
Abstract
Background
Specific coupling of de novo designed recombinant protein polymers for the construction of precisely structured nanomaterials is of interest for applications in biomedicine, pharmaceutics and diagnostics. An attractive coupling strategy is to incorporate specifically interacting peptides into the genetic design of the protein polymers. An example of such interaction is the binding of particular proline-rich ligands by so-called WW-domains. In this study, we investigated whether these domains can be produced in the yeast Pichia pastoris as part of otherwise non-interacting protein polymers, and whether they bring about polymer coupling upon mixing.
Results
We constructed two variants of a highly hydrophilic protein-based polymer that differ only in their C-terminal extensions. One carries a C-terminal WW domain, and the other a C-terminal proline-rich ligand (PPxY). Both polymers were produced in P. pastoris with a purified protein yield of more than 2 g L−1 of cell-free broth. The proline-rich module was found to be O-glycosylated, and uncommonly a large portion of the attached oligosaccharides was phosphorylated. Glycosylation was overcome by introducing a Ser → Ala mutation in the PPxY peptide. Tryptophan fluorescence monitored during titration of the polymer containing the WW domain with either the glycosylated or nonglycosylated PPxY-containing polymer revealed binding. The complementary polymers associated with a Kd of ~3 µM, regardless of glycosylation state of the PPxY domain. Binding was confirmed by isothermal titration calorimetry, with a Kd of ~9 µM.
Conclusions
This article presents a blueprint for the production in P. pastoris of protein polymers that can be coupled using the noncovalent interaction between WW domains and proline-rich ligands. The availability of this highly specific coupling tool will hereafter allow us to construct various supramolecular structures and biomaterials.
Similar content being viewed by others

Background
Protein-based block copolymers (or protein polymers, for short) are de novo designed polypeptides that consist of different blocks, where each of the blocks adopts a specific conformation and fulfills a specific function [1]. Self-assembling protein polymers are being intensively explored for use as biomaterials for drug encapsulation, controlled drug release, tissue engineering, tissue augmentation, and biosensors [2–8]. Protein polymers are produced via recombinant DNA technology, and thus, aside from possible problems with the biological production, in principle have a defined sequence, mass, and chemical composition. They are biodegradable, often biocompatible, and easy to functionalize by inclusion of bioactive sequences in the genetic design [1, 7].
In early work, the sequences of protein polymers were typically inspired by naturally occurring self-assembling structural proteins such as silk, elastin, collagen, and resilin, with inherent attractive properties as soft materials [9, 10]. The repertoire of useful motifs for self-assembly has been expanded over the years with sequences designed or modified using molecular modelling. Several self-assembling modules can be combined into a multifunctional polypeptide block copolymer that can spontaneously self-organize via non-covalent interactions into nanostructures such as micelles, fibrils, or hydrogels [11–13]. However, peptide blocks that can facilitate the assembly of supramolecular structures with particularly high precision are still highly sought after in biomaterial science [14–16].
Our group has successfully produced several protein polymers at high yield using the yeast Pichia pastoris [17–23]. Many of these are block copolymers that self-organize into stimulus-responsive supramolecular structures. We currently aim to expand our library of functional protein modules with blocks that allow highly precise heterodimerization, in an effort to gain more control over the self-assembly of supramolecular structures. A pair of block copolymers fitted with complementary interacting modules could then self-assemble into higher order structures upon mere mixing of the two [24]. Besides providing crosslinks in self-assembled structures, heterodimerizing modules can also be used for incorporation of functionalities such as growth factors, antimicrobial peptides, and cell-adhesive peptides [25].
The so-called WW domain is found in various natural proteins and binds particular proline-rich peptides with a high degree of specificity [26]. As its name suggests, the amino acid sequence of the WW domain contains two highly conserved tryptophans. The domain consists of a slightly bent three-stranded antiparallel β-sheet, the concave side of which forms a binding pocket for the proline-rich ligand [27]. Wong Po Foo et al. successfully used the interaction between two different WW domains (CC43 and Nedd4.3) and a so-called group I proline-rich peptide (PPxY) to generate two-component hydrogels [28]. Here, we investigate the separate incorporation of this same PPxY peptide derived from p53-binding protein-2 [29] and another WW domain (WWP1-1; one of three WW domains present in the human ubiquitin ligase homolog WWP1 [29]) at the C-terminus of the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) protein polymer previously developed by us [18]. The \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) polymer (formerly referred to as ‘P4’) consists of four identical copies of a 99 amino acid long, highly hydrophilic random coil polypeptide [18]. We report the high-yield secretory production of these polymers in Pichia pastoris and their characterization. Undesired O-glycosylation of the PPxY peptide was overcome, and the polymers interacted as intended. We have thus expanded our toolkit towards the creation of well-defined supramolecular topologies for new biomaterials.
Results and discussion
Protein production and purification
The WW and PPxY domains are referred to hereafter as D WW and D PPxY, respectively (D for Dimerization). For amino acid sequences see Table 1. The domains were cloned at the 3’ end of the gene encoding the previously reported \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) protein polymer [18]. The encoded proteins \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) were produced in secretory fashion using genetically modified P. pastoris, grown in methanol fed-batch mode. Proteins were purified from the cell-free broth by differential ammonium sulfate precipitation, and subsequently dialyzed and lyophilized. Previous studies showed that ammonium sulfate precipitation of protein polymers containing the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) block typically results in a purity of ~99 % at the protein level [19, 30]. The gravimetrically determined yields, expressed in g per L of cell-free broth, were 2.2 g L−1 for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\), and 2.3 g L−1 for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\). Although several reports describe the production of proteins with WW domains and proline-rich ligands in Escherichia coli, yields have not been reported [28, 31, 32]. To our knowledge, production of WW and PPxY domains in P. pastoris has not been reported before. The non-optimized yields are in the same g L−1 range as for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) [18], showing that the D WW and D PPxY modules are not a significant bottle-neck. This offers good prospects towards their further use in the construction of complex supramolecular architectures and biomaterials.
Protein characterization
\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) were characterized by SDS–PAGE (Fig. 1). Based on the well-established aberrant migration behavior of the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) block in SDS-PAGE, the proteins were expected to migrate much more slowly in SDS-PAGE than would be expected on the basis of their theoretical molecular weights of ~41 and ~39 kDa, respectively [18, 19, 23, 30]. This is due to the highly hydrophilic character and consequent low SDS-binding capacity of the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) block [18]. The 13-residue D PPxY block resembles the proline-rich nature of the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) block, and as such is not expected to much affect the mobility of the protein in SDS-PAGE relative to that of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) alone. On the other hand, the 37-residue hydrophobic D WW block likely will, because attachment to \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) of peptides that improve SDS binding is known to increase the mobility of the polymer [23, 30]. Indeed, the proteins migrated accordingly in SDS-PAGE (Fig. 1): \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) migrates as slowly as the control \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) protein, and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) migrates faster than \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\). Although SDS-PAGE is not informative about the molecular mass of the polymers, it does show the proteins are relatively pure and intact.

SDS-PAGE analysis of the purified protein polymers. Lane 1, \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\); lane 2, \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\); lane 3, \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\); lane M, protein molecular weight marker; lane 4, control protein \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\). Samples were loaded at 13 µg of protein per lane
The molecular weight distribution of purified \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) was further investigated by MALDI-TOF mass spectrometry. A peak at m/z 41,440 was observed in the spectrum of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) (Fig. 2a). This peak corresponds, within experimental error, to the expected molecular weight of the intact protein (41,437 Da). The small shoulder represents a minor fraction of the molecules with an N-terminal (Glu-Ala)2 extension. Such extensions commonly occur in P. pastoris due to incomplete processing of the α-factor prepro secretory signal [17]. The MALDI-TOF analysis confirms the conclusion from SDS-PAGE that the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) protein is pure and intact. The MALDI-TOF spectrum for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) (Fig. 2b), however, showed several peaks. The minor low mass peak at m/z 38,553 is in accordance with the expected molecular weight of the intact protein (38,552 Da). It seems likely that the other peaks of higher molecular mass represent glycosylated species. Indeed, SDS-PAGE followed by Periodic acid-Schiff staining [33] confirmed that \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) is glycosylated (Fig. 3). Although the sequence of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) contains no N-x-[ST] N-glycosylation motifs, P. pastoris is also capable of O-glycosylation [34]. Because \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\), as a separate protein, is known to be nonglycosylated [18] (see also Fig. 3) most likely the single Ser residue in the added D PPxY block has been modified with O-glycans.

MALDI-TOF analysis of the purified protein polymers. \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) (a), \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) (b), \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) (c)

Detection of glycosylation by SDS-PAGE. Lane 1, \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) control; lane 2, \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\); lane M: protein molecular weight marker. Stained with Coomassie Brilliant Blue (a). Stained using Periodic acid-Schiff staining (b). Equal amounts of protein were loaded for all samples
Construction of a Ser12 → Ala PPxY mutant
Glyosylation of therapeutic proteins by P. pastoris may cause adverse immune responses [35]. Also from a nanomaterials point of view, the polydispersity resulting from glycosylation is undesirable, and desired interactions may well be hindered by the presence of oligosaccharides. To abolish the observed glycosylation of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\), we constructed a variant with a Ser12→Ala mutation in the PPxY module (Table 1). This variant, denoted \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\), was produced in P. pastoris and purified as described above. SDS-PAGE shows a single band (Fig. 1), and the yield of purified protein was 2.5 g L−1 of cell-free broth. MALDI-TOF of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) no longer showed the extensive pattern of glycosylated species (Fig. 2c). Instead, a major peak at m/z 38,536 is seen, matching the expected molecular weight of 38,536 Da. The shoulder represents a minor fraction of the protein not fully processed by P. pastoris dipeptidylaminopeptidase, as mentioned above.
The absence of glycosylation in \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) shows that indeed Ser12 of the PPxY module in \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) was glycosylated. Although there is no known consensus sequence for O-glycosylation, serine/threonine rich sequences appear relatively susceptible, particularly when prolines are in the vicinity of the hydroxyl residues [36]. Although both \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) and D PPxY are rich in proline and serine, interestingly, all serines in \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) are followed by proline, while in D PPxY the single serine is preceded by proline. The observed exclusive glycosylation of serine in the PPxY module seems to agree with the reported enhancement of mannosyl transfer in S. cerevisiae for peptide substrates with proline N-terminal to the hydroxyl amino acid [37], and with its inhibition when proline is the C-terminal neighbor [38]. However, this should not be taken to imply that such simple motifs are sufficient to determine O-glycosylation.
Limited glycan characterization
The fact that exclusively Ser12 in the PPxY module was glycosylated allows a direct interpretation of the mass spectrum of the glycosylated protein in terms of the oligosaccharide composition of the population of attached glycans. Although relatively few studies have reported O-glycosylation in P. pastoris, it is clear that O-glycans in this host usually consist of up to five mannose units [34], with dimers and trimers being the most abundant species [39, 40]. However, longer glycan chains of up to nine mannose residues have been described [41], as well as a phosphorylated Man6 O-glycan [40]. The glycosidic links between the mannose units are mostly in α1-2 arrangement [39, 40], although also α1-3 and α1-6 links have been reported [41, 42], as well as ß1-2 links [40].
For closely studying the glycan mass distribution, the MALDI-TOF analysis of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) was repeated with optimal settings for the m/z range of the relevant [M + H]+ ions (Fig. 4a). Table 2 provides an interpretation of the observed peaks, all of which can be explained in terms of mannose units (+162 Da mass shift) and phosphorylation (+80 Da mass shift). Applying Occam’s razor, we assumed maximally one phosphate per glycan structure.

MALDI-TOF of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) treated with α(1-2,1-3,1-6) mannosidase and phosphatase. Untreated \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) (a). The protein was digested with α-mannosidase (b), then with phosphatase (c), and finally digested again with α-mannosidase (d). Enzymes were thermally inactivated between consecutive digestions. See Table 2 for observed m/z values and theoretical masses of the indicated glycoforms
Hypothetically, the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) protein itself may have been phosphorylated, rather than the oligosaccharides attached to it. Because a nonglycosylated peak shifted by +80 Da is not observed in the MALDI-TOF spectrum of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\), and because both \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) [18] and the nonglycosylated \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}^ * }}\) are not phosphorylated, such hydroxyl amino acid phosphorylation would need to have occurred specifically in the glycosylated fraction of the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) molecules. This unlikely notion can be excluded because treatment with alkaline phosphatase did not result in significant changes to the MALDI-TOF spectrum (Additional file 1: Fig. S1). This finding furthermore indicates that the phosphorylated oligosaccharides do not contain phosphomonoesters, but rather contain phosphorylated Man in diester linkage, in agreement with the findings by Trimble et al. for the above-mentioned phosphorylated Man6 O-glycan [40].
When \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) was treated with the exoglycosidase jack bean α(1-2,1-3,1-6) mannosidase, a clearly altered mass distribution was obtained in MALDI-TOF (Fig. 4b; Table 2). Repeated digestions using increasing amounts of enzyme and incubation time all resulted in similar spectra (not shown), suggesting that the glycan structure is partially resistant to the α(1-2,1-3,1-6) mannosidase. The observed phosphorylation provides a likely explanation. Nonphosphorylated species were hardly detectable after digestion with jack bean mannosidase (Fig. 4b; Table 2). Furthermore, a peak corresponding to Man2 + P is absent in the undigested sample, but appears upon α-mannosidase digestion at the apparent expense of Man3 + P and larger phosphorylated forms. Although jack bean α-mannosidase can trim terminal mannoses linked to phosphate, it cannot thereafter proceed further [43–45]. Thus, the digestion likely halted upon generation of the phosphorylated species observed in Fig. 4b (Man2 + P, Man3 + P, and Man6 + P). Treatment of the mannosidase-digested sample with alkaline phosphatase resulted in a shift by −80 Da for these three phosphorylated species (Fig. 4c; Table 2), confirming that they were present as phosphodiesters prior to α-mannosidase digestion, and that indeed, as assumed above, each glycoform contains only one phosphate. Moreover, dephosphorylation rendered the remaining glycan structures susceptible to further digestion by α-mannosidase (Fig. 4d; Table 2). Possibly, the minor amount of remaining Man6 is capped with a resistant β1-2 Man disaccharide, as has been described for P. pastoris [40].
A detailed study of O-glycans chemically released from \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) would be needed to determine their precise structure with certainty, but is beyond the scope of this work. Nonetheless, it is clear that the heterogeneous mixture of glycans attached to Ser12 in the PPxY module contains phosphorylated species in diester linkage. Similarly phosphorylated O-glycans have been described for several proteins in S. cerevisiae [44–46]. To our knowledge, the present study represents the first confirmation of the occurrence of phosphorylated O-glycans in P. pastoris as reported by Trimble et al. [40]. Interestingly, the phosphorylated Man6 previously reported was described as only a minor component [40], whereas most of the oligosaccharides on C 4 -D PPxY are phosphorylated.
Binding of proline-rich ligands by the WW domain
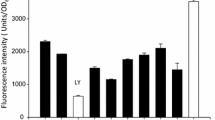
Because tryptophans are exclusively present in the WW domain, tryptophan fluorescence can be used to monitor binding of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) to its proline-rich ligands. Upon binding, the local environment of the tryptophans becomes more hydrophobic, causing a blue-shift of the emission maximum and increased fluorescence. Tryptophan fluorescence of a fixed amount of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) (10 µM) was thus followed during titration with a concentrated stock solution of either \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) or \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\).
An excitation wavelength of 295 nm was used to prevent excitation of tyrosines [47], which are present in the PPxY domain. With increasing concentration of the ligand, the maximum emission wavelength of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) decreased, indicating the transition of at least one tryptophan of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) from a solvent-exposed to a more hydrophobic environment (Fig. 5). As expected, this was accompanied by an increased fluorescence quantum yield. From the titration plots in Fig. 5, a Kd of 2.5 µM was calculated by non-linear regression for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\), and a Kd of 2.7 µM for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\). Apparently, the WW domain binds equally well to glycosylated and nonglycosylated \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\).

Steady-state tryptophan fluorescence of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) upon titration with \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) (a) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) (b). The top graphs present tryptophan fluorescence spectra obtained for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) upon addition of different concentrations of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\). The bottom graphs show titration of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) with \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\), where fluorescence was monitored at 340 nm. Error bars represent s.d. (n = 3)
Binding affinities were further established using isothermal titration calorimetry. A fixed amount of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) (200 µM) was titrated with concentrated ligand stock solution (Fig. 6). Control experiments where buffer was titrated with ligand, resulted in a relatively small and constant heat of dilution (Additional file 1: Fig S2A, B). Also \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) titrated with control protein \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) showed only heat of dilution (Additional file 1: Fig. S2C). The binding isotherms of both \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) showed an immediate decrease in differential power for each consecutive injection (Fig. 6). The Kd values derived from the integrated heat plots in Fig. 6 are 9.3 and 9.2 µM for \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\), respectively. Both values are similar, and in reasonable agreement with the fluorescence spectroscopy results. In general, the Kd of various WW-domains and their proline-rich ligands are in the high nM to low µM range [48]. According to Russ et al., the PPxY peptide, also used in our D PPxY block, binds the CC43 WW domain with Kd = 1.7 µM, and the Nedd4.3 WW domain with Kd = 11.2 µM [49]. Wong Po Foo et al. used the same couples in the context of protein polymers and found relatively high Kd values of 4.6 µM and 62 µM, respectively, for polymers containing three PPxY motifs, interacting with polymers containing three CC43 or Nedd4.3 domains. The combination of PPxY (p53BP-2) and WWP1-1 used by us was among the best performing pairs tested by Porozzi et al. [29], but to our knowledge no Kd values have been published. In our protein polymer context, the Kd of this combination (~3 to 9 µM) is in a similar range as the above-mentioned literature values for other WW/PPxY combinations. This range is quite sufficient for various supramolecular systems, and multiple D blocks could be introduced into the polymer for applications that require even lower working concentrations. We did attempt to produce the CC43 domain as well, but encountered proteolytic degradation in P. pastoris that could not be readily resolved. The stoichiometry (N) determined for both \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) is around 0.9, which, given unavoidable inaccuracies in preparing stock solutions from lyophilized proteins, is in good agreement with the expected 1:1 stoichiometry.

Binding study using isothermal titration calorimetry. \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) titrant (a), \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) titrant (b). The top graphs show the heat response upon titration of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) with the proline-rich ligands. Bottom graphs represent the integrated peak areas per mole of ligand
Conclusion
We have shown that polymers containing the WW domain and a proline-rich ligand can be efficiently produced in Pichia pastoris. The PPxY module was found to be O-glycosylated, and remarkably a considerable fraction of the oligomannose structures was phosphorylated. O-glycosylation was abolished by changing the serine in the PPxY sequence to alanine. The WW domain effectively bound both the glycosylated and nonglycosylated PPxY modules, with similar binding affinities. This work provides proof-of-concept that otherwise noninteracting protein polymers can be brought together using the specifically interacting WW and PPxY modules. This will allow us to create protein materials with still better controlled structures at the nanoscale, and hence biomedical materials with more precisely defined interactions with living cells.
Methods
Construction of expression vectors and strains
The double-stranded gene fragments encoding D WW and D PPxY were assembled via overlap extension PCR [50] from the oligonucleotides shown in Additional file 1: Table S1. The gene fragments were digested with XhoI/EcoRI and cloned into XhoI/EcoRI-digested vector pMTL23ΔBsaI [51], in order to obtain two vectors pMTL23ΔBsaI-D WW and pMTL23ΔBsaI-D PPxY. The vector pMTL23-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) contains the sequence encoding \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) (previously referred to as ‘P4’) [18], and was opened at the 3’ end of the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) gene with Van91I/EcoRI. The newly prepared constructs pMTL23ΔBsaI-D WW and pMTL23ΔBsaI-D PPxY were digested with DraIII/EcoRI to release inserts D WW and D PPxY. The inserts were ligated into the opened pMTL23-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) vector, resulting in pMTL23-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) and pMTL23-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\).
A Ser12 → Ala mutant of the D PPxY module was prepared by annealing of a pair of largely complementary oligos (Additional file 1: Table S1), and is referred to as D PPxY*. This double-stranded adapter with DraIII/EcoRI overhangs was ligated into vector pMTL23-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) previously digested with Van91I/EcoRI (at the 3’ end of the \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}}\) gene), resulting in pMTL23ΔBsaI-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\).
The \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\), \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\), and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) inserts were then released with XhoI/EcoRI and cloned into the likewise-digested Pichia pastoris expression vector pPIC9 (ThermoFisher, Bleiswijk, The Netherlands). This resulted in the construction of the vectors: pPIC9-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\), pPIC9-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) and pPIC9-\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}^* }}\), respectively. The vectors were linearized with SalI to target for integration at the his4 locus. Transformation of P. pastoris GS115 by electroporation and selection of Mut+ transformants were performed as described previously [17].
Fermentation
The fermentation setup consisted of a 2.5-L Bioflo 3000 stirred-tank bioreactor (New Brunswick Scientific, Nijmegen, The Netherlands) interfaced with BioCommand Software (New Brunswick Scientific, Nijmegen, The Netherlands) and a homebuilt methanol sensor-controller. The fermentations were performed as described previously [19], as follows. A starting volume of 1.25 L minimal basal salts medium [52] was used. The cultures were always inoculated with precultures grown to similar OD600. Growth temperature was 30 °C, and the pH was controlled at 3.0 throughout the entire fermentation. The air was supplemented with 20 % (v/v) oxygen during the glycerol fed-batch phase and the methanol fed-batch phase. During the latter protein production phase, lasting two days, methanol levels were kept at 0.2 % (w/v). Wet biomass was typically ~150 g L−1 at the end of the glycerol fed-batch phase, and ~500 g L−1 at the end of the fermentation. After harvesting, cells were removed from the broth by centrifugation for 20 min. at 15,000×g (RT), followed by microfiltration.
Protein purification
Purification of all protein polymers was done by ammonium sulfate precipitation essentially as described [19], except that heating of the supernatant and acetone precipitation were omitted. Shortly, medium salts were removed by raising the pH of the cell-free broth to 8.0 with sodium hydroxide, followed by 30 min. of centrifugation at 20,000×g (RT). The protein was precipitated from the supernatant by addition of ammonium sulfate to 40 % of saturation, followed by incubation on ice for 30 min. and centrifugation for 30 min. at 20,000×g (4 °C). The protein pellet was resuspended in Milli-Q water and precipitated using ammonium sulfate at 40 % of saturation as before. The pellet was then resuspended in Milli-Q water, desalted by extensive dialysis against Milli-Q water, and finally lyophilized.
SDS-PAGE
SDS-PAGE was performed using the NuPAGE Novex System (ThermoFisher, Bleiswijk, The Netherlands) with 10 % Bis–Tris gels, MES SDS running buffer, and SeeBlue Plus2 pre-stained molecular mass markers. Prior to electrophoresis, all samples were heated for 10 min. at 70 °C in NuPAGE LDS Sample Buffer with NuPAGE Sample Reducing Agent, as per manufacturer’s recommendations for denaturing and reducing PAGE. Gels were stained using Coomassie SimplyBlue SafeStain (ThermoFisher, Bleiswijk, The Netherlands).
For detection of glycosylated proteins, SDS-PAGE gels were stained using Periodic acid-Schiff staining [33]. The gel was incubated for 1 h in 12.5 % TCA, 1 h in 1 % periodic acid/3 % acetic acid, 1 h in 15 % acetic acid (replaced every 10 min.), and 1 h at 4 °C in the dark in Schiff’s reagent (Sigma-Aldrich, Zwijndrecht, The Netherlands). The gel was then washed two times for 5 min. in 0.5 % sodium bisulfite and destained in 7 % acetic acid.
Treatment of proteins with α-mannosidase or phosphatase
For α-mannosidase digestions, 30 µg of glycoprotein was incubated for 24 h at 37 °C under mild agitation with 0.9 U of jack bean α(1-2,1-3,1-6) mannosidase (Sigma-Aldrich, Zwijndrecht, The Netherlands) in 60 µL of 20 mM sodium acetate, 0.4 mM zinc chloride, pH 5. Dephosphorylation involved incubation of 30 µg of glycoprotein for 24 h at 37 °C under mild agitation with 60 U of calf intestinal alkaline phosphatase (NEB, Ipswich, MA) in 60 µL of 50 mM Tris–HCl, 10 mM magnesium chloride, pH 8.5.
For consecutive α-mannosidase/phosphatase digestions, the enzyme after each step was inactivated by heating for 15 min. at 100 °C, followed by desalting using Micro Bio-Spin P-6 columns (Bio-Rad, Veenendaal, The Netherlands). To allow mass spectrometric analysis of the reaction products, it was verified that such analysis of enzyme-only digestions revealed no significant peaks in the relevant ~38–41 kDa range (not shown).
Mass spectrometry
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry was performed using an ultrafleXtreme mass spectrometer (Bruker, Leiderdorp, The Netherlands). Proteins were desalted using Micro Bio-Spin P-6 columns (Bio-Rad, Veenendaal, The Netherlands), and samples were prepared by the dried droplet method on a 600 µm AnchorChip target (Bruker, Leiderdorp, The Netherlands), using 5 mg mL−1 2,5-dihydroxyacetophenone, 1.5 mg mL−1 diammonium hydrogen citrate, 25 % (v/v) ethanol and 3 % (v/v) trifluoroacetic acid as matrix. Spectra were derived from ten 500-shot (1000 Hz) acquisitions taken at non-overlapping locations across the sample. Wide mass-range measurements were made in the positive linear mode, with ion source 1, 25.0 kV; ion source 2, 23.3 kV; lens, 6.5 kV; pulsed ion extraction, 680 ns. Detailed analyses of glycoproteins in the ~38-41 kDa range were done with ion source 1, 20.0 kV; ion source 2, 18.4 kV; lens, 6.2 kV; pulsed ion extraction, 450 ns, and spectra were derived from ten 1000-shot (1000 Hz) acquisitions. Protein Calibration Standard II (Bruker, Leiderdorp, The Netherlands) was used for external calibration.
Steady-state fluorescence spectroscopy
Steady-state fluorescence spectroscopy was performed on a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies, Amstelveen, The Netherlands), monitoring the intrinsic fluorescence emission of the tryptophan residues in the WW domain at 340 nm, with excitation at 295 nm. Proteins were dissolved overnight in 10 mM sodium phosphate buffer (pH 7) at RT. The binding assays for both couples \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\)/\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\), and \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\)/\({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) were conducted in triplicate at RT. A 500 μL aliquot of 10 μM \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) was pipetted into a quartz fluorescence cuvette (Sigma-Aldrich, Zwijndrecht, The Netherlands). A 500 µM solution of ligand was stepwise added to the cuvette, up to a final ligand concentration of ~24 µM. The time interval between additions was 30 min. The final volume of added ligand solution did not exceed 5 % of the starting volume of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\). Curve fitting for Kd determination was done on averaged data of triplicate titrations.
Isothermal titration calorimetry
ITC was conducted on a MicroCal VP-ITC (Malvern Instruments, Malvern, United Kingdom) at 25 °C. All purified protein polymers were dissolved in 10 mM sodium phosphate buffer, pH 7 and filtrated with 0.2 μm Minisart NML Syringe Filters (Sigma-Aldrich, Zwijndrecht, The Netherlands). Prior to titration, each protein polymer solution was degassed under vacuum for 60 min. at RT. The ligand concentration of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) or \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}} ^* }}\) in the titration syringe was 2.9 mM. Each titration consisted of 63 injections at an interval of 250 s. Ligand aliquots of 4 µL were titrated into 1.4 mL of a 200 µM \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{WW}}}}\) protein solution inside the calorimeter cell under continuous stirring at 329 rpm. Data obtained from the injection of ligand molecules into 1.4 mL of 10 mM PBS buffer (Additional file 1: Fig. S2) were subtracted as blanks from the experimental data before the data were analyzed using MicroCal Origin Software (Malvern Instruments, Malvern, United Kingdom). Titrations were performed in triplicate and Kd values were averaged after curve fitting.
Abbreviations
- ITC:
-
isothermal titration calorimetry
- Kd :
-
dissociation constant
- LDS:
-
lithium dodecyl sulfate
- MALDI-TOF:
-
matrix-assisted laser desorption/ionization time-of-flight
- Man:
-
mannose
- MES:
-
2-(N-Morpholino)ethanesulfonic acid
- m/z :
-
mass-to-charge ratio
- OD600 :
-
optical density at 600 nm
- P:
-
phosphate
- PAGE:
-
polyacrylamide gel electrophoresis
- PBS:
-
phosphate-buffered saline
- PCR:
-
polymerase chain reaction
- rpm:
-
revolutions per minute
- RT:
-
room temperature
- SDS:
-
sodium dodecyl sulfate
- TCA:
-
trichloroacetic acid
- Tris:
-
tris(hydroxymethyl)aminomethane
References
Rabotyagova OS, Cebe P, Kaplan DL. Protein-based block copolymers. Biomacromolecules. 2011;12:269–89.
Ghandehari H, Cappello J. Genetic engineering of protein-based polymers: potential in controlled drug delivery. Pharm Res. 1998;15:813–5.
Tessmar JK, Göpferich AM. Matrices and scaffolds for protein delivery in tissue engineering. Adv Drug Deliv Rev. 2007;59:274–91.
Brea RJ, Reiriz C, Granja JR. Towards functional bionanomaterials based on self-assembling cyclic peptide nanotubes. Chem Soc Rev. 2010;39:1448–56.
Matson JB, Stupp SI. Self-assembling peptide scaffolds for regenerative medicine. Chem Commun. 2012;48:26–33.
Price R, Poursaid A, Ghandehari H. Controlled release from recombinant polymers. J Control Release. 2014;190:304–13.
Gagner JE, Kim W, Chaikof EL. Designing protein-based biomaterials for medical applications. Acta Biomater. 2014;10:1542–57.
Shi P, Gustafson JA, MacKay JA. Genetically engineered nanocarriers for drug delivery. Int J Nanomedicine. 2014;9:1617–26.
Heslot H. Artificial fibrous proteins: a review. Biochimie. 1998;80:19–31.
Annabi N, Mithieux SM, Camci-Unal G, Dokmeci MR, Weiss AS, Khademhosseini A. Elastomeric recombinant protein-based biomaterials. Biochem Eng J. 2013;77:110–8.
Liu L, Busuttil K, Zhang S, Yang Y, Wang C, Besenbacher F, Dong M. The role of self-assembling polypeptides in building nanomaterials. Phys Chem Chem Phys. 2011;13:17435–44.
Kopeček J, Yang J. Smart Self-Assembled Hybrid Hydrogel Biomaterials. Angew Chem Int Ed. 2012;51:7396–417.
Fichman G, Gazit E. Self-assembly of short peptides to form hydrogels: design of building blocks, physical properties and technological applications. Acta Biomater. 2014;10:1671–82.
Kim H, Siu K-H, Raeeszadeh-Sarmazdeh M, Sun Q, Chen Q, Chen W. Bioengineering strategies to generate artificial protein complexes. Biotechnol Bioeng. 2015;112:1495–505.
Walper SA, Turner KB, Medintz IL. Enzymatic bioconjugation of nanoparticles: developing specificity and control. Curr Opin Biotechnol. 2015;34:232–41.
Domeradzka NE, Werten MWT, de Wolf FA, de Vries R. Protein cross-linking tools for the construction of nanomaterials. Curr Opin Biotechnol. 2016;39:61–7.
Werten MWT, van den Bosch TJ, Wind RD, Mooibroek H, de Wolf FA. High-yield secretion of recombinant gelatins by Pichia pastoris. Yeast. 1999;15:1087–96.
Werten MWT, Wisselink WH, Jansen-van den Bosch TJ, de Bruin EC, de Wolf FA. Secreted production of a custom-designed, highly hydrophilic gelatin in Pichia pastoris. Protein Eng. 2001;14:447–54.
Werten MWT, Teles H, Moers APHA, Wolbert EJH, Sprakel J, Eggink G, de Wolf FA. Precision gels from collagen-inspired triblock copolymers. Biomacromolecules. 2009;10:1106–13.
Schipperus R, Eggink G, de Wolf FA. Secretion of elastin-like polypeptides with different transition temperatures by Pichia pastoris. Biotechnol Progress. 2012;28:242–7.
Beun LH, Storm IM, Werten MWT, de Wolf FA, Cohen Stuart MA, de Vries R. From micelles to fibers: balancing self-assembling and random coiling domains in pH-responsive silk-collagen-like protein-based polymers. Biomacromolecules. 2014;15:3349–57.
Włodarczyk-Biegun MK, Werten MWT, de Wolf FA, van den Beucken JJJP, Leeuwenburgh SCG, Kamperman M, Cohen Stuart MA. Genetically engineered silk–collagen-like copolymer for biomedical applications: production, characterization and evaluation of cellular response. Acta Biomater. 2014;10:3620–9.
Domeradzka NE, Werten MWT, de Vries R, de Wolf FA. Production in Pichia pastoris of protein-based polymers with small heterodimer-forming blocks. Biotechnol Bioeng. 2015;113:953–60.
Wang H, Heilshorn SC. Adaptable hydrogel networks with reversible linkages for tissue engineering. Adv Mater. 2015;27:3717–36.
Chan G, Mooney DJ. New materials for tissue engineering: towards greater control over the biological response. Trends Biotechnol. 2008;26:382–92.
Sudol M, Chen HI, Bougeret C, Einbond A, Bork P. Characterization of a novel protein-binding module—the WW domain. FEBS Lett. 1995;369:67–71.
Macias MJ, Hyvonen M, Baraldi E, Schultz J, Sudol M, Saraste M, Oschkinat H. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature. 1996;382:646–9.
Wong Po Foo CTS, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Two-component protein-engineered physical hydrogels for cell encapsulation. PNAS. 2009;106:22067–72.
Pirozzi G, McConnell SJ, Uveges AJ, Carter JM, Sparks AB, Kay BK, Fowlkes DM. Identification of novel human WW domain-containing proteins by cloning of ligand targets. J Biol Chem. 1997;272:14611–6.
Moers APHA, Wolbert EJH, de Wolf FA, Werten MWT. Secreted production of self-assembling peptides in Pichia pastoris by fusion to an artificial highly hydrophilic protein. J Biotechnol. 2010;146:66–73.
Bao W-J, Gao Y-G, Chang Y-G, Zhang T-Y, Lin X-J, Yan X-Z, Hu H-Y. Highly efficient expression and purification system of small-size protein domains in Escherichia coli for biochemical characterization. Protein Expr Purif. 2006;47:599–606.
Martinez-Rodriguez S, Bacarizo J, Luque I, Camara-Artigas A. Crystal structure of the first WW domain of human YAP2 isoform. J Struct Biol. 2015;191:381–7.
Zacharius RM, Zell TE, Morrison JH, Woodlock JJ. Glycoprotein staining following electrophoresis on acrylamide gels. Anal Biochem. 1969;30:148–52.
Bretthauer RK, Castellino FJ. Glycosylation of Pichia pastoris—derived proteins. Biotechnol Appl Biochem. 1999;30:193–200.
Daly R, Hearn MTW. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit. 2005;18:119–38.
Herscovics A, Orlean P. Glycoprotein biosynthesis in yeast. FASEB J. 1993;7:540–50.
Lehle L, Bause E. Primary structural requirements for N- and O-glycosylation of yeast mannoproteins. Biochim Biophys Acta. 1984;799:246–51.
Strahl-Bolsinger S, Tanner W. Protein O-glycosylation in Saccharomyces cerevisiae Purification and characterization of the dolichyl-phosphate-d-mannose-protein o-d-mannosyltransferase. FEBS J. 1991;196:185–90.
Duman JG, Miele RG, Liang H, Grella DK, Sim KL, Castellino FJ, Bretthauer RK. O-Mannosylation of Pichia pastoris cellular and recombinant proteins. Biotechnol Appl Biochem. 1998;28:39–45.
Trimble RB, Lubowski C, Hauer CR, Stack R, McNaughton L, Gemmill TR, Kumar SA. Characterization of N- and O-linked glycosylation of recombinant human bile salt–stimulated lipase secreted by Pichia pastoris. Glycobiology. 2004;14:265–74.
Gustafsson A, Sjöblom M, Strindelius L, Johansson T, Fleckenstein T, Chatzissavidou N, Lindberg L, Ångström J, Rova U, Holgersson J. Pichia pastoris-produced mucin-type fusion proteins with multivalent O-glycan substitution as targeting molecules for mannose-specific receptors of the immune system. Glycobiology. 2011;21:1071–86.
Boraston AB, Sandercock LE, Warren RAJ, Kilburn DG. O-glycosylation of a recombinant carbohydrate-binding module mutant secreted by Pichia pastoris. J Mol Microbiol Biotechnol. 2003;5:29–36.
Hernández LM, Ballou L, Alvarado E, Gillece-Castro BL, Burlingame AL, Ballou CE. A new Saccharomyces cerevisiae mnn mutant N-linked oligosaccharide structure. J Biol Chem. 1989;264:11849–56.
Bergwerff AA, Stark W, Fendrich G, Knecht R, Blommers MJJ, Maerki W, Kragten EA, van Oostrum J. Identification of Manα1-3Manα1-2Man and Man-linked phosphate on O-mannosylated recombinant leech-derived tryptase inhibitor produced by Saccharomyces cerevisiae and determination of the solution conformation of the mannosylated polypeptide. Eur J Biochem. 1998;253:560–75.
Nakayama K-I, Feng Y, Tanaka A, Jigami Y. The involvement of mnn4 and mnn6 mutations in mannosylphosphorylation of O-linked oligosaccharide in yeast Saccharomyces cerevisiae. Biochim Biophys Acta Gen Subj. 1998;1425:255–62.
Jars MU, Osborn S, Forstrom J, MacKay VL. N- and O-Glycosylation and phosphorylation of the bar secretion leader derived from the barrier protease of Saccharomyces cerevisiae. J Biol Chem. 1995;270:24810–7.
Weljie AM, Vogel HJ. Steady-state fluorescence spectroscopy. In: Vogel HJ, editor. Methods in Molecular Biology, vol. 2. Totowa: Springer New York; 2002. p. 75–87.
Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–41.
Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R. Natural-like function in artificial WW domains. Nature. 2005;437:579–83.
Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–9.
Golinska MD, Włodarczyk-Biegun MK, Werten MWT, Cohen Stuart MA, de Wolf FA, de Vries R. Dilute self-healing hydrogels of silk-collagen-like block copolypeptides at neutral pH. Biomacromolecules. 2014;15:699–706.
Zhang W, Bevins MA, Plantz BA, Smith LA, Meagher MM. Modeling Pichia pastoris growth on methanol and optimizing the production of a recombinant protein, the heavy-chain fragment C of botulinum neurotoxin, serotype A. Biotechnol Bioeng. 2000;70:1–8.
Authors’ contributions
NED and MWTW designed and performed all experiments, analyzed the data, wrote most of the manuscript, and should be considered joint first authors. RdV and FdW initiated the study, and contributed to data interpretation and writing. MWTW, RdV and FdW coordinated the study. All authors have read and approved the final manuscript.
Acknowledgements
We thank Ésio Bessa Ramos for his help in the design of the steady-state fluorescence spectroscopy experiment.
Availability of data and materials
Data and materials available on request for non-commercial purposes.
Competing interests
The authors declare that they have no competing interests.
Funding
The work of N.E.D. is supported by NanoNextNL, a micro and nanotechnology consortium of the government of The Netherlands and 130 partners. Financial support to M.W.T.W. and F.A.W. is provided by ERC Advanced Grant 267254 “BioMate”. The funding bodies played no role in the design and execution of the study, nor in the writing of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Natalia E. Domeradzka and Marc W. T. Werten contributed equally to this work
Additional file
12934_2016_498_MOESM1_ESM.pdf
Additional file 1. This file consists of one supplemental table and two supplemental figures. Table S1. Oligonucleotides used in gene construction. Figure S1. MALDI-TOF of \({\mathbf{C}}_{{\mathbf{4}}}^{{\mathbf{P}}} - {\mathbf{D}}^{{{\mathbf{PPxY}}}}\) before and after phosphatase treatment. Figure S2. Control ITC measurements.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Domeradzka, N.E., Werten, M.W.T., de Vries, R. et al. Production in Pichia pastoris of complementary protein-based polymers with heterodimer-forming WW and PPxY domains. Microb Cell Fact 15, 105 (2016). https://doi.org/10.1186/s12934-016-0498-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0498-3