enzymes shown in the adenosylcobalamin biosynthetic pathway originate from P. denitrificans or S. typhimurium, which either use the aerobic pathway or the anaerobic pathway, respectively
Biosynthetic pathways of tetrapyrrole compounds: ALA is synthesized by either C4 or C5 pathway and adenosylcobalamin is synthesized either via de novo or salvage pathways. The enzymes shown in the adenosylcobalamin biosynthetic pathway originate from P. denitrificans or S. typhimurium, which use either the aerobic pathway or anaerobic pathway, respectively.
The first committed precursor of the tetrapyrrole synthesis pathway is ALA. ALA is synthesized by either the C4 pathway or the C5 pathway. In the C4 pathway, the enzyme ALA synthase from glycine and succinyl-CoA catalyzes the formation of ALA. In the C5 pathway, ALA is synthesized from glutamate through three enzymatic reactions [20]. Two molecules of ALA are condensed to form monopyrrole porphobilinogen by porphobilinogen synthase and four porphobilinogen molecules are then polymerized and cyclized to form uroporphyrinogen III. This reaction is catalyzed by the enzymes porphobilinogen deaminase and uroporphyrinogen III synthase. Methylation of uroporphyrinogen III at C-2 and C-7 results in the synthesis of precorrin-2 (which is a common precursor of cobalamin), siroheme, and coenzyme F430 [7, 21]. In P. denitrificans, CobA catalyzes this methylation step. In S. typhimurium and E. coli, the fusion enzyme CysG is shared between siroheme and cobalamin synthesis. The N-terminus of CysG has dehydrogenase/ferrochelatase activity and the C-terminus has uroporphyrinogen III methyltransferase activity. In S. cerevisiae, MET1p functions as a uroporphyrinogen III methyltransferase [22].
The aerobic and anaerobic pathways diverge at precorrin-2 and converge at coby(II)rinic acid a, c-diamide. Eight peripheral methylation reactions occur during de novo cobalamin biosynthesis, within identical temporal and spatial orders in both the aerobic and anaerobic pathways. Many of the methyltransferase enzymes involved in these reactions show high degrees of sequence similarity [23].
Cob(I)yrinic acid a,c-diamide is adenosylated to form adenosyl cobyrinic acid a,c-diamide. Cob(I)yrinic acid a,c-diamide adenosyltransferase can also adenosylate other corrinoids, where at least the a and c positions of the carboxyl groups are amidated. Adenosyl cobyrinic acid a,c-diamide is subjected to four stepwise amidation reactions at carboxyl groups at positions b, d, e, and g to yield adenosyl cobyric acid. Two separate methods have evolved to attach (R)-1-amino-2-propanol or (R)-1-amino-2-propanol phosphate at the f position of the carboxyl group of adenosyl cobyric acid in the aerobic and anaerobic pathways. In the anaerobic pathway, the linker between the corrinoid ring and the lower axial ligand is phosphorylated prior to attachment of the corrinoid ring. The enzyme PduX from S. enterica is an l-threonine kinase used in the de novo synthesis of coenzyme B12; however, it is not involved in the cobinamide salvage pathway [17]. l-threonine O-3-phosphate is then decarboxylated to yield (R)-1-amino-2-propanol O-2-phosphate via CobD in S. typhimurium LT2 [15]. However, in P. denitrificans it is most likely (although proof remains to be published), that (R)-1-amino-2-propanol is directly attached to the corrinoid ring via protein α and β. Currently, protein α remains as yet unknown and protein β is a complex of CobC and CobD. The molecule is then phosphorylated by CobP, which is a bifunctional enzyme possessing ATP:AdoCbi (adenosylcobinamide) kinase and GTP:AdoCbi-P guanylyltransferase activity [24]. Two additional reactions transfer lower axial ligands onto AdoCbi-GDP, thus producing adenosylcobalamin (AdoCbl). There are two alternative views of the last step in vitamin B12 biosynthesis. One view is that the last reaction in the biosynthesis of AdoCbl involves the addition of α-ribazole, catalyzed via cobalamine synthase. However, in S. typhimurium, α-ribazole 5′-phosphate is added to AdoCbi-GDP and thus, the last reaction would be the dephosphorylation of AdoCbl 5′-phosphate to AdoCbl, catalyzed by an AdoCbl-5-P phosphatase (CobC) [25]. The nucleotide loop assembly pathway is the last characterized reaction in the vitamin B12 biosynthesis. BluB from S. meliloti is a member of the reduced form of nicotinamide-adenine dinucleotide (NADH)/flavin mononucleotide (FMN)-dependent nitroreductase family, which can convert FMNH2 to DMB (5, 6-dimethylbenzimidazole) [26, 27]. In the anaerobic bacterium E. limosum, 5-aminoimidazole ribotide is converted to DMB by enzymes encoded in the bza operon [28] and subsequently, CobT can activate a range of lower ligand substrates including DMB, which determine cobamide diversity [29].
Salvage pathway
The salvage pathway is a cost-effective way (in terms of energy) for bacteria and archaea to obtain cobalamin. In gram-negative bacteria, exogenous corrinoids are transported into the cell via an ATP-binding cassette (ABC) transport system, consisting of BtuC, BtuD, and BtuF, which are membrane permease, ATPase, and periplasmic-binding protein components, respectively. BtuB is a TonB-dependent transporter located in the outer membrane, delivering corrinoid to the periplasmic corrinoid-binding protein BtuF. The latter then delivers corrinoid to the BtuCD complex located in the inner membrane [30]. Archaea also use ABC transporters for corrinoid uptake. Archaeal orthologs of the bacterial BtuC, BtuD, and BtuF have been found in Halobacterium sp. strain NRC-1 [31]. Subsequent to transport through the membrane, cobinamide is adenosylated by ATP:co(I)rrinoid adenosyltransferases (ACATs). Three families of ACATs exist, namely: CobA, EutT, and PduO [32]. In bacteria, AdoCbi is the substrate for a bifunctional enzyme (CobU in S. typhimurium or CobP in P. denitrificans) with kinase and guanylyltransferase activities. In archaea, the cbiZ gene encodes an amidohydrolase that converts AdoCbi to adenosylcobyric acid, which is condensed with 1-aminopropanol-O-2-phosphate by an AdoCbi-P synthase (CbiB) to yield AdoCbi-P. Since the archaeal enzyme lacks AdoCbi kinase activity, CobY, which has GTP:AdoCbi-P guanylyltransferase activity, is used to transfer guanylyl to AdoCbi-P [30, 33]. Identical to the de novo pathway, two additional reactions transfer lower axial ligands onto AdoCbi-GDP to produce AdoCbl in the salvage pathway.
Metabolic regulation of cobalamin synthesis
The relationship between cobalamin, heme, and chlorophyll
Photosynthetic bacteria possess all three tetrapyrrole compounds including cobalamin, heme, and bacteriochlorophyll (Fig. 1), which share the same biosynthetic pathway from ALA to uroporphyrinogen III. Excess heme inhibits HemA via feedback inhibition, reducing the flux of cobalamin and bacteriochlorophyll, while heme limitation weakly regulates hemCD, hemH, and hemA expression in E. coli [34]. However, not just a competitive relationship exists among the tetrapyrrole compounds, as these compounds also share a complex interdependent and interactional relationship [2]. Hydrogenobyrinic acid synthase from Rhodobacter capsulatus possesses two Fe-S centers, a flavin and a heme group [35]. Rhodobacter capsulatus requires a cobalamin cofactor to form the isocyclic ring of chlorophyll [36]. Synthesis of heme is also cobalamin dependent, as heme synthesis requires S-adenosylmethionine as a methyl group donor, while S-adenosylmethionine synthesis involves a cobalamin-dependent enzyme [37]. This complex relationship is also manifest at the transcriptional level. E.g., cobalamin participates in the regulation of bacteriochlorophyll synthesis via the AerR-CrtJ regulatory pair in R. capsulatus. CrtJ is responsible for the repression of bacteriochlorophyll during aerobic growth [38], while AerR functions as an anti-repressor of CrtJ, inhibiting CrtJ binding to the bchC promoter when cobalamin is bound to the conserved histidine (His145) of AerR [39]. In R. sphaeroides, heme affects the ability of PpsR to regulate genes that are involved in the tetrapyrrole biosynthesis by binding PpsR and changing its DNA binding pattern. Thus, PpsR functions as both a redox and a heme sensor to coordinate cellular heme and bacteriochlorophyll levels [38]. This complex relationship between tetrapyrrole compounds is a consequence of natural evolution.
Regulation of the key enzyme S-Adenosyl-l-methionine: uroporphyrinogen III methyltransferase (SUMT)
Feedback inhibition of a key enzyme located at the branch point of a biosynthetic pathway is a common method for metabolic regulation in microbes. In many microorganisms, SUMT regulates cobalamin flux. In P. denitrificans, SUMT is sensitive to feedback inhibition by cobalamin and corrinoid intermediates, and exhibits substrate inhibition at uroporphyrinogen III concentrations above 2 µM [40]. Bacillus megaterium SUMT exhibits substrate inhibition at uroporphyrinogen concentrations above 0.5 µM [41], while P. denitrificans SUMT is inhibited by uroporphyrinogen concentrations above 0.2 µM [40]. Fortunately, uroporphyrinogen III methyltransferase shows limited substrate inhibition. As an example, the uroporphyrinogen III methyltransferase of Methanobacterium ivanovii and Paracoccus pantotrophus were not inhibited by uroporphyrinogen III, even at concentrations of up to 20 µM [42, 43]. The identification of new SUMT enzymes that are insensitive to feedback inhibition to replace the native enzyme of industrial strains may be an effective method to improve vitamin B12 yield.
Cobalamin riboswitches
The cobalamin riboswitch is the predominant form of metabolic regulation to control vitamin B12 concentration in microbes. Riboswitches are evolutionarily conserved non-coding regions that are situated in the 5′-untranslated region of mRNAs that regulate gene expression in response to direct binding of intracellular metabolites by the RNA itself [44]. Riboswitches are composed of two functional domains: one domain serves as an evolutionarily conserved natural aptamer, binding the target metabolite with high selectivity and affinity, while the other domain harnesses allosteric changes in RNA structure, caused by aptamer-ligand complex formation, to control expression of an adjacent gene or operon. Johnson et al. solved the crystal structures of two different classes of cobalamin riboswitches, which share a common four-way junction (P3–P6 helices), forming the core receptor domain that is responsible for cobalamin binding, but use distinct peripheral extensions (P8–P11) to recognize different cobalamin derivatives [45]. The P6 extension is present for the AdoCbl binding class, while it is absent for the methylcobalamin and the aquocobalamin binding classes [45–47]. A kissing-loop interaction between loop L5 of the cobalamin-binding core and L13 of the regulatory domain regulates the expression machinery [46]. Holmstrom et al. identified the conformational mechanism responsible for the regulation of gene expression by a hydroxocobalamin binding riboswitch (env8HyCbl). The authors utilized single-molecule fluorescence resonance energy transfer techniques [48]. Binding of hydroxocobalamin promotes the formation of the L5–L13 kissing-loop, which sequesters the Shine-Dalgarno sequence via base pairing, thus preventing translation initiation. Cobalamin riboswitches participate in the regulation of cobalamin biosynthesis and transport at the transcriptional or translational levels, such as the btuB gene of E. coli and the cob operon of S. typhimurium [49]. In case of transcription inhibition and for high cobalamin concentrations, an alternative Rho-independent termination hairpin or Rho binding site cause premature transcriptional termination. High cobalamin concentrations can also promote sequestration of ribosome binding sites (RBS) and blockage of translation initiation. When cobalamin concentrations are low, an anti-terminator hairpin forms, enabling RNA polymerase to complete transcription of the downstream gene. Low cobalamin concentrations facilitate anti-sequester hairpin formation that releases the RBS, thus enabling translation initiation [50, 51]. Cobalamin riboswitches also have a particular function in ethanolamine utilization. In Listeria monocytogenes, a cobalamin riboswitch controls the expression of a noncoding regulatory RNA named Rli55, which controls the expression of the eut genes that requires vitamin B12 as a cofactor and determines ethanolamine utilization [52].
Synthetic biology to improve cobalamin production
Design of a heterologous biosynthetic pathway for the vitamin B12 production
Apart from altering native microbial hosts or identifying novel microbial hosts to produce cobalamin, the construction of heterologous biosynthetic pathways in model organisms that can be easily genetically manipulated is a promising strategy. Synthetic biology is an efficient tool that can be used to reconstruct pathways or genetic networks to produce compounds in a heterologous host. The construction of a vitamin B12 biosynthetic pathway in a heterologous host includes selection of a suitable host, building the biosynthetic pathway with functional components, and pathway tuning (Fig. 2). Several points should be noted in the selection of an ideal host. (1) The host should have the ability to supply precursors (e.g. ALA) and cofactors (e.g. S-adenosylmethionine) for the production of the desired chemical. E.g., the heterologous C4 pathway in E. coli has been reported to generate high ALA production [13], avoiding addition of exogenous ALA to the medium; (2) there need to be sufficient genetic engineering tools such as transformation protocols, expression vectors, and chromosomal gene knockout/integration systems to manipulate the host [53]; (3) the host is suitable for industrial-scale fermentation, utilizing cheap and readily available carbon sources such as glucose, xylose, and arabinose. When the host is selected, various candidate genes from diverse native vitamin B12 producers can be expressed in the host. The use of in vitro kinetic analysis is an efficient approach to detect ideal enzymes. Most of the intermediates of the vitamin B12 synthetic pathway are not available; consequently, to prepare substrates for in vitro kinetic analysis, the desired chemicals need to be separated from the products of corresponding strains or be prepared enzymatically. The products of an in vitro assay can then be detected by spectroscopic analysis, mass spectrometry, or microbiological assays. Sometimes, the heterologous production of enzymes does not work and it is necessary to screen for novel enzymes from various sources. For heterologous expression studies, all native forms of regulation, such as riboswitches, should be removed. After the addition of transcriptional and translational elements, such as promoters, ribosome binding sequences, and terminators, the structural genes for cobalamin synthesis can be expressed either monocistronically or polycistronically on plasmids or be integrated into the genome. The rapid development of synthetic biology tools has facilitated the simplified construction of heterologous pathways. Heterogeneous genes can be assembled by a number of different techniques such as SLIC, CPEC, Gibson assembly, golden gate cloning, DNA assembler, and LCR [54]. When too many heterogeneous genes need to be transferred to a heterologous host, it is difficult to build the metabolic pathway one gene at a time. The metabolic route can be split into multiple modules. After sequential validation of the function of these modules, their assembly becomes possible. The final construct can then be transferred to the chosen host for heterologous expression, thus allowing the host to synthesize cobalamin. To evaluate the capacity of vitamin B12 production, the engineered strains are then cultured under optimal conditions.

Design of a heterologous biosynthetic pathway. a A host for the heterologous biosynthetic pathway is selected considering the capability of precursor and cofactor supply, genetic engineering tools, and industrial-scale fermentation capability, utilizing cheap and readily available carbon sources. b Enzyme activity is verified in vitro and subsequently in vivo. Products of the in vitro assay or intracellular reaction products are detected via spectroscopic analysis, mass spectrometry, or microbiological assays. c Heterogeneous genes and other functional elements are assembled on plasmids via gene assembly methods such as SLIC, CPEC, Gibson, golden gate, DNA assembler and LCR, or integrated into the genome. To decrease the difficulty of building the metabolic pathway, it is divided into separate modules. These modules are verified sequentially in a heterologous host and then assembled. d Based on the quantification of metabolites, bottlenecks should be removed and metabolic flux should be integrated to target compound maximization. To optimize gene expression in the metabolic pathway, promoters, RBS, and gene copy number are designed and implemented at the transcriptional or translational levels. e The characteristics of the engineered strains are verified via fermentation. Various substrates (e.g., ALA, cobalt ions, betaine and DMB) and varying conditions (e.g., dissolved oxygen concentration, pH, and temperature) can be optimized to improve yield and productivity
Design of a heterologous biosynthetic pathway. (A) A host for the heterologous biosynthetic pathway is selected considering capabilities of precursor and cofactor supply, genetic engineering tools, and industrial-scale fermentation capability using cheap and readily available carbon sources. (B) Enzyme activity is verified in vitro and thereafter in vivo. Products of in vitro assay or intracellular reaction products are detected via spectroscopic analysis, mass spectrometry, or microbiological assays. (C) Heterogeneous genes and other functional elements are assembled on plasmids via gene assembly methods such as SLIC, CPEC, Gibson, golden gate, DNA assembler, and LCR, or integrated into the genome. To decrease the difficulty of building the metabolic pathway, it is divided into separate modules. These modules are sequentially verified in a heterologous host, and then assembled. (D) Based on the quantification of metabolites, bottlenecks should be removed and metabolic flux to target compound maximization. To optimize gene expression in the metabolic pathway, promoters, RBS, and gene copy number are designed and implemented at the transcriptional or translational levels. (E) The characteristics of engineered strains are verified via fermentation. Various substrates (e.g., ALA, cobalt ions, betaine and DMB) and varying conditions (e.g., dissolved oxygen concentration, pH, and temperature) can be optimized to improve yield and productivity.
Pathway tuning
Biologists face the challenge to direct the flux of intermediates to generate the desired product. To avoid the accumulation of toxic intermediates and to drive flux to the desired end product, each step in the pathway needs to be balanced. In addition, gene overexpression may cause an undesirable metabolic burden on the host, and the level of gene expression needs to be accurately adjusted to coordinate both metabolic flux and cell growth. Sometimes chromosomal expression of heterologous metabolic pathways can avoid plasmid instability and thus reduce the metabolic burden on the cell. Vitamin B12 biosynthesis is a highly evolved pathway, with more than twenty steps, including corrinoid ring construction and peripheral modifications, and the enzymes involved have high substrate specificity. Therefore, it is necessary to optimize gene expression in existing metabolic pathways.
Gene expression in a given pathway can be altered at the level of transcription or translation [55]. Promoter strength affects gene expression at the transcriptional level, while DNA sequence of the RBS affects gene expression at the translational level. The “Ribosome binding site calculator” and RBSDesigner have been used to predict and design synthetic RBSs to yield the desired level of protein expression in bacteria [56]. The expression of multiple genes in synthetic operons can be optimized to enhance production levels via engineering of promoters [57] and RBSs [58], or via generation of libraries of tunable intergenic regions [59]. Tuning operon expression can be achieved via different plasmid replicons to control gene copy number, different promoters to control the rate of transcription initiation, and different RBSs for controlling the level of translation [60]. Multiplex automated genome engineering (MAGE) can be used for genome-scale optimization of gene expression on the chromosome [61].
The translational efficiency of each gene in an operon is affected by intergenic sequence regions and involves post-transcriptional processes such as transcription termination, mRNA stability, and translation initiation. Therefore, achieving the desired expression level of each gene within an operon remains challenging. Pfleger et al. generated libraries of tunable intergenic regions, recombining various post-transcriptional control elements to optimize the expression of multiple genes in a synthetic operon [62]. Tian et al. also designed synthetic operons that utilize translational coupling to achieve the desired expression level ratios [63].
Scaffolding is a way to rapidly increase overall pathway flux and is complementary to these conventional methods. Scaffolding can increase the effective concentration of intermediates in the pathway of interest, via the recruitment of enzymes to synthetic protein scaffolds. Three mevalonate biosynthetic enzymes (acetoacetyl-CoA thiolase, hydroxy-methylglutaryl-CoA synthase, and hydroxymethylglutaryl-CoA reductase) were recruited to a synthetic protein scaffold through interactions between the GTPase binding domain, the SH3 domain, the PSD95/DlgA/Zo-1 domain and their respective ligands, increasing mevalonate yields 77-fold compared to cells without the scaffold [64]. Protein scaffolds simulate natural multienzyme complexes and have been used to solve problems of toxic pathway intermediate accumulation, competing metabolic reactions, and imbalances in flux [65]. Protein scaffolds have been successfully applied in various other pathways, including the glucaric acid pathway, where three pathway enzymes were joined in a protein scaffold, increasing titers by approximately five-fold [66]. In another example, heterologous butyrate pathway enzymes were co-localized in a protein scaffold, resulting in a threefold improvement in butyrate production [67]. Cobalamin synthesis is a complex pathway with several competing intermediates, such as heme and siroheme. Co-localization of enzymes to the same subcellular organelle or compartment can increase the local intermediate concentration and exclude competing cytosolic pathways [68]. Substrate channeling is a process of transferring the product of one enzyme to an adjacent enzyme within the cascade or cell without complete mixing with the bulk phase [69]. This phenomenon occurs for at least some of the enzymes involved in hydrogenobyrinic acid (HBA) biosynthesis. An enzyme-trap approach has been used to isolate intermediates of the cobalamin biosynthetic pathway based on this mechanism [70]. Therefore, protein scaffolds may offer a promising alternative to balance multiple enzymes within the vitamin B12 biosynthetic pathway, thus improving flux to cobalamin.
All these approaches optimize gene expression or increase pathway flux in vivo. In vitro steady-state analysis is an efficient approach to identify bottlenecks in metabolic pathways and to balance flux to the desired compounds. It has been successfully used to increase the production of fatty acids, fatty acid short-chain esters, fatty alcohols, farnesenes, alkenes, and alkanes [71–75]. Previously, we have optimized the concentration of enzymes involved in precorrin-2 synthesis in vitro via response surface methodology [76]. This approach minimizes the effects of substrate inhibition and feedback inhibition by SUMT and increases the production of precorrin-2 approximately five-fold.
Construction of a heterologous biosynthetic pathway for cobalamin in E. coli
Escherichia coli is a well-characterized prokaryote that has been used as a microbial cell factory for many chemicals. Although it has lost the de novo cobalamin synthesis pathway during evolution, E. coli can synthesize cobalamin through the salvage pathway, thus saving resources and energy. Escherichia coli has been used to produce ALA in many studies. A sufficient ALA supply is necessary for the vitamin B12 biosynthesis. This implies that E. coli may be a suitable host for vitamin B12 production. Other common bacteria such as Corynebacterium glutamicum and Bacillus subtilis lack the genes involved in the cobalamin synthesis pathway after precorrin-2, which may explain why the construction of a heterologous de novo cobalamin synthesis pathway in bacteria other than E. coli has yet to be reported.
For a long time, cobalamin production was restricted to native bacterial producers, with the exception of several studies that focused on the determination of gene function in the cobalamin synthesis pathway in vivo [16, 35, 70, 77] or by using cell extracts from recombinant E. coli in vitro (Table 2) [78–82]. After insertion of the B. megaterium cob I operon and S. typhimurium cbiP, E. coli was able to synthesize cobyric acid de novo [77]. Co-expression of the cobA gene from Propionibacterium freudenreichii and the cbiAP, cbiCDETFGHJ and cbiJKL genes from S. typhimurium equipped E. coli with the ability to produce cobyrinic acid a,c-diamide [16]. Apart from the genes involved in the anaerobic cobalamin biosynthetic pathway, genes involved in aerobic cobalamin biosynthetic pathway have also been shown to work in E. coli. Furthermore, E. coli possesses enzymes that perform the transformation of uroporphyrinogen III into HBA, thus potentially producing HBA [70]. McGoldrick et al. conducted a similar experiment, where nine genes from R. capsulatus and Brucella melitensis, encoding enzymes for the transformation of uroporphyrinogen III into HBA, were constructed as an operon in pET14b and introduced into E. coli, allowing the host to produce HBA [35]. To the best of our knowledge, there are only two reports on de novo cobalamin synthesis in E. coli: Firstly, an E. coli strain harboring a plasmid with a 21.5-kb native operon (pduBAF-pocR-cbiABCDETFGHJKLMNQOP-cobUST) from S. typhimurium resulted in de novo cobalamin biosynthesis [18]. Recently, Ko et al. also accomplished cobalamin biosynthesis in E. coli [83]. Twenty-two genes located in six different operons of P. denitrificans were cloned via traditional restriction and ligation into three compatible plasmids under the control of a T7 promoter. The recombinant E. coli strain harboring these three plasmids produced vitamin B12 under both anaerobic and aerobic conditions. Via optimizing the culture conditions, the engineered strain produced 0.65 ± 0.03 μg/g cdw of coenzyme B12. These examples show that E. coli can be utilized to produce vitamin B12 or pathway intermediates de novo through the aerobic or anaerobic pathway.
Metabolic engineering for cobalamin production
Scheme of metabolic engineering
When a micro-organism possesses its own or a heterologous cobalamin synthesis pathway, efforts should be directed towards engineering the metabolic network to enhance vitamin B12 production and yield. Classical metabolic engineering involves an iterative process of synthesis and analysis, where increasingly refined strains are designed and constructed based on gathered knowledge [84]. However, systems metabolic engineering allows microbes to be engineered at the whole-organism level for the production of valuable chemicals far beyond their native capabilities [85]. Metabolic design based on in silico simulations and experimental validation of the metabolic state in the engineered strain facilitates systematic metabolic engineering [86]. Many genome-scale metabolic flux models have been developed to design microbial cell factories. In silico simulations based on genome-scale metabolic models have provided valid guidance for rational design. Computational tools used in metabolic flux analysis and gene manipulation studies have been systematically reviewed [87]. Depending on the design of the specific strain, experimental implementation can involve a combination of gene over-expression, introduction of foreign enzymes, gene deletions, or knockdowns and the modification of enzyme properties.
Gene over-expression and heterologous expression
Gene over-expression is used to enhance or redirect flux to the desired chemical reaction. Expression of the target gene can be increased through the use of multi-copy plasmids, or the insertion of transcriptional and translational control elements, such as strong promoters, highly efficient RBSs, and terminator sequences. The introduction of heterologous genes is used to overcome the comparatively low activity of native genes in a host. The structural genes required for cobalamin synthesis are typically targets for gene over-expression to boost cobalamin production. In a recombinant P. freudenreichii strain, which harbors an expression vector containing cobA, cbiLF, or cbiEGH, vitamin B12 production was increased 1.7-, 1.9-, and 1.5-fold, respectively, compared to P. freudenreichii harboring the expression vector pPK705 [88]. Expression of cobU and cobS in the same P. freudenreichii strain led to a slight increase in the production of vitamin B12. ALA is a precursor, restricting cobalamin synthesis and a direct strategy to overcome this is to up-regulate the availability of ALA. A recombinant P. freudenreichii strain with heterologous hemA from R. sphaeroides, hemB, and cobA homologues, revealed a 2.2-fold increase in vitamin B12 production [88]. To improve the production of tetrapyrrole compounds, the hemA gene from R. sphaeroides and the hemB gene from P. freudenreichii subsp. shermanii IFO12424 were expressed either monocistronically or polycistronically in the strain P. freudenreichii subsp. shermanii IFO12426. The recombinant strains accumulated larger amounts of ALA and PBG, with a resultant 28- to 33-fold increase in the production of porphyrinogens, compared to strain P. freudenreichii subsp. shermanii IFO12426, harboring the vector pPK705 [89]. A cobalamin-riboswitch can be used to inhibit excess vitamin B12 in microbes. To bypass the effects of the cobalamin-riboswitch, a cbi operon without the cobalamin-riboswitch was cloned. Growth of B. megaterium, harboring a plasmid expressing the cbi operon on minimal media supplemented with glycerol as a carbon source, resulted in a significant increase in cobalamin production (up to 200 μg/l) [90]. All these strategies focus on the over-expression of structural genes that are directly involved in the vitamin B12 biosynthesis. However, to the best of our knowledge, no published reports exist on other genes, such as those responsible for the S-adenosylmethionine (SAM) or DMB metabolism. This suggests that research into the metabolic engineering of vitamin B12 production is still in its infancy.
Inactivation or down-regulation of genes
A further general method of metabolic engineering is to knockout genes to improve precursor supply, or to eliminate by-products or competing chemical synthesis reactions. Genes that encode enzymes at specific branch points in the metabolic pathway are good targets for this strategy. For essential genes that cannot be deleted, clustered regularly interspaced short palindromic repeats (CRISPR), CRISPR interference (CRISPRi), sRNA, or regulatory RNA can be used to repress the desired genes [91, 92]. Many genome editing tools such as, traditional homologous recombination systems and ZFN (zinc finger nucleases), TALEN (transcription activator-like effector nucleases), and CRISPR/Cas-based methods readily enable genetic modifications. Heme and siroheme are competitive byproducts of the vitamin B12 synthesis. To reduce flux to the heme branch of the tetrapyrrole pathway, antisense RNA was used to silence hemZ (which encodes coproporphyrinogen III oxidase) in B. megaterium. This approach led to a 20% increase in the intracellular vitamin B12 concentration [93]. We also inhibited hemE and hemH to improve flux to precorrin-2 via an sRNA approach (unpublished data).
Protein engineering
During the engineering of native or heterologous hosts to produce a desired chemical, a common scenario is that a substance or product inhibits an enzyme or it may not function heterologously. Protein engineering is a useful approach to improve the specificity, solubility, and stability of enzymes. It includes directed evolution, semi rational design, and rational design. The enzyme glutamyl-tRNA reductase (HemA) catalyzes the first committed step in the heme biosynthetic pathway. Expression of this gene is inhibited in a negative feedback loop by excess heme. The addition of two positively charged lysine residues to the third and fourth positions of the N terminus of HemA resulted in a complete stabilization of the protein. Cells expressing the stabilized HemA showed a substantial decrease in heme inhibition [94]. Engineering of CobA to remove substrate inhibition or the identification of novel genes without substrate inhibition may improve the yield of vitamin B12.
13C-metabolic flux analysis
13C-metabolic flux analysis is an efficient method to determine flux distribution based on experimental data and has been used to accurately estimate flux in the central carbon metabolism of P. denitrificans in response to different specific oxygen uptake rates under oxygen limiting conditions [95]. Metabolic flux analysis revealed that glucose was predominantly catabolized by the Entner–Doudoroff and pentose phosphate pathways. A higher specific oxygen uptake rate accelerated the supply of precursors, methyl groups, and NADPH to increase vitamin B12 production [95].
Other strategies to improve cobalamin production
Random mutagenesis
To produce strains with a high cobalamin yield, a conventional strategy is random mutagenesis. Specifically, UV light or chemical mutagens can both be used to treat the respective microorganisms and then, strains with the desired phenotype, such as improved productivity, genetic stability, reasonable growth rates, or resistance to high concentrations of toxic intermediates can be selected [7]. High throughput screening methods based on signals, such as survival and fluorescence, have been used to obtain desired mutants from large libraries. Moreover, random mutagenesis can also be applied to inverse metabolic engineering for targeted transformation.
Genome-scale engineering
Genome shuffling, an approach that combines random mutagenesis and protoplast fusion, has been used to improve vitamin B12 production in P. shermanii. The engineered P. shermanii strain produced approximately 61% more vitamin B12 than the parent strain after 96 h. Comparative proteome analysis revealed that the expression of 38 proteins varied significantly [96]. Compared to genome shuffling, which results in random mutations, MAGE provides an efficient method to simultaneously modify multiple genomic locations [61]. MAGE is based on the λ red recombination system. The repetitive introduction of ssDNA targeting multiple loci in the E. coli genome results in various mutants. Combined with standard high-throughput screening methods, MAGE may represent a rapid and efficient tool to obtain “ideal” bacterial producers. In addition, another versatile method, trackable multiplex recombineering (TRMR), enables rapid and simultaneous modification and mapping of thousands of genes. By changing functional regions such as promoters, translation sites, switches, oscillators, or sensors, a broad range of studies can be performed [97].
Biosensors
Biosensors have been extensively applied in high-throughput assays. For chemicals that cannot be easily detected via measurement of absorption or fluorescence, biosensors indirectly reflect chemical concentrations. A vitamin B12 riboswitch sensor has been used to study synthesis and import of coenzyme B12, and was shown to detect intracellular vitamin B12 concentration using colorimetric, fluorescent, or luminescent reporters with high sensitivity [98]. Reporter genes integrated with the btuB riboswitch have been used to monitor intracellular AdoCbl concentrations [98]. A combination of evolutionary strategies applied to metabolic pathways, whole genomes, or biosensors may be a useful approach to advance high-throughput screening for “ideal” strains.
Fermentation process optimization
The addition of precursors of the vitamin B12 biosynthetic pathway, such as cobalt ions, ALA, DMB, glycine, threonine, or compatible solutes like betaine and choline has been frequently described to be beneficial [7]. Propionic acid is a byproduct of the vitamin B12 cultivation process in P. freudenreichii and causes feedback inhibition of microbial cell growth. Propionic acid production and DMB addition were controlled via expanded bed adsorption bioreactors, which were shown to improve vitamin B12 biosynthesis [99]. Mixed culture of Propionibacterium and Ralstonia eutropha have also been used to solve this problem, as the latter can assimilate propionic acid produced by the former [100]. Betaine is an important methyl-group donor for the formation of methionine, which is further converted to SAM by the activity of methionine adenosyltransferase. S-adenosylmethionine is an important precursor during corrinoid ring formation. Despite the fact that betaine delays cell growth, betaine feeding during fermentation has been revealed as an effective strategy to increase vitamin B12 production [101]. To reduce medium and fermentation costs, cheap carbon sources such as maltose syrup and corn steep liquor can be used to replace refined sucrose [102].
Conclusions
Vitamin B12 is widely used in medical and food industries and microbes produce vitamin B12 via an intricate pathway. Tetrapyrrole compounds, including heme, cobalamin, and siroheme are not simple competitive compounds, but have both interdependent and interactive relationships in several bacterial species. To maintain stable vitamin B12 concentrations, its biosynthesis and transport are regulated at the transcriptional or translational level via riboswitches. Vitamin B12 is produced by microbial fermentation, using strains such as P. denitrificans and P. shermanii. Since relatively few genetic tools are available for these strains, and the fermentation process is complicated, strain engineering has focused on traditional strategies such as random mutagenesis and the optimization of the fermentation process. It is imperative to introduce new engineering tools, such as systems metabolic engineering, to manipulate these strains. Apart from native producers of vitamin B12, E. coli has also been used as a heterologous host. To provide guidance for the construction of microbial cell factories for vitamin B12 production, we summarized synthetic biology and metabolic engineering strategies, as well as other traditional strategies that either have been or could be applied to vitamin B12 production. These strategies have been extensively applied in microbial strain engineering to improve the production of many other chemicals. Based on a clear understanding of the vitamin B12 metabolism in microbes, the utilization of these strategies should promote an improved microbial vitamin B12 production.
Abbreviations
- ALA:
-
δ-aminolevulinate
- UTR:
-
untranslated regions
- ACAT:
-
ATP co(I)rrinoid adenosyltransferase
- AdoCbi:
-
adenosylcobinamide
- AdoCbl:
-
adenosylcobalamin
- NADH:
-
reduced form of nicotinamide-adenine dinucleotide
- FMN:
-
flavin mononucleotide
- NADPH:
-
reduced nicotinamide adenine dinucleotide phosphate
- ABC:
-
ATP-binding cassette
- SUMT:
-
S-Adenosyl-l-methionine:uroporphyrinogen III methyltransferase
- HBA:
-
hydrogenobyrinic acid
- RBS:
-
ribosome-binding site
- MAGE:
-
multiplex automated genome engineering
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- CRISPRi:
-
CRISPR interference
- TRMR:
-
trackable multiplex recombineering
- ZFN:
-
zinc finger nucleases
- TALEN:
-
transcription activator-like effector nucleases
References
Roth JR, Lawrence JG, Rubenfield M, Kieffer-Higgins S, Church GM. Characterization of the cobalamin (vitamin B12) biosynthetic genes of Salmonella typhimurium. J Bacteriol. 1993;175:3303–16.
Yin L, Bauer CE. Controlling the delicate balance of tetrapyrrole biosynthesis. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120262.
Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J Biol Chem. 2003;278:41148–59.
Lawrence JG, Roth JR. Evolution of coenzyme B12 synthesis among enteric bacteria: evidence for loss and reacquisition of a multigene complex. Genetics. 1996;142:11–24.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Swithers KS, Petrus AK, Secinaro MA, Nesbo CL, Gogarten JP, Noll KM, Butzin NC. Vitamin B12 synthesis and salvage pathways were acquired by horizontal gene transfer to the Thermotogales. Genome Biol Evol. 2012;4:730–9.
Martens JH, Barg H, Warren MJ, Jahn D. Microbial production of vitamin B12. Appl Microbiol Biotechnol. 2002;58:275–85.
Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol. 2003;21:796–802.
Zhang K, Sawaya MR, Eisenberg DS, Liao JC. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc Natl Acad Sci USA. 2008;105:20653–8.
Choi SY, Park SJ, Kim WJ, Yang JE, Lee H, Shin J, Lee SY. One-step fermentative production of poly(lactate-co-glycolate) from carbohydrates in Escherichia coli. Nat Biotechnol. 2016;34:435–40.
Lee SK, Chou H, Ham TS, Lee TS, Keasling JD. Metabolic engineering of microorganisms for biofuels production: from bugs to synthetic biology to fuels. Curr Opin Biotechnol. 2008;19:556–63.
Jarboe LR, Zhang X, Wang X, Moore JC, Shanmugam KT, Ingram LO. Metabolic engineering for production of biorenewable fuels and chemicals: contributions of synthetic biology. J Biomed Biotechnol. 2010;2010:761042.
Zhang L, Chen J, Chen N, Sun J, Zheng P, Ma Y. Cloning of two 5-aminolevulinic acid synthase isozymes HemA and HemO from Rhodopseudomonas palustris with favorable characteristics for 5-aminolevulinic acid production. Biotechnol Lett. 2013;35:763–8.
Kang Z, Wang Y, Gu P, Wang Q, Qi Q. Engineering Escherichia coli for efficient production of 5-aminolevulinic acid from glucose. Metab Eng. 2011;13:492–8.
Brushaber KR. CobD, a novel enzyme with l-Threonine-O-3-phosphate decarboxylase activity, is responsible for the synthesis of (R)-1-Amino-2-propanol O-2-phosphate, a proposed new intermediate in cobalamin biosynthesis in Salmonella typhimurium LT2. J Biol Chem. 1998;273:2684–91.
Roessner CA, Williams HJ, Scott AI. Genetically engineered production of 1-desmethylcobyrinic acid, 1-desmethylcobyrinic acid a, c-diamide, and cobyrinic acid a, c-diamide in Escherichia coli implies a role for CbiD in C-1 methylation in the anaerobic pathway to cobalamin. J Biol Chem. 2005;280:16748–53.
Fan C, Bobik TA. The PduX enzyme of Salmonella enterica is an l-threonine kinase used for coenzyme B12 synthesis. J Biol Chem. 2008;283:11322–9.
Raux E, Lanois A, Levillayer F, Warren MJ, Brody E, Rambach A, Thermes C. Salmonella typhimurium cobalamin (vitamin B12) biosynthetic genes: functional studies in S. typhimurium and Escherichia coli. J Bacteriol. 1996;178:753–67.
Moore SJ, Warren MJ. The anaerobic biosynthesis of vitamin B12. Biochem Soc Trans. 2012;40:581–6.
Avissar Y, Ormerod J, Beale S. Distribution of δ-aminolevulinic acid biosynthetic pathways among phototrophic bacterial groups. Arch Microbiol. 1989;151:513–9.
Zappa S, Li K, Bauer CE. The tetrapyrrole biosynthetic pathway and its regulation in Rhodobacter capsulatus. Adv Exp Med Biol. 2010;675:229–50.
Raux E, Mcveigh T, Peters SE, LeustekMcveigh T, Warren MJ. The role of Saccharomyces cerevisiae Met1p and Met8p in sirohaem and cobalamin biosynthesis. Biochem J. 1999;338:701–8.
Escalante-Semerena J, Warren M. Biosynthesis and use of cobalamin (B12). EcoSal Plus. 2008;3:1.
Cohen GN. Biosynthesis of cobalamins including vitamin B12. In Microbial biochemistry. Dordrecht: Springer; 2014. p. 555–565.
Zayas CL, Escalante-Semerena JC. Reassessment of the late steps of coenzyme B12 synthesis in Salmonella enterica: evidence that dephosphorylation of adenosylcobalamin-5′-phosphate by the CobC phosphatase is the last step of the pathway. J Bacteriol. 2007;189:2210–8.
Taga ME, Larsen NA, Howard-Jones AR, Walsh CT, Walker GC. BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature. 2007;446:449–53.
Campbell GR, Taga ME, Mistry K, Lloret J, Anderson PJ, Roth JR, Walker GC. Sinorhizobium meliloti bluB is necessary for production of 5,6-dimethylbenzimidazole, the lower ligand of B12. Proc Natl Acad Sci USA. 2006;103:4634–9.
Mehta AP, Abdelwahed SH, Fenwick MK, Hazra AB, Taga ME, Zhang Y, Ealick SE, Begley TP. Anaerobic 5-hydroxybenzimidazole formation from aminoimidazole ribotide: an unanticipated intersection of thiamin and vitamin B12 biosynthesis. J Am Chem Soc. 2015;137:10444–7.
Hazra AB, Tran JL, Crofts TS, Taga ME. Analysis of substrate specificity in CobT homologs reveals widespread preference for DMB, the lower axial ligand of vitamin B12. Chem Biol. 2013;20:1275–85.
Escalante-Semerena JC. Conversion of cobinamide into adenosylcobamide in bacteria and archaea. J Bacteriol. 2007;189:4555–60.
Woodson JD, Reynolds AA, Escalante-Semerena JC. ABC transporter for corrinoids in Halobacterium sp. strain NRC-1. J Bacteriol. 2005;187:5901–9.
Moore TC, Newmister SA, Rayment I, Escalante-Semerena JC. Structural insights into the mechanism of four-coordinate Cob(II)alamin formation in the active site of the Salmonella enterica ATP:Co(I)rrinoid adenosyltransferase enzyme: critical role of residues Phe91 and Trp93. Biochemistry. 2012;51:9647–57.
Newmister SA, Otte MM, Escalante-Semerena JC, Rayment I. Structure and mutational analysis of the archaeal GTP:AdoCbi-P guanylyltransferase (CobY) from Methanocaldococcus jannaschii: insights into GTP binding and dimerization. Biochemistry. 2011;50:5301–13.
McNicholas PM, Javor G, Darie S, Gunsalus RP. Expression of the heme biosynthetic pathway genes hemCD, hemH, hemM and hemA of Escherichia coli. FEMS Microbiol Lett. 1997;146:143–8.
McGoldrick HM, Roessner CA, Raux E, Lawrence AD, McLean KJ, Munro AW, Santabarbara S, Rigby SEJ, Heathcote P, Scott AI, Warren MJ. Identification and characterization of a novel vitamin B12 (cobalamin) biosynthetic enzyme (CobZ) from Rhodobacter capsulatus, containing flavin, heme, and Fe-S cofactors. J Biol Chem. 2004;280:1086–94.
Gough SP, Petersen BO, Duus JØ. Anaerobic chlorophyll isocyclic ring formation in Rhodobacter capsulatus requires a cobalamin cofactor. Proc Natl Acad Sci. 2000;97:6908–13.
Layer G, Moser J, Heinz DW, Jahn D, Schubert W-D. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J. 2003;22:6214–24.
Ponnampalam SN, Buggy JJ, Bauer CE. Characterization of an aerobic repressor that coordinately regulates bacteriochlorophyll, carotenoid, and light harvesting-II expression in Rhodobacter capsulatus. J Bacteriol. 1995;177:2990–7.
Cheng Z, Li K, Hammad LA, Karty JA, Bauer CE. Vitamin B12 regulates photosystem gene expression via the CrtJ antirepressor AerR in Rhodobacter capsulatus. Mol Microbiol. 2014;91:649–64.
Blanche F, Debussche L, Thibaut D, Crouzet J, Cameron B. Purification and characterization of S-adenosyl-l-methionine: uroporphyrinogen III methyltransferase from Pseudomonas denitrificans. J Bacteriol. 1989;171:4222–31.
Robin C, Blanche F, Cauchois L, Cameron B, Coude M, Crouzet J. Primary structure, expression in Escherichia coli, and properties of S-adenosyl-l-methionine-uroporphyrinogen III methyltransferase from Bacillus megaterium. J Bacteriol. 1991;173:4893–6.
Blanche F, Robin C, Couder M, Faucher D, Cauchois L, Cameron B, Crouzet J. Purification, characterization, and molecular cloning of S-adenosyl-l-methionine-uroporphyrinogen III methyltransferase from Methanobacterium ivanovii. J Bacteriol. 1991;173:4637–45.
Zajicek RS, Bali S, Arnold S, Brindley AA, Warren MJ, Ferguson SJ. d(1) haem biogenesis—assessing the roles of three nir gene products. FEBS J. 2009;276:6399–411.
Nahvi A, Barrick JE, Breaker RR. Coenzyme B12 riboswitches are widespread genetic control elements in prokaryotes. Nucleic Acids Res. 2004;32:143–50.
Johnson JE Jr, Reyes FE, Polaski JT, Batey RT. B12 cofactors directly stabilize an mRNA regulatory switch. Nature. 2012;492:133–7.
Souliere MF, Haller A, Santner T, Micura R. New insights into gene regulation—high-resolution structures of cobalamin riboswitches. Angew Chem Int Ed Engl. 2013;52:1874–7.
Choudhary PK, Duret A, Rohrbach-Brandt E, Holliger C, Sigel RKO, Maillard J. Diversity of cobalamin riboswitches in the corrinoid-producing organohalide respirer Desulfitobacterium hafniense. J Bacteriol. 2013;195:5186–95.
Holmstrom ED, Polaski JT, Batey RT, Nesbitt DJ. Single-molecule conformational dynamics of a biologically functional hydroxocobalamin riboswitch. J Am Chem Soc. 2014;136:16832–43.
Vitreschak AG. Regulation of the vitamin B12 metabolism and transport in bacteria by a conserved RNA structural element. RNA. 2003;9:1084–97.
Mandal M, Breaker RR. Gene regulation by riboswitches. Nat Rev Mol Cell Biol. 2004;5:451–63.
Serganov A, Nudler E. A decade of riboswitches. Cell. 2013;152:17–24.
Mellin JR, Koutero M, Dar D, Nahori MA, Sorek R, Cossart P. Riboswitches. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science. 2014;345:940–3.
Lee SY, Kim HU, Park JH, Park JM, Kim TY. Metabolic engineering of microorganisms: general strategies and drug production. Drug Discov Today. 2009;14:78–88.
de Kok S, Stanton LH, Slaby T, Durot M, Holmes VF, Patel KG, Platt D, Shapland EB, Serber Z, Dean J, et al. Rapid and reliable DNA assembly via ligase cycling reaction. ACS Synth Biol. 2014;3:97–106.
Fong SS. Computational approaches to metabolic engineering utilizing systems biology and synthetic biology. Comput Struct Biotechnol J. 2014;11:28–34.
Salis HM. Chapter two—the ribosome binding site calculator. In: Christopher V, editor. Methods in enzymology volume, vol. 498. Cambridge: Academic Press; 2011. p. 19–42.
Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci. 2006;103:3006.
Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–50.
Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotech. 2006;24:1027–32.
Arpino JA, Hancock EJ, Anderson J, Barahona M, Stan GB, Papachristodoulou A, Polizzi K. Tuning the dials of synthetic biology. Microbiology. 2013;159:1236–53.
Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–8.
Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol. 2006;24:1027–32.
Tian T, Salis HM. A predictive biophysical model of translational coupling to coordinate and control protein expression in bacterial operons. Nucleic Acids Res. 2015;43:7137–51.
Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KL, Keasling JD. Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol. 2009;27:753–9.
Chen AH, Silver PA. Designing biological compartmentalization. Trends Cell Biol. 2012;22:662–70.
Moon TS, Dueber JE, Shiue E, Prather KL. Use of modular, synthetic scaffolds for improved production of glucaric acid in engineered E. coli. Metab Eng. 2010;12:298–305.
Baek JM, Mazumdar S, Lee SW, Jung MY, Lim JH, Seo SW, Jung GY, Oh MK. Butyrate production in engineered Escherichia coli with synthetic scaffolds. Biotechnol Bioeng. 2013;110:2790–4.
Boyle PM, Silver PA. Parts plus pipes: synthetic biology approaches to metabolic engineering. Metab Eng. 2012;14:223–32.
Zhang YH. Substrate channeling and enzyme complexes for biotechnological applications. Biotechnol Adv. 2011;29:715–25.
Deery E, Schroeder S, Lawrence AD, Taylor SL, Seyedarabi A, Waterman J, Wilson KS, Brown D, Geeves MA, Howard MJ, et al. An enzyme-trap approach allows isolation of intermediates in cobalamin biosynthesis. Nat Chem Biol. 2012;8:933–40.
Yu X, Liu T, Zhu F, Khosla C. In vitro reconstitution and steady-state analysis of the fatty acid synthase from Escherichia coli. Proc Natl Acad Sci USA. 2011;108:18643–8.
Guo D, Zhu J, Deng Z, Liu T. Metabolic engineering of Escherichia coli for production of fatty acid short-chain esters through combination of the fatty acid and 2-keto acid pathways. Metab Eng. 2014;22:69–75.
Liu R, Zhu F, Lu L, Fu A, Lu J, Deng Z, Liu T. Metabolic engineering of fatty acyl-ACP reductase-dependent pathway to improve fatty alcohol production in Escherichia coli. Metab Eng. 2014;22:10–21.
Zhu F, Zhong X, Hu M, Lu L, Deng Z, Liu T. In vitro reconstitution of mevalonate pathway and targeted engineering of farnesene overproduction in Escherichia coli. Biotechnol Bioeng. 2014;111:1396–405.
Liu Q, Wu K, Cheng Y, Lu L, Xiao E, Zhang Y, Deng Z, Liu T. Engineering an iterative polyketide pathway in Escherichia coli results in single-form alkene and alkane overproduction. Metab Eng. 2015;28:82–90.
Fang H, Dong H, Cai T, Zheng P, Li H, Zhang D, Sun J. In vitro optimization of enzymes involved in precorrin-2 synthesis using response surface methodology. PLoS ONE. 2016;11:e0151149.
Evelyne RA, Rambach A, Warren MJ, Thermes C. Cobalamin (vitamin B12) biosynthesis: functional characterization of the Bacillus megaterium cbi genes required to convert uroporphyrinogen III into cobyrinic acid a, c-diamide. Biochem J. 1998;335:167–73.
Roessner CA, Spencer JB, Stolowich NJ, Wang J, Nayar GP, Santander PJ, Pichon C, Min C, Holderman MT, Scott AI. Genetically engineered synthesis of precorrin-6x and the complete corrinoid, hydrogenobyrinic acid, an advanced precursor of vitamin B12. Chem Biol. 1994;1:119–24.
Roessner CA, Spencer JB, Ozaki S, Min CH, Atshaves BP, Nayar P, Anousis N, Stolowich NJ, Holderman MT, Scott AI. Overexpression in Escherichia coli of 12 vitamin B12 biosynthetic enzymes. Protein Expr Purif. 1994;6:155–63.
Stamford NPJ, Duggan S, Li Y, Alanine AID, Crouzet J, Battersby AR. Biosynthesis of vitamin B12: the multi-enzyme synthesis of precorrin-4 and factor IV. Chem Biol. 1997;4:445–51.
Lundqvist J, Elmlund D, Heldt D, Deery E, Soderberg CA, Hansson M, Warren M, Al-Karadaghi S. The AAA(+) motor complex of subunits CobS and CobT of cobaltochelatase visualized by single particle electron microscopy. J Struct Biol. 2009;167:227–34.
Lawrence AD, Deery E, McLean KJ, Munro AW, Pickersgill RW, Rigby SE, Warren MJ. Identification, characterization, and structure/function analysis of a corrin reductase involved in adenosylcobalamin biosynthesis. J Biol Chem. 2008;283:10813–21.
Ko Y, Ashok S, Ainala SK, Sankaranarayanan M, Chun AY, Jung GY, Park S. Coenzyme B12 can be produced by engineered Escherichia coli under both anaerobic and aerobic conditions. Biotechnol J. 2014;9:1526–35.
Tee TW, Chowdhury A, Maranas CD, Shanks JV. Systems metabolic engineering design: fatty acid production as an emerging case study. Biotechnol Bioeng. 2014;111:849–57.
Choi KR, Shin JH, Cho JS, Yang D, Lee SY. Systems metabolic engineering of Escherichia coli. EcoSal Plus. 2016;7:1.
Toya Y, Shimizu H. Flux analysis and metabolomics for systematic metabolic engineering of microorganisms. Biotechnol Adv. 2013;31:818–26.
Park JM, Kim TY, Lee SY. Constraints-based genome-scale metabolic simulation for systems metabolic engineering. Biotechnol Adv. 2009;27:979–88.
Piao Y, Yamashita M, Kawaraichi N, Asegawa R, Ono H, Murooka Y. Production of vitamin B12 in genetically engineered Propionibacterium freudenreichii. J Biosci Bioeng. 2004;98:167–73.
Piao Y, Kiatpapan P, Yamashita M, Murooka Y. Effects of expression of hemA and hemB genes on production of porphyrin in Propionibacterium freudenreichii. Appl Environ Microbiol. 2004;70:7561–6.
Moore SJ, Mayer MJ, Biedendieck R, Deery E, Warren MJ. Towards a cell factory for vitamin B12 production in Bacillus megaterium: bypassing of the cobalamin riboswitch control elements. N Biotechnol. 2014;31:553–61.
Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013;8:2180–96.
Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 2013;31:170–4.
Biedendieck R, Malten M, Barg H, Bunk B, Martens JH, Deery E, Leech H, Warren MJ, Jahn D. Metabolic engineering of cobalamin (vitamin B12) production in Bacillus megaterium. Microb Biotechnol. 2010;3:24–37.
Wang L, Wilson S, Elliott T. A mutant HemA protein with positive charge close to the N terminus is stabilized against heme-regulated proteolysis in Salmonella typhimurium. J Bacteriol. 1999;181:6033–41.
Wang Z-J, Wang P, Liu Y-W, Zhang Y-M, Chu J, Huang MZ, Zhuang YP, Zhang SL. Metabolic flux analysis of the central carbon metabolism of the industrial vitamin B12 producing strain Pseudomonas denitrificans using 13C-labeled glucose. J Taiwan Inst Chem Eng. 2012;43:181–7.
Zhang Y, Liu J-Z, Huang J-S, Mao Z-W. Genome shuffling of Propionibacterium shermanii for improving vitamin B12 production and comparative proteome analysis. J Biotechnol. 2010;148:139–43.
Warner JR, Reeder PJ, Karimpour-Fard A, Woodruff LB, Gill RT. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat Biotechnol. 2010;28:856–62.
Fowler CC, Brown ED, Li Y. Using a riboswitch sensor to examine coenzyme B12 metabolism and transport in E. coli. Chem Biol. 2010;17:756–65.
Wang P, Zhang Z, Jiao Y, Liu S, Wang Y. Improved propionic acid and 5,6-dimethylbenzimidazole control strategy for vitamin B12 fermentation by Propionibacterium freudenreichii. J Biotechnol. 2015;193:123–9.
Ken-ichiro Miyano KY, Shimizu K. Improvement of vitamin B12 fermentation by reducing the inhibitory metabolites by cell recycle system and a mixed culture. Biochem Eng J. 2000;6:207–14.
Li KT, Liu DH, Li YL, Chu J, Wang YH, Zhuang YP, Zhang SL. Improved large-scale production of vitamin B12 by Pseudomonas denitrificans with betaine feeding. Bioresour Technol. 2008;99:8516–20.
Xia W, Chen W, Peng WF, Li KT. Industrial vitamin B12 production by Pseudomonas denitrificans using maltose syrup and corn steep liquor as the cost-effective fermentation substrates. Bioprocess Biosyst Eng. 2015;38:1065–73.
Authors’ contributions
HF and DZ conceived the manuscript. HF and DZ wrote the manuscript. JK wrote the “Cobalamin riboswitches” section. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Funding
Funding was provided by the National Nature Science Foundation of China (31370089), the State Key Development 973 Program for Basic Research of China (2013CB733600), the Nature Science Foundation of Tianjin City (CN) (16JCYBJC23500), and the Key Projects in the Tianjin Science & Technology Pillar Program (11ZCZDSY08400). All funding providers mentioned above had no role in the design of the study, collection, analysis, and interpretation of data nor in the writing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fang, H., Kang, J. & Zhang, D. Microbial production of vitamin B12: a review and future perspectives. Microb Cell Fact 16, 15 (2017). https://doi.org/10.1186/s12934-017-0631-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0631-y