
Abstract
Background
Bacterial spores have been proposed as vehicles to display heterologous proteins for the development of mucosal vaccines, biocatalysts, bioremediation and diagnostic tools. Two approaches have been developed to display proteins on the spore surface: a recombinant approach, based on the construction of gene fusions between DNA molecules coding for a spore surface protein (carrier) and for the heterologous protein to be displayed (passenger); and a non-recombinant approach based on spore adsorption, a spontaneous interaction between negatively charged, hydrophobic spores and purified proteins. The molecular details of spore adsorption have not been fully clarified yet.
Results
We used the monomeric Red Fluorescent Protein (mRFP) of the coral Discosoma sp. and Bacillus subtilis spores of a wild type and an isogenic mutant strain lacking the CotH protein to clarify the adsorption process. Mutant spores, characterized by a strongly altered coat, were more efficient than wild type spores in adsorbing mRFP but the interaction was less stable and mRFP could be in part released by raising the pH of the spore suspension. A collection of isogenic strains carrying GFP fused to proteins restricted in different compartments of the B. subtilis spore was used to localize adsorbed mRFP molecules. In wild type spores mRFP infiltrated through crust and outer coat, localized in the inner coat and was not surface exposed. In mutant spores mRFP was present in all surface layers, inner, outer coat and crust and was exposed on the spore surface.
Conclusions
Our results indicate that different spores can be selected for different applications. Wild type spores are preferable when a very tight protein-spore interaction is needed, for example to develop reusable biocatalysts or bioremediation systems for field applications. cotH mutant spores are instead preferable when the heterologous protein has to be displayed on the spore surface or has to be released, as could be the case in mucosal delivery systems for antigens and drugs, respectively.
Similar content being viewed by others


Background
Bacterial endospores (spores) are quiescent cells produced in response to harsh environments by Gram-positive bacteria mainly belonging to the Bacillus and Clostridium genera [1]. Spores can survive in their dormant state for long periods, resisting to a vast range of stresses such as high temperature, dehydration, absence of nutrients and presence of toxic chemicals. When the environmental conditions ameliorate, the spore germinates originating a vegetative cell able to grow and sporulate [2]. The ability of the spore to survive non-physiological conditions is, in part, due to its surface structures. In Bacillus subtilis, the model system for spore formers, the spore surface is organized in a multilayered coat and in a crust. The latter has been recently discovered and described as the outermost layer of the spore [2]. When observed by thin-section electron microscopy the spore coat appears formed by a lamellar inner coat and a more coarsely layered outer coat. The crust is only visible after a ruthenium red staining and appears as an amorphous layer surrounding the outer coat [2]. Coat and crust together are formed by at least 70 different proteins (Cot proteins) that spontaneously assemble into the multilayered structures, as recently reviewed [2]. B. subtilis spores are negatively charged [3, 4] and have a relative hydrophobicity, due in part to the glycosylation of some spore surface proteins [5].
The bacterial spore has been proposed as a platform to display heterologous proteins, with potential applications ranging from the development of mucosal vaccine to re-usable biocatalysts, diagnostic tools and bioremediation devices for field use [1, 6–8]. Various reasons support the use of the spore as a display system: (i) the remarkable and well documented resistance of the spore [2] that ensures high stability of the display system; (ii) the availability of genetic tools [9] that allows an easy manipulation; (iii) the safety record of several endospore-forming species [10, 11], that makes spores of those species ideal candidates also to deliver displayed molecules to mucosal surfaces [1, 8].
Two strategies have been so far developed to display heterologous proteins on the spore surface. A recombinant strategy based on the construction of gene fusions between the gene coding for a selected spore surface protein (carrier) and the heterologous DNA coding for the protein to be displayed was first developed to display an antigen, the C fragment of the tetanus toxin [12]. By this recombinant approach a variety of heterologous proteins have been displayed and recombinant spores proposed for several potential applications, as extensively reviewed [8]. More recently, a non-recombinant approach based on the spontaneous adsorption between purified spores and purified proteins has been also proposed [3]. Enzymes [13–15] and antigens [3, 16] have been efficiently displayed also by this approach and the system has been recently reviewed [17].
Spore adsorption is more efficient when the pH of the binding buffer is acidic (pH 4) and less efficient or totally inhibited at pH values of 7 or 10 [3, 15, 16]. A combination of electrostatic and hydrophobic interactions between spores and antigens has been suggested to drive the adsorption, which is not dependent on specific spore coat components [3, 17]. However, some mutant spores with severely altered spore surface were shown to interact more efficiently than isogenic wild type spores with the model enzyme beta-galactosidase [15]. In addition, heat-inactivated spores have been also shown to be able to efficiently display heterologous proteins [18, 19].
We used a fluorescent protein, the monomeric form of the Red Fluorescent Protein (mRFP) of the coral Discosoma sp. [20] and B. subtilis spores of a wild type and an isogenic mutant lacking the CotH protein [21] to analyze in more details the spore adsorption process. Spores lacking CotH have been previously shown to be more efficient than wild type spores in adsorbing the model enzyme beta-galactosidase [15] and to have a strongly altered coat: the inner coat is thinner than in wild type spores, the outer coat is much less electron-dense, and the two coat layers do not tightly adhere to each other [22]. These strong structural effects do not cause a strong phenotype in laboratory conditions and only a minor germination defect could be associated to the mutant spore [21]. CotH is a coat component with a regulatory role on other coat components [21], therefore spores lacking CotH also lack at least nine other proteins in the outer coat and crust [23]. Recently, it has been observed that the assembly of some CotH-dependent proteins, such as CotC, CotU, CotS and CotZ, is restored in spores lacking both CotH and CotG, another outer coat protein, and therefore that CotH counteracts CotG negative effects on spore coat assembly [24].
Results and discussion
mRFP of Discosoma sp. adsorbs to B. subtilis spores
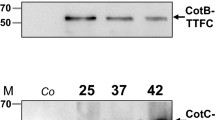
In an initial experiment 5 μg of mRFP of the coral Discosoma sp., over-expressed in E. coli and purified by affinity chromatography with Ni–Nta columns ("Methods" section), was incubated with 2.0 × 109 purified spores of the B. subtilis strain PY79 [25] or of the isogenic strain ER209, lacking CotH [21]. The adsorption reactions were performed at pH 4.0, as previously described [15]. After the adsorption reaction spores were extensively washed, spore surface proteins were extracted as previously described [26] and analyzed by western blot with monoclonal anti-polyHistidine–peroxidase antibody (Sigma), able to recognize the his tagged N terminus of mRFP. As shown in Fig. 1a, mRFP was extracted from both wild type and cotH mutant spores, indicating that it was absorbed by both types of spores, retained during the washing steps and released by the extraction treatment. mRFP adsorption was extremely stable over time and the protein was still extracted by SDS–DTT treatment after 2 weeks of storage at room temperature of the adsorbed spores (Additional file 1: Figure S1).

Western (a) and dot (b) blot of proteins extracted from wild type and cotH mutant spores after mRFP-spore adsorption. Purified recombinant mRFP is used as marker. In panel b the amount of purified recombinant mRFP (ng) and the number of spores present in each dilution are indicated. Immuno reactions were performed with anti-His primary antibody conjugated with horseradish peroxidase ("Methods'' section)
Although the western blot of Fig. 1a is not quantitative, it suggested that cotH mutant spores released more mRFP than wild type spores. To confirm this suggestion by a quantitative approach we performed a dot blot experiment using purified mRFP as a standard. After the adsorption reaction spores were collected by centrifugation, serially diluted and analyzed by dot blotting (Fig. 1b). A densitometric analysis of the dot blot (Additional file 2: Table S1) showed that when 5 µg of mRFP were used in the adsorption reaction, wild type and mutant spores were able to adsorb, respectively, 3.40 µg (68 %) and 4.43 µg (88 %) of the total protein. (P = 0.00132). Therefore, indicating that mutant spores lacking CotH are more efficient than isogenic wild type spores in adsorbing mRFP.
To analyze in more details the efficiency of adsorption, increasing amounts of mRFP (2, 5, 10 and 20 µg) were used in the reaction with the same amount of wild type or cotH mutant spores. In all cases the supernatant fraction of the adsorption reaction was serially diluted and analyzed by dot blotting with mRFP-recognizing anti-His antibody (Fig. 2a). Results of the densitometric analysis of the dot blots (Additional file 3: Table S2) are reported in Fig. 2b and indicate that with all amounts of mRFP tested, cotH mutant spores were more efficient than wild type spores in adsorbing the fluorescent protein.

(a) Dot blot of serial dilutions of unbound mRFP after adsorption reactions with wild type and cotH mutant spores. The amount of purified recombinant mRFP (ng) and the volume of supernatant loaded are indicated. Immuno reactions were performed with anti-His primary antibody conjugated with the horseradish peroxidase (“Methods” section). b Efficiency of adsorption (%) of wild type and mutant spores (black and white squares, respectively) with various amounts of mRFP. The graph is designed on the base of the averages from densitometric analysis of three independent dot blots, reported in Additional file 2: Table S1. Error bars show the standard errors of the mean from the three experiments (P = 0.0025)
Adsorbed mRFP is tightly bound to spores
Upon adsorption to spores, mRFP is not easily released even after multiple washes. Spores adsorbed with mRFP were washed three times with 100 μl PBS buffer at pH 3.0 or pH 7.0 or extracted with 1 M NaCl, 0.1 % Triton X-100 [3]. With wild type spores no mRFP was released by any of these treatments, while with cotH mutant spores some mRFP was released by the washes at pH 7.0 and by the NaCl−Triton extraction (Fig. 3a). To evaluate the kinetic of mRFP-release adsorbed mutant spores were washed two times, resuspended in PBS pH 7.0 or 1 M NaCl, 0.1 % Triton X-100 for 5, 10, 15 or 30 min and the supernatant fractions analyzed by dot blot (not shown). A densitometric analysis of the dot blot (Additional file 4: Table S3) was performed and Fig. 3b reports the percentage of mRFP released by the two treatments at the different time points. The pH 7.0 buffer extracted about 5 % of the adsorbed mRFP within the first 5 min of incubation and did not extract more protein over time. The NaCl–Triton solution extracted about 3 % of mRFP in the first 5 min and the amount of extracted mRFP increased over time in an almost linear way reaching over 9 % of mRFP released after 30 min of treatment (Fig. 3b).

(a) Dot blot of released mRFP after three washes (W1, W2 and W3) with PBS buffer at pH 3 or pH 7 or after extraction with 1 M NaCl, 0.1 % Triton X-100 (Ext) (“Methods” section). Purified recombinant mRFP (mRFP) and unbound mRFP (u-mRFP) after the adsorption reaction were used as markers. b Kinetic of mRFP release based on the densitometric analysis (Additional file 4: Table S3) of mRFP released upon incubation either in PBS buffer pH 7.0 (triangles) or 1 M NaCl, 0.1 % Triton X-100 (squares). c Western blot of mRFP extracted by a SDS–DTT or after consecutive SDS–DTT and urea treatments of wild type and cotH mutant spores. Immuno reactions were performed with anti-His primary antibody anti-His primary antibody conjugated with the horseradish peroxidase (“Methods” section)
When spores were extracted with an SDS–DDT treatment, the standard protocol for extract spore coat proteins [26], mRFP was released by both wild type and mutant spores (Fig. 3c). The release of some mRFP after washes at pH 7.0 or 1 M NaCl, 0.1 % Triton X-100 treatment (Fig. 3a) and the high amount of mRFP released after SDS–DTT extraction by mutant spores may reflect the higher amount of mRFP adsorbed by mutant than wild type spores (Fig. 2). However, if spores previously extracted with SDS–DTT were re-extracted with Urea, a procedure used to completely remove the spore coat [27], additional mRFP molecules were released but, in this case, in higher amount from wild type than from mutant spores (Fig. 3c). Taken together the experiments of Figs. 2 and 3 suggest that wild type spores adsorb mRFP less efficiently but more tightly than cotH mutant spores.
Localization of adsorbed mRFP on wild type and mutant spores
To assess whether spore-adsorbed mRFP molecules retained their fluorescence properties and investigate their distribution around the spore we performed a fluorescence microscopy analysis. With both wild type and mutant spores red fluorescent signals were observed all around the spore (Fig. 4), and in agreement with results of Figs. 2 and 3, the fluorescence signal appeared stronger with mutant than with wild type spores (Fig. 4). In all cases the fluorescent signal was stronger at the spore poles, indicating that adsorbed mRFP molecules were not evenly distributed around the spore and accumulated at the poles (Fig. 4). We used Image J software (v1.48, NIH) to perform a quantitative fluorescence image analysis and the corrected spore fluorescence was calculated as described in the “Methods” section. The analysis of 80 spores of each strain indicated an average fluorescence intensity, in arbitrary units, of 7816 ± 2712 and of 11541 ± 2573 for wild type spores and mutant spores, respectively (Fig. 5, P < 0.0001), confirming that cotH mutant spores adsorb more mRFP than wild type spores.

Fluorescence of mRFP upon adsorption to wild type and mutant spores. For each strain the same microscopy field was observed by phase contrast and fluorescence microscopy. Merge panels are reported. The exposure time was of 500 ms for both strains. Scale bar 1 μm

Fluorescence intensity profiles (Fl. Int.; scale in arbitrary units) of wild type and mutant spores adsorbed with mRFP. The profiles were generated from fluorescence microscopy images using the 3D Surface plotter function of Image J (http://imagej.nih.gov/ij/). a Representative fluorescence intensity profiles of a wild type (left) and cotH mutant spore (right). The fluorescence intensity is reported in arbitrary units. b Box plots displaying the total corrected cellular fluorescence (TCCF) for 80 different spores of each strain. Limits of each box represent the first and the third quartile (25 and 75 %) and the values outside the boxes represent the maximum and the minimum values. The line dividing the box indicates the median value for each strain. P value is less than 0.0001
Adsorbed mRFP is not exposed on the surface of wild type spores
To assess whether adsorbed mRFP molecules were exposed on the spore surface we followed an immuno-fluorescence microscopy approach. Wild type and mutant spores adsorbed with mRFP were reacted with monoclonal mRFP-recognizing anti-His antibody, then with fluorescent anti-mouse secondary antibody (Santa Cruz Biotechnology, Inc.) and observed under the fluorescence microscope. While mRFP molecules adsorbed to mutant spores were accessible to the specific antibody all around the spore surface, mRFP molecules adsorbed to wild type spores were only poorly accessible to the antibody and only in specific spots mainly at the spore poles (Fig. 6). A fluorescent signal was observed with wild type spores only with an exposure time fivefold longer that used with mutant spores (Fig. 6). Results of Fig. 6, then indicate that mRFP molecules are mostly not accessible to the antibody upon adsorption on wild type spores. Since mRFP is abundantly present at the poles of wild type spores (Fig. 4) but it is only accessible to anti-mRFP antibody in few specific spots at the spore poles (Fig. 6) we hypothesize that either mRFP molecules bind wild type spores in an orientation that precludes the interaction with the antibody or that mRFP molecules infiltrate inside the spore coat layers of wild type spores.

Immuno-fluorescence microscopy of wild type and mutant spores adsorbed with mRFP. Immuno reactions were performed with anti-His primary antibody and fluorescent anti-mouse secondary antibody conjugated with FITC (“Methods” section). For each strain the same microscopy field was observed by phase contrast and fluorescence microscopy. Merge panels are reported. The exposure time is indicated as ms. The numbers in the panels are the percentage of spores found with a similar mRFP-localization pattern in three different microscopy fields. Scale bar 1 μm
Adsorbed mRFP infiltrates in the inner coat of wild type spores
In order to verify whether mRFP molecules infiltrate inside the spore we used a fluorescence microscopy approach (“Methods” section). In particular we followed the red fluorescence of mRFP and the green fluorescence of GFP fused to B. subtilis proteins known to localize in the spore core (SpoIIQ [28]), cortex (YhcN [29, 30]), inner coat (CotS [31]), outer coat (CotC [30, 32]) or crust (CotZ [30]). Spores purified from isogenic strains carrying the spoIIG::gfp, yhcN::gfp, cotS::gfp, cotC::gfp or cotZ::gfp gene fusion were used to adsorb mRFP and observed by fluorescence microscopy (Fig. 7A). In order to determine the relative position of red and green signals quantitatively, more than 20 free spores for each strain were used to measure the intensity of GFP and RFP fluorescence along the spore axis by using image J software (“Methods” section). Averages of red and green fluorescence intensities of the various strains were plotted in Fig. 7B. With spores carrying the SpoIIQ–GFP (core) or the YhcN–GFP (cortex) fusion the green signal was internal with respect to the red one indicating that mRFP molecules were localized outside of the spore core (SpoIIQ) and of the cortex (YhcN). With spores carrying the CotS–GFP (inner coat) fusion the intensity peaks of green and red signals were coincident, indicating a co-localization of the two fluorescence signals. With spores carrying the CotC–GFP (outer coat) or the CotZ–GFP (crust) fusion, the intensity peaks of the green signals were slightly external to the red ones, indicating that in both cases mRFP molecules were inside the outer coat (CotC) and of the crust (CotZ) (Fig. 7). These results then indicate that mRFP molecules cross crust and outer coat layers and localize at the level of the inner coat of wild type spores, explaining why mRFP is not exposed on the surface of the spore (Fig. 6).

Fluorescence localization of coat protein-GFP fusions and adsorbed-mRFP (A) and plots of fluorescence intensities along the long axis of the spore (B). Spores of otherwise wild type B. subtilis strains carrying GFP fused to core, cortex, inner coat, outer coat or crust proteins were adsorbed with mRFP and observed by fluorescence microscopy. Phase-contrast images (a), GFP fluorescence images (b), mRFP fluorescence images (c) merged images (d) are shown
Adsorbed mRFP localizes in all surface layers of cotH mutant spores
The same approach was used to localize mRFP molecules inside mutant spores. Spores purified from isogenic strains carrying the cotH mutation and the spoIIG::gfp, yhcN::gfp, cotS::gfp, cotC::gfp or cotZ::gfp gene fusion were used to adsorb mRFP and observed by fluorescence microscopy (Fig. 8A). Since CotS (inner coat marker) and CotC (outer coat marker) fail to assemble within the coat of a cotH mutant [23, 33] but assemble normally in a strain lacking both CotH and CotG [24], we used a double mutant strain carrying null mutations in cotH and cotG genes for those two GFP fusions. As above, more than 20 free spores for each strain were used to measure the intensity of GFP and RFP fluorescence signals along the spore axis by using image J software (“Methods” section). Averages of red and green fluorescence intensities of the various strains were plotted in Fig. 8B. With spores carrying the SpoIIQ–GFP (core) or the YhcN–GFP (cortex) fusion the green signal was internal with respect to the red one indicating that mRFP molecules were localized outside of the spore core (SpoIIQ) and of the cortex (YhcN). With spores carrying the CotS–GFP (inner coat) and CotC–GFP (outer coat) fusion the red peaks were slightly outside the green ones and with the CotZ–GFP (crust) fusion the intensity peaks of green and red signals were coincident (Fig. 8). However, with mutant spores the red peaks did not appear sharp but rather broad suggesting that the red fluorescent molecules were not concentrated in a specific point but were diffused in a large space. Taken together these observations suggest a localization of mRFP molecules in all surface layers of cotH mutant spores that explains why mRFP is exposed on the surface of the spore (Fig. 6).

Fluorescence localization of coat protein-GFP fusions and adsorbed-RFP (A) and plots of fluorescence intensities along the long axis of the spore (B). Spores of cotH mutant strains carrying GFP fused to core, cortex or crust proteins and of cotH cotG mutant strains carrying GFP fused to inner and outer coat were adsorbed with mRFP and analyzed by fluorescence microscopy. Phase-contrast images (a), GFP fluorescence images (b), RFP fluorescence images (c) and merged images (d) are shown
The experiments of Figs. 7 and 8 were performed using standard adsorption conditions: 5 μg of purified mRFP were incubated with 2.0 × 109 purified spores for 1 h. To evaluate whether the amount of mRFP and/or the incubation time had an effect on mRFP localization we incubated 2.0 × 109 wild type spores either with 50 μg of purified mRFP for 1 h or with 5 μg of mRFP for 4 h. In both cases mRFP localized in the inner coat of wild type spores as under standard conditions (Additional file 5: Figure S2), indicating that neither the amount of mRFP or the incubation time affected mRFP localization.
Conclusions
Wild type spores of B. subtilis adsorb and tightly bind mRFP that is not displayed on the spore surface. We propose that it infiltrates through crust and outer coat layers, localizes in the inner coat and appears more abundantly concentrated at the spore poles (Fig. 9a). The observation that mRFP crosses crust and outer coat indicates these structures as permeable to a 27 kDa protein. Permeability of the spore surface is not totally surprising since germinants (small molecules with molecular masses typically <200 Da) present in the environment have to cross the external layers of the spore to reach their receptors. In addition, studies conducted on B. megaterium suggested that the spore surface is permeable to the passage of molecules with masses somewhere between 2 and 40 kDa [34, 35].

Cartoon model of mRFP localization in wild type (a) and cotH mutant (b) spores. Wild type and mutant spores are over imposed with red dots representing mRFP molecules
Mutant spores lacking CotH are strongly defective: (i) their outer coat is diffuse, lacks the characteristic multilayered pattern, electron density, and a defined outer edge; (ii) the typical lamellar structure of the inner coat, in which three to five lamellae can usually be recognized, is reduced to one or two lamellae; (iii) the inner and outer coats are not tightly associated [22]; at least nine spore coat proteins are not present [23]. These mutant spores are more efficient than wild type spores in adsorbing mRFP that, although more abundant at the spore poles is present all around the spore and exposed on its surface. However, the interaction between mRFP and cotH spores is less stable than with wild type spores and mRFP is partially released by washes at pH 7.0 (Fig. 3). We propose that mRFP molecules adsorb better to cotH mutant than to wild type spores because they not only localize in the inner coat but also in the outer coat and in the crust (Fig. 9b).
The observation that neither the amount of heterologous protein or the time of incubation affect mRFP localization in wild type spores (Additional file 1: Figure S1) argue that in those spores crust and outer coat are compact structures that allow mRFP transit but not accumulation. In cotH spores the outer coat is rather diffuse [22] and allows mRFP to localize and accumulate.
These findings shed light on the mechanism of spore adsorption and indicate that different spores, wild type or mutant, can potentially have different applications. As a carrier of enzymes to be re-used several times a surface display of the enzyme may be not strictly required while a tight adhesion to the carrier could desirable making wild type spores preferable over the mutant spores. On the contrary, as a drug/vaccine delivery vehicle a surface display could be essential and the release of the drug/antigen in some cases an useful property, making mutant spores preferable over the wild type.
Methods
Bacterial strains and transformation
Bacillus subtilis strain PY79 strain [25] was used as a wild type. Strain ER220 (cotH null mutant [21] and all other mutant strains of B. subtilis used in this study were isogenic derivatives of PY79. B. subtilis strains used in this study are listed in Additional file 6: Table S4. Isolation of plasmid DNA, restriction digestion, ligation of DNA and transformation of E. coli competent cells were carried out by standard methods [36].
Construction of gfp fusions
The coding sequence of yhcN (564 bp) and the promotor region of spoIIQ (500 bp) were PCR amplified using B. subtilis chromosomal DNA as a template and synthetic oligonucleotide pairs spoIIQ-F/spoIIQ-R and yhcN-F/yhcN-R (Additional file 7: Table S5) respectively to prime the reactions. Purified DNA fragments were cloned in frame to the 5′ end of the gfp gene [37]. Plasmids were used to transform competent cells of strain PY79 yielding strain RH2466 (P spoIIQ ::gfp) and RH282 (yhcN::gfp). Chromosomal DNA of the two strains and of AZ573 (cotZ::gfp) was used to transform competent cells of isogenic strains RH220 (cotH::spc) (Additional file 6: Table S4), yielding strains RH285 (P spoIIQ ::gfp cotH::spc) and RH284 (yhcN::gfp cotH::spc) and RH278 (cotZ::gfp cotH::spc). Chromosomal DNA of strains DS127 (cotC::gfp) was used to transform competent cells of isogenic strains AZ603 (ΔcotG ΔcotH::neo), yielding strains AZ636 (cotC::gfp ΔcotG ΔcotH::neo).
Purification of spores and RFP
Sporulation of wild type and recombinant strains was induced by the exhaustion method. After 30 h of growth in Difco Sporulation medium at 37 °C with vigorous shaking [36] spores were collected, washed three times with distilled water and purified on a step gradient of 20–50 % of Gastrografin (Bayer) as described before [38, 39]. The number of purified spores obtained was measured by direct counting with a Bürker chamber under an optical microscope (Olympus BH-2 with 40× lens).
Competent cells of E. coli strain BL21(DE3) (Invitrogen) was transformed with the expression vector pRSET-A carrying an in-frame fusion of the 5′ end of the rfp coding region to six histidine codons under the transcriptional control of a T7 promoter (generous gift of Dr. A. Storlazzi). The new strain RH161 was grown for 18 h at 37 °C in 100 ml of autoinduction medium to express the heterologous protein [40]. The six-His-tagged RFP protein was purified under native conditions using a His-Trap column as recommended by the manufacturer (GE Healthcare Life Science). Purified protein was desalted using a PD10 column (GE Healthcare Life Science) to remove high NaCl and imidazole concentrations and mRFP purity analyzed by SDS-PAGE.
Adsorption reaction
Unless otherwise specified 5 μg of purified recombinant mRFP were added to a suspension 2 × 109 spores in 0.15 M PBS pH 4.0 at 25 °C in a final volume of 200 μl. After 1 h of incubation, the binding mixture was centrifuged (10 min at 13,000g) to fractionate pellet and supernatant. The pellet was resuspended in 0.15 M PBS at pH 4.0 to a final concentration of 2 × 105 spores/μl and stored at 4 °C. The supernatant was stored at −20 °C.
Western and dot-blot analysis
2 × 108 mRFP-adsorbed spores were resuspended in 20 μl of spore coat extraction buffer [26], incubated at 68 °C for 1 h to solubilize spore coat proteins and loaded onto a 12 % SDS-PAGE gel. The proteins were then electro transferred to nitrocellulose filters (Amersham Pharmacia Biotech) and used for Western blot analysis as previously reported [39] using monoclonal mRFP-recognizing anti-His antibody (Sigma). A quantitative determination of the amount of mRFP was obtained by dot blot experiments analyzing serial dilutions of purified RFP, binding assay supernatant and RFP-adsorbed spores. Filters were then visualized by the ECL-prime (Amersham Pharmacia Biotech) method and subjected to densitometric analysis by Quantity One 1-D Analysis Software (Bio-Rad).
Fluorescence and immunofluorescence microscopy
Post adsorption spores were resuspended in 50 μl of 1× PBS and 5 μl of the suspension placed on microscope slides and covered with a coverslip previously treated for 30 s with poly-l-lysine (Sigma). Immunofluorescence was performed as described by Isticato et al. [16], with the following modifications: 2.0 × 106 RFP-adsorbed spores of wild type and cotH mutant spores were pretreated with 1 % bovine serum albumin (BSA)—1× PBS, pH 4.0 for 30 min prior to 2 h-incubation a 4 °C with the monoclonal anti-polyHistidine antibodies (mouse) (Sigma) diluted 1:20 in 1× PBS−1 % BSA. As a control of the specificity of this technique, non-adsorbed spores were directly treated with anti-His antibodies. After three washes, the samples were incubated with a 64-fold diluted anti-mouse secondary antibody conjugated with Fluorescein isothiocyanate, FITC (Santa Cruz Biotechnology, Inc.) and washed four times with PBS. Washed samples were resuspended in 20 μl of 1× PBS and 10 μl were analyzed. All samples were observed with an Olympus BX51 fluorescence microscope fitted with a 100× objective UPlanF1; U-MNG or U-MWIBBP cube-filters were used to detect the red and green fluorescence emission respectively. Images were captured using an Olympus DP70 digital camera equipped with Olympus U-CA Magnification Changer and processed with Image Analysis Software (Olympus) for minor adjustments of brightness, contrast and color balance and for creation of merged images of GFP and RFP [41]. Exposure times were in the range between 200 and 2000 ms for image capture of GFP fusions and for adsorbed RFP. Fluorescence intensities and the distance between two fluorescent peaks were measured using unadjusted merged images with Image J processing software (version 1.48, NIH) as previously describe by Imamura and collaborators [42]. One pixel corresponds to 1.18 nm in our detection system. Image J was also used to draw an outline around 80 spores for each strain, prior to area, integrated density and the mean fluorescence measurements being recorded, together with several adjacent background readings. The total corrected cellular fluorescence (TCCF) was calculated by subtracting (area of selected cell × mean fluorescence of background readings) from integrated density values. Minimum, maximum and mean value of TCCF were displayed as box-plots with 5–95 % confidence intervals [42]. Fluorescence intensity profiles were generated from the microscopy images using the 3D Surface plotter function of Image J as reported by Serrano and collaborators [43].
Statistical analysis
Results from dot blot and fluorescence microscopy analysis are the averages from three independent experiments. Statistical significance was determined by the Student t test, and the significance level was set at P < 0.01.
Abbreviations
- mRFP:
-
monomeric Red Fluorescent Protein
- RT:
-
room temperature
- GFP:
-
green fluorescent protein
- SDS-DDT:
-
sodium dodecyl sulfate–dithiothreitol
- NIH:
-
National Institute of Health
- PBS:
-
phosphate saline buffer
- TCCF:
-
total corrected cellular fluorescence
References
Cutting SM, Hong HA, Baccigalupi L, Ricca E. Oral vaccine delivery by recombinant spore probiotics. Int Rev Immunol. 2009;28:487–505.
McKenney PT, Driks A, Eichemberger P. The Bacillus subtilis endospore: assembly and functions of the multilayered coat. Nat Rev Microbiol. 2012;11:33–44.
Huang JM, Hong HA, Van Tong H, Hoang TH, Brisson A, Cutting SM. Mucosal delivery of antigens using adsorption to bacterial spores. Vaccine. 2010;28:1021–30.
Pesce G, Rusciano G, Sirec T, Isticato R, Sasso A, Ricca E. Surface charge and hydrodynamic coefficient measurements of Bacillus subtilis spore by optical tweezers. Colloids Surf B. 2014;116C:568–75.
Cangiano G, Sirec T, Panarella C, Isticato R, Baccigalupi L, De Felice M, Ricca E. The sps gene products affect germination, hydrophobicity and protein adsorption of Bacillus subtilis spores. Appl Environ Microbiol. 2014;80:7293–302.
Knecht LD, Pasini P, Daunert S. Bacterial spores as platforms for bioanalytical and biomedical applications. Anal Bioanal Chem. 2011;400:977–89.
Hinc K, Ghandili S, Karbalaee G, Shali A, Noghabi K, Ricca E, Ahmadian G. Efficient binding of nickel ions to recombinant Bacillus subtilis spores. Res Microbiol. 2010;161:757–64.
Isticato R, Ricca E. Spore surface display. Microbiol Spectr. 2014. doi:10.1128/microbiolspecTBS-0011-2012.
Harwood C, Cutting S. Molecular biological methods for Bacillus. Chichester: Wiley; 1990.
Cutting SM. Bacillus probiotics. Food Microbiol. 2011;28:214–20.
Baccigalupi L, Ricca E, Ghelardi E. Non-LAB probiotics: spore formers. In: Probiotics and prebiotics: current research and future trends, Norfolk: Caister Academic Press; 2015. p. 93–103.
Isticato R, Cangiano G, Tran HT, Ciabattini A, Medaglini D, Oggioni MR, De Felice M, Pozzi G, Ricca E. Surface display of recombinant proteins on Bacillus subtilis spores. J Bacteriol. 2001;183(21):6294–301.
Yim S-K, Jung H-C, Yun C-H, Pan J-G. Functional expression in Bacillus subtilis of mammalian NADPH-cytochrome P450 oxidoreductase and its spore-display. Protein Expr Purif. 2009;63:5–11.
Cho EA, Kim EJ, Pan JG. Adsorption immobilization of Escherichia coli phytase on probiotic Bacillus polyfermenticus spores. Enzyme Microb Technol. 2011;49:66–71.
Sirec T, Strazzulli A, Isticato R, De Felice M, Moracci M, Ricca E. Adsorption of beta-galactosidase of Alicyclobacillus acidocaldarius on wild type and mutants spores of Bacillus subtilis. Microb Cell Fact. 2012;11:100.
Isticato R, Sirec T, Treppiccione L, Maurano F, De Felice M, Rossi M, Ricca E. Non-recombinant display of the B subunit of the heat labile toxin of Escherichia coli on wild type and mutant spores of Bacillus subtilis. Microb Cell Fact. 2013;12:98.
Ricca E, Baccigalupi L, Cangiano G, De Felice M, Isticato R. Mucosal vaccine delivery by non-recombinant spores of Bacillus subtilis. Microb Cell Fact. 2014;13:115.
Reljic R, Sibley L, Huang JM, Pepponi I, Hoppe A, Hong HA, Cutting SM. Mucosal vaccination against tuberculosis using inert bioparticles. Infect Immun. 2013;81:4071–80.
Nguyen VA, Huynh HA, Hoang TY, Ninh NT, Pham AT, Nguyen HA, Phan TN, Cutting SM. Killed Bacillus subtilis spores expressing streptavidin: a novel carrier of drugs to target cancer cells. J Drug Targ. 2013;21:528–41.
Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci. 2002;99:7877–82.
Naclerio G, Baccigalupi L, Zilhao R, De Felice M, Ricca E. Bacillus subtilis spore coat assembly requires cotH gene expression. J Bacteriol. 1996;178:4375–80.
Zilhao R, Naclerio G, Baccigalupi L, Henriques A, Moran C, Ricca E. Assembly requirements and role of CotH during spore coat formation in Bacillus subtilis. J Bacteriol. 1999;181:2631–3.
Kim H, Hahn M, Grabowski P, McPherson D, Otte MM, Wang R, Ferguson CC, Eichenberger P, Driks A. The Bacillus subtilis spore coat protein interaction network. Mol Microbiol. 2006;59:487–502.
Saggese A, Scamardella V, Sirec T, Cangiano G, Isticato R, Pane F, Amoresano A, Ricca E, Baccigalupi L. Antagonistic role of CotG and CotH on spore germination and coat formation in Bacillus subtilis. PLoS ONE. 2014;9(8):e104900.
Youngman P, Perkins JB, Losick R. A novel method for the rapid cloning in Escherichia coli of Bacillus subtilis chromosomal DNA adjacent to Tn917 insertion. Mol Gen Genet. 1984;195:424–33.
Cutting S, Vander Horn PB. Genetic analysis. In: Harwood C, Cutting S, editors. Molecular biological methods for Bacillus. Chichester: Wiley; 1990. p. 27–74.
Tennen R, Setlow B, Davis KL, Loshon CA, Setlow P. Mechanisms of killing of spores of Bacillus subtilis by iodine, glutaraldehyde and nitrous acid. J Appl Microbiol. 2000;89:330–8.
Rubio A, Pogliano K. Septal localization of forespore membrane proteins during engulfment in Bacillus subtilis. EMBO J. 2004;23:1636–46.
Bagyan I, Noback M, Bron S, Paidhungat M, Setlow P. Characterization of yhcN, a new fore spore-specific gene of Bacillus subtilis. Gene. 1998;212:179–88.
Imamura D, Kuwana R, Takamatsu H, Watabe K. Localization of proteins to different layers and regions of Bacillus subtilis spore coats. J Bacteriol. 2010;192:518–24.
Takamatsu H, Chikahiro Y, Kodama T, Koide H, Kozuka S, Tochikubo K, Watabe K. A spore coat protein, CotS, of Bacillus subtilis is synthesized under the regulation of Sigma K and GerE during development and is located in the inner coat layer of spores. J Bacteriol. 1998;180:2968–74.
Isticato R, Pelosi A, De Felice M, Ricca E. CotE binds to CotC and CotU and mediates their interaction during spore coat formation in Bacillus subtilis. J Bacteriol. 2010;192:949–54.
Isticato R, Esposito G, Zilhão R, Nolasco S, Cangiano G, De Felice M, Henriques AO, Ricca E. Assembly of Multiple CotC Forms into the Bacillus subtilis Spore Coat. J Bacteriol. 2004;186:1129–35.
Koshikawa T, Beaman TC, Pankratz HS, Nakashio S, Corner TR, Gerhardt P. Resistance germination and permeability correlates of Bacillus megaterium spores successively divested of integument layers. J Bacteriol. 1984;159:624–32.
Nishihara T, Takubo Y, Kawamata E, Koshikawa T, Ogaki J, Kondo M. Role of outer coat in resistance of Bacillus megaterium spore. J Biochem. 1989;106:270–3.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, laboratory manual. 2nd ed. Cold Spring Harbor Laboratory Press; 1989.
Isticato R, Mase DS, Mauriello EMF, De Felice M, Ricca E. Amino terminal fusion of heterologous proteins to CotC increases display efficiencies in the Bacillus subtilis spore system. BioTechniques. 2007;42:151–6.
Nicholson WL, Setlow P. Sporulation, germination and out-growth. In: Harwood C, Cutting S, editors. Molecular biological methods for Bacillus. Chichester: Wiley; 1990. p. 391–450.
Rusciano G, Zito G, Isticato R, Sirec T, Ricca E, Bailo E, Sasso A. Nanoscale chemical imaging of Bacillus subtilis spores by combining tip-enhanced Raman scattering and advanced statistical tools. ACS Nano. 2014;8:12300–9.
Isticato R, Pelosi A, Zilhão R, Baccigalupi L, Henriques AO, De Felice M, Ricca E. CotC–CotU heterodimerization during assembly of the Bacillus subtilis spore coat. J Bacteriol. 2008;190:1267–75.
Manzo N, Di Luccia B, Isticato R, D’Apuzzo E, De Felice M, Ricca E. Pigmentation and sporulation are alternative cell fates in Bacillus pumilus SF214. PLoS ONE. 2013;8:e62093.
McCloy R, Rogers S, Caldon E, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle. 2014;13:1400–12.
Serrano M, Crawshaw AD, Dembek M, Monteiro JM, Pereira FC, Pinho MG, Fairweather NF, Salgado PS, Henriques AO. The SpoIIQ–SpoIIIAH complex of Clostridium difficile controls forespore engulfment and late stages of gene expression and spore morphogenesis. Mol Microbiol. 2016;100:204–28.
Authors’ contributions
GD and ML performed most of the experiments; TS performed immunofluorescence experiments; ER contributed discussions and suggestions during the work and contributed to write the manuscript; RI led the work and contributed to write the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data supporting the conclusions of this article are included within the manuscript and in the additional files. The content of the manuscript has not been published, or submitted for publication elsewhere.
Ethics approval
The manuscript does not report data collected from humans or animals.
Funding
This work was supported by a grant (STAR PROGRAMME 2014) from the Federico II University of Naples, Italy to RI.
Author information
Authors and Affiliations
Corresponding author
Additional information
Giuliana Donadio and Mariamichela Lanzilli equally contributed to this work
Additional files
12934_2016_551_MOESM1_ESM.pdf
Additional file 1. Stability of spore adsorption over time. Purified suspensions of wt spores were mixed with purified recombinant mRFP (5μg) for 1 h at RT. Spores were centrifuged, washed two times, resuspended in PBS, pH3 and stored at RT. After 1, 2, 7 and 14 days, the adsorption mixtures were fractionated by centrifugation and the pellet fractions used for protein extraction by SDS-DTT treatment. Extracted proteins were fractionated on SDS-PAGE and analyzed by western blot. Immuno-reactions were performed with anti-His antibody conjugated with horseradish peroxidase.
12934_2016_551_MOESM2_ESM.pdf
Additional file 2. Densitometric analysis of dot blot experiments reported in Fig 1 performed with the pellets of the adsorption reaction with wild type and cotH mutant spores.
12934_2016_551_MOESM3_ESM.pdf
Additional file 3. Densitometric analysis of dot blot experiments reported in Fig 2 performed with the supernatants of the adsorption reaction performed with different amounts of mRFP and with wild type and cotH mutant spores.
12934_2016_551_MOESM4_ESM.pdf
Additional file 4. Densitometric analysis of dot blot experiments with the supernatants of adsorption reaction and with washes after various time of incubation with 1× PBS pH7.0 or 1M NaCl–0.1% Triton 100.
12934_2016_551_MOESM5_ESM.pdf
Additional file 5. Fluorescence microscopy analysis of spores of otherwise wild type B. subtilis strains carrying GFP fused to inner coat, outer coat or crust proteins adsorbed with 50 μg of purified mRFP for 1 h (A) or with 5 μg of purified mRFP for 4 h (B). For each strain the same microscopy fields was observed by phase contrast and fluorescence microscopy (red and green). Merge and overlay (red on green or viceversa) are also shown.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Donadio, G., Lanzilli, M., Sirec, T. et al. Localization of a red fluorescence protein adsorbed on wild type and mutant spores of Bacillus subtilis . Microb Cell Fact 15, 153 (2016). https://doi.org/10.1186/s12934-016-0551-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0551-2