
Abstract
Background
Glucokinase (GK) plays a key role in glucose metabolism. In the liver, GK is regulated by GK regulatory protein (GKRP) with nuclear sequestration at low plasma glucose level. Some GK activators (GKAs) disrupt GK-GKRP interaction which increases hepatic cytoplasmic GK level. Excess hepatic GK activity may exceed the capacity of glycogen synthesis with excess triglyceride formation. It remains uncertain whether hypertriglyceridemia associated with some GKAs in previous clinical trials was due to direct GK activation or impaired GK-GKRP interaction.
Methods
Using publicly available genome-wide association study summary statistics, we selected independent genetic variants of GCKR and GCK associated with fasting plasma glucose (FPG) as instrumental variables, to mimic the effects of impaired GK-GKRP interaction and direct GK activation, respectively. We applied two-sample Mendelian Randomization (MR) framework to assess their causal associations with lipid-related traits, risks of metabolic dysfunction-associated steatotic liver disease (MASLD) and cardiovascular diseases. We verified these findings in one-sample MR analysis using individual-level statistics from the Hong Kong Diabetes Register (HKDR).
Results
Genetically-proxied impaired GK-GKRP interaction increased plasma triglycerides, low-density lipoprotein cholesterol and apolipoprotein B levels with increased odds ratio (OR) of 14.6 (95% CI 4.57–46.4) per 1 mmol/L lower FPG for MASLD and OR of 2.92 (95% CI 1.78–4.81) for coronary artery disease (CAD). Genetically-proxied GK activation was associated with decreased risk of CAD (OR 0.69, 95% CI 0.54–0.88) and not with dyslipidemia. One-sample MR validation in HKDR showed consistent results.
Conclusions
Impaired GK-GKRP interaction, rather than direct GK activation, may worsen lipid profiles and increase risks of MASLD and CAD. Development of future GKAs should avoid interfering with GK-GKRP interaction.
Similar content being viewed by others


Background
Diabetes is a pressing global healthcare concern. Many patients have suboptimal glycemic control with high risk for multiple complications [1], calling for new treatment strategies. Glucokinase (GK) is the first and rate-limiting enzyme of glycolysis that senses glucose levels in the pancreas and liver [2]. In the pancreatic β-cells, GK phosphorylates glucose to Glucose-6-Phosphate (G-6-P), which is essential for ATP production and insulin secretion. In the liver, GK enhances hepatic glucose uptake and glycogen synthesis. Hepatic GK is modulated by glucokinase regulatory protein (GKRP), a GK regulator which is exclusively expressed in hepatocytes. GKRP binds to GK and forms a complex sequestered in the nucleus when plasma glucose (PG) and fructose levels are low to avoid excessive hepatic glucose uptake and hypoglycemia. In response to rising PG levels, GK can be rapidly released from the complex to promote glycogen synthesis [3]. Dysregulation of GK function due to genetic variants can cause abnormal glucose metabolism. For example, glucokinase-maturity-onset diabetes of the young (GK-MODY) and persistent hyperinsulinemic hypoglycemia of infancy (PHHI) are caused by inactivating and activating GCK mutations, respectively [4, 5]. Individuals with GK-MODY due to inactivating mutations have impaired glucose sensing with a higher setpoint which triggers increased insulin secretion which may lead to mild and asymptomatic fasting hyperglycemia (5.5–8 mmol/L) [6]. Until the recent introduction of a GK activator known to restore the glucose-sensing and insulin-secreting setpoints [7], conventional oral antidiabetic drugs (OADs) are not effective among these patients [6].
GK activators (GKAs) are a new class of OADs targeting GK since 2003 [8,9,10,11,12]. These compounds bind to GK, allosterically modulate enzymatic kinetics, enhance glucose sensitivity in beta-cells with improved insulin response. GKAs can be divided into liver-selective and dual-acting (liver and pancreas) activators. In addition to directly activating GK by stabilizing its active conformation, some GKAs, although not intentionally designed, can impair the protein–protein interaction of GK-GKRP complex in hepatocytes. The latter can lead to increased GK translocation from the nucleus to the cytoplasm [13, 14] with excessive hepatic glucose uptake. When excessive glucose hepatic flux exceeds the capacity of glycogen synthesis, hypertriglyceridemia may ensue due to increased glucose-fatty acid cycle [15]. This adverse effect has led to the discontinuation of development of some GKAs in the past [16]. Likewise, GK-GKRP disruptors had been developed as OADs with reduced GK nuclear sequestration and increased hepatic GK activity [17, 18]. Although GK-GKRP disruptors exhibited low hypoglycemic risk [17], potential hypertriglyceridemia due to excess liver glycogen deposition and de novo lipogenesis are significant concerns [19, 20]. Enhanced hepatic de novo lipogenesis may contribute to hepatic lipid accumulation and result in metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as non-alcoholic fatty liver disease (NAFLD), with dyslipidaemia and MASLD being both independent risk factors for cardiovascular diseases [21, 22]. With better design of the molecules, the more recently developed GKAs such as dorzagliatin (a dual-acting GKA) and TTP399 (a liver-selective GKA) [11, 23,24,25] did not cause hypertriglyceridemia in the clinical studies. Notably, dorzagliatin, the first-in-class GKA, has been approved in China in 2022, showing a placebo-subtracted HbA1c reduction of 0.5% in phase 3 studies in patients with type 2 diabetes (T2D) with a good safety profile [23, 24].
In our previous work, we investigated the influence of GK activation on cardiovascular disease risk using Mendelian randomization (MR) analysis and reported that target-specific glucose-lowering through GK activation might confer protection against coronary artery disease (CAD) and heart failure (HF) [26]. Importantly, the cardiovascular benefits of GK-targeted glucose-lowering exceeded that due to non-targeted glucose-lowering [26]. In the present study, we sought to discern how different GK activating mechanisms might influence long-term cardiovascular outcomes via their impacts on lipid-related traits. We hypothesized that disruption of GK-GKRP interaction, rather than GK activation per se, could worsen lipid profiles and contribute to long-term complications. We tested our hypothesis through MR analyses and corroborated our primary findings using data from a prospective cohort.
Methods
Study design
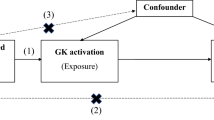
We first conducted a two-sample MR analysis using publicly available summary statistics from genome-wide association studies (GWAS), followed by a one-sample MR validation using in-house data from the Hong Kong Diabetes Register (HKDR). The rise in plasma glucose is sensed by GK expressed in beta-cells in pancreatic islets. This is followed by release of insulin which promotes hepatic glycogen synthesis by GK in the liver. The hepatic activity of GK is regulated by GKRP which is exclusively expressed in the liver. Here, GK-GKRP is formed as a complex in the nucleus. Influx of glucose in the liver will trigger release of GK from the complex to the cytoplasm to promote glycogen synthesis. Impaired GK-GKRP interaction can lead to excessive GK activity with increased glucose influx into the liver causing low fasting plasma glucose (FPG) and if the capacity of glycogen synthesis is exceeded, de novo lipogenesis can follow. This provides the basis of utilizing GCKR single nucleotide polymorphisms (SNPs) to mimic the hepatic GK-GKRP disruption and GCK SNPs to mimic the GK activation in the pancreas and liver with low FPG. The low FPG level due to mimicked impaired GK-GKRP interaction or GK activation was regarded as the exposure. Lipid-related traits, MASLD and cardiovascular complications were the outcomes of interest. Figure 1 compares the conceptual framework of our analyses versus that of randomized controlled trial. All contributing studies in the MR analyses had received appropriate ethical approval and patient consent.

Conceptual framework of study design. GK, glucokinase; GKRP, glucokinase regulatory protein
Instrumental variables selection
We identified SNPs in GCKR gene (genomic position chromosome 2: 27719706–27746551 on build GRCh37.p13) associated (P ≤ 5 × 10−8) with FPG in the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) [27] as proxies for pharmacological disruption of GK-GKRP interaction. We used linkage disequilibrium (LD) clumping algorithm in the PLINK (window size = 1000 KB, r2 threshold = 0.01, European LD reference panel from the 1000 Genomes Project) to filter and retain SNPs with the strongest significance in the MAGIC. For IVs selection, we limited the GWAS data from MAGIC to European population (N = 200,622) to avoid biases from population difference.
Outcome data sources
In the two-sample MR study, we investigated the impact of genetically-proxied impaired GK-GKRP interaction on lipid-related traits including plasma triglyceride (TG), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and apolipoprotein B (ApoB) levels as well as MASLD and cardiovascular diseases including CAD, peripheral arterial disease (PAD), stroke and HF, as defined by the respective Consortiums and the original studies. We employed incident T2D as positive control and insulin level as negative control given that GK-GKRP disruptors were designed to treat diabetes without influencing insulin secretion [17].
Additional file 1: Table S1 shows the sources of summary statistics from publicly available data used in our study: (1) TG (n = 441,016), LDL-C (n = 440,546), HDL-C (n = 403,943), ApoB (n = 439,214) levels from the UK BioBank [28]; (2) MASLD (reported as NAFLD in the original GWAS) from a meta-analysis published by Ghodsian et al. (8434 cases and 770,180 controls) [29]; (3) CAD from a meta-analysis of the CARDIOGRAMplusC4D Consortium and UK BioBank (122,733 cases and 424,528 controls) [30]; (4) PAD from the FinnGen Consortium (7098 cases and 206,541 controls); (5) stroke from the MEGASTROKE Consortium (40,585 cases and 406,111 controls) [31]; (6) HF from the Heart Failure Molecular Epidemiology for Therapeutic Targets (HERMES) Consortium (47,309 cases and 930,014 controls) [32]; (7) T2D from the 70KforT2D study (12,931 cases and 57,196 controls) [33] and (8) plasma insulin level from a GWAS published by Sun et al. (3301 samples) [34]. These summary statistics were all derived from subjects of European ancestry and the populations did not overlap with those in the MAGIC. Details of disease definitions and diagnoses are available in the respective Consortium websites or original publications and are briefly summarized in Additional file 1: Supplementary Method.
One-sample MR validation
We performed a one-sample MR study of impaired GK-GKRP interaction in the HKDR for internal validation. The HKDR was established in 1995 as a quality improvement initiative at the Prince of Wales Hospital, the teaching hospital of the Chinese University of Hong Kong. The register consecutively enrolled patients referred to the Diabetes Mellitus and Endocrine Centre with documentation of detailed phenotypes at baseline and outcome data during follow-up. Once enrolled, the patients were monitored for the development of diabetic complications until their death. Using a unique identifier, clinical data on drug dispensing, laboratory results and hospitalization diagnosis were regularly retrieved from the electronic medical record (EMR) system of Hong Kong Hospital Authority. Patients provided written informed consent for allowing the anonymous use of their clinical data for the purpose of conducting clinical research. A portion of patients also consented to the collection of blood samples for future genetic and biomedical analyses. This project received ethical approval from the Clinical Research Ethics Committee of the Chinese University of Hong Kong. Details of the HKDR have been previously described [35] (CREC 2009-421 CREC 2019-080).
In the two-sample MR analyses, we only identified one GCKR SNP (rs1260326) as the IV, which was applied to the one-sample MR in a sub-cohort of the HKDR [36]. Patients with rs1260326 T allele were considered to have impaired GK-GKRP interaction because the T allele was associated with lower FPG in the MAGIC. The exposure was FPG-lowering secondary to impaired GK-GKRP interaction. The outcomes included lipid traits, prevalent MASLD, CAD, PAD, stroke and HF as defined in the HKDR. The definitions of these diseases were summarized in Additional file 1: Table S2.
Evaluating causal effects of GK activation
To mimic GK activation, we selected SNPs located in GCK gene (genomic position chromosome 7: 44182812–44229038 on build GRCh37.p13) as IVs. These variants were associated (P ≤ 5 × 10−8) with FPG in the MAGIC and were not in LD (r2 = 0.01) with other variants. The outcomes were the same as those investigated for impaired GK-GKRP interaction.
Colocalization analysis
We conducted colocalization analysis to assess the IV assumptions validity to mitigate the possibility that the exposure and outcome might be causally influenced by different variants which happen to be correlated with each other (in LD) [37]. We utilized the “coloc” package in R software to determine the probability of shared causal variants between exposure and outcomes which presented significant causality. The posterior probabilities (PP) were generated using approximate Bayes factor (ABF) computation with 5 exclusive hypotheses as following: (i) no genetic association for either trait (PPH0); (ii) a genetic association exists for only the first trait (PPH1); (iii) a genetic association exists for only the second trait (PPH2); (iv) both traits exhibit associations but with distinct causal variants (PPH3); (v) both traits exhibit associations and share a single causal variant (PPH4). We set the prior probability (p1) of the variant being associated with trait 1 to 1 × 10−4; the probability (p2) of the variant being associated with trait 2 to 1 × 10−4; and the probability (p12) of the variant being associated with both traits to 1 × 10−5. The exposure and the outcome were considered to have strong evidence of colocalization if PPH4 ≥ 0.8 and medium evidence if 0.6 ≤ PPH4 < 0.8. Colocalization analysis was carried out by generating ± 100 kb windows from the GCKR and GCK gene regions.
Statistical analysis
We computed the F-statistic for IVs used in two-sample MR analyses to assess the robustness of instruments [38]. We defined the FPG-reducing allele as the effect allele of each SNP in the MAGIC, in line with the expected effects of impaired GK-GKRP interaction and direct GK activation. The genetic associations of IVs with the exposures and outcomes were then harmonized by aligning the effect alleles. The causal estimates of each SNP were calculated using Wald-ratio method [39]. The causal relationships were assessed by combining the Wald-ratio using the random-effects inverse variance-weighted (IVW) method if multiple SNPs were selected as IVs. Heterogeneity and horizontal pleiotropy for IVs which contained multiple SNPs were detected using Cochran’s Q statistic [40] and MR-Egger regression [41], respectively.
For the one-sample MR, the Wald-ratio method was used to calculate the causal estimates [39]. Firstly, the association of rs1260326 genotype with FPG was tested using multivariable linear regression to obtain the β-coefficient of exposure on IV. The associations of rs1260326 genotype with lipid traits and complications were assessed using multivariable linear or logistic regressions to obtain the β-coefficients of outcomes on IV. We only adjusted for covariates including age, sex and diabetes duration. Further adjustment was not implemented as it might bias estimates if the variable was on the causal pathway, or the adjustment induced collider bias [42, 43]. The Wald-ratio of one-sample MR was calculated using the formula: βiv-outcome/βiv-exposure, and the standard error (SE) was calculated via the delta method [44].
All analyses were performed using R 4.1.2 software with packages “TwoSampleMR” and “coloc”. A P-value of < 0.05 was deemed statistically significant.
Results
Two-sample MR for impaired GK-GKRP interaction
Only one SNP (Additional file 1: Table S3) in GCKR gene was identified from the MAGIC (European ancestry) as the IV (F-statistic = 275) to mimic impaired GK-GKRP interaction. Genetically-proxied impaired GK-GKRP interaction was associated with reduced risk of T2D (odds ratio [OR] 0.09 per 1 mmol/L lower FPG, 95% confidence interval [CI] 0.03–0.28, P = 2.53 × 10−5) and did not influence plasma insulin level (β − 1.43 per 1 mmol/L lower FPG, 95% CI − 3.19 to 0.33, P = 0.111) (Tables 1 and 2). These expected associations confirmed the validity of our IV. We found that genetically-proxied impaired GK-GKRP interaction significantly increased TG level (β 3.54 per 1 mmol/Llower FPG, 95% CI 3.26–3.82, P = 2.29 × 10−132), LDL-C level (β 1.23 per 1 mmol/L lower FPG, 95% CI 1.08–1.38, P = 5.13 × 10−60) and ApoB level (β 1.76 per 1 mmol/L lower FPG, 95% CI 1.61–1.90, P = 1.93 × 10−121), while decreased HDL-C level (β − 0.16 per 1 mmol/L lower FPG, 95% CI − 0.30 to − 0.03, P = 0.018) (Table 1).
Table 2 shows the causal relationships with binary outcomes of interest. Genetically-proxied impaired GK-GKRP interaction was associated with increased risk of MASLD (OR 14.6 per 1 mmol/L lower FPG, 95% CI 4.57–46.4, P = 5.98 × 10−6), as well as increased risk of CAD (OR 2.92 per 1 mmol/L lower FPG, 95% CI 1.78–4.81, P = 2.44 × 10−5). We did not observe any significant causal association between genetically-proxied impaired GK-GKRP interaction and risks of PAD (OR 3.17 per 1 mmol/L lower FPG, 95% CI 0.83–12.15, P = 0.091), stroke (OR 1.25 per 1 mmol/L lower FPG, 95% CI 0.63–2.51, P = 0.522), or HF (OR 1.36 per 1 mmol/L lower FPG, 95% CI 0.78–2.39, P = 0.283).
One-sample MR validation
The SNP (GCKR rs1260326) identified in the two-sample MR analyses was utilized as the IV in our one-sample MR validation. In additional file 1, Table S4 shows the patients’ characteristics stratified by the number of GCKR rs1260326 effect allele (allele T). A total of 6072 patients were included in this one-sample MR analysis. FPG level was lower in patients with rs1260326 T allele, confirming the validity of IV selection. Plasma TG level and use of lipid-lowering drugs were higher in T allele carriers, consistent with the expected downstream effects of GK-GKRP disruption. The IV was associated with decreased FPG (βIV-exposure − 0.22 mmol/L per T allele, SE 0.06, 95% CI − 0.10 to − 0.33, P = 0.0003) in an additive model. Each copy of T allele was also associated with higher level of TG (βIV-outcome 0.15 mmol/L per T allele, SE 0.03, 95% CI 0.08–0.22, P = 1.56 × 10−5) (Additional file 1: Table S5). Other associations of IV with outcome were not significant (Additional file 1: Table S5).
The causal effects of genetically-proxied impaired GK-GKRP interaction on outcomes were estimated using the Wald-ratio method. Genetically-proxied impaired GK-GKRP interaction was causally associated with TG level (βwald 0.69 mmol/L per 1 mmol/L lower FPG, 95% CI 0.20–1.18, P = 0.005). Though not significant, the causal effects on MASLD and cardiovascular outcomes also tended toward increased risk (Table 3).
Causal effects of GK activation
Six GCK SNPs (Additional file 1: Table S6) associated with FPG were identified from the MAGIC as IVs (F-statistic = 171) to mimic GK activation. Genetically-proxied GK activation was associated with reduced risk of T2D (OR 0.31 per 1 mmol/L lower FPG, 95% CI 0.17–0.56, P = 9.88 × 10−5) and higher plasma insulin level (β 0.84 per 1 mmol/L lower FPG, 95% CI − 0.01 to 1.69, P = 0.051) (Tables 1 and 2). These expected associations confirmed the validity of the IV selection. Genetically-proxied GK activation was associated with decreased TG level (β − 0.17 mmol/L per 1 mmol/L lower FPG, 95% CI − 0.31 to − 0.02, P = 0.025) and increased HDL-C level (β 0.09 mmol/L per 1 mmol/L lower FPG, 95% CI 0.02–0.16, P = 0.007) (Table 1) but not with increased LDL-C and ApoB levels (Table 1). As for complications, genetically-proxied GK activation was associated with reduced risk of CAD (OR 0.69 per 1 mmol/L lower FPG, 95% CI 0.54–0.88, P = 0.003) and a tendency for reduced risk of HF (OR 0.77 per 1 mmol/L lower FPG, 95% CI 0.58–1.02, P = 0.071). Genetically-proxied GK activation was not causally associated with risks of MASLD (OR 0.76, 95% CI 0.42–1.39, P = 0.374), PAD (OR 0.86, 95% CI 0.42–1.76, P = 0.681) or stroke (OR 1.00, 95% CI 0.70–1.43, P = 0.995).
Posterior probabilities of colocalization analysis
We carried out colocalization analysis to test potential confounding due to LD between SNPs linked to exposure and outcomes (Table 4). The posterior probabilities suggested that genetically-proxied impaired GK-GKRP interaction shared common causal variants with most outcomes (PPH4 = 0.991 for TG; PPH4 = 1.00 for ApoB; PPH4 = 1.00 for LDL-C; PPH4 = 0.985 for MASLD; PPH4 = 0.954 for CAD), providing strong evidence for colocalization. Genetically-proxied GK activation also shared common causal variant with CAD (PPH4 = 0.685), but distinct causal variants might exist between GK activation and TG (PPH3 = 0.886).
Discussion
In this study, we utilized MR frameworks to evaluate the causal associations of impaired GK-GKRP interaction as well as GK activation with lipid traits and complications. We provided genetic evidence suggesting that impaired GK-GKRP interaction could worsen lipid profiles with increased risks of MASLD and CAD. In contrast, GK activation had minimal effects on lipid profiles with slightly lowered TG and increased HDL-C levels. GK activation also decreased the risk of CAD. These results suggested that disruption of GK-GKRP interaction, rather than GK activation per se, could worsen lipid profiles and increase risk of MASLD and cardiovascular complications.
In 2022, the first GKA (dorzagliatin) had been approved for treatment of T2D in China and so far, no data (more than 52 weeks) are available on its long-term effects. A previous MR study investigated the impact of GK activation (instrumented by GCK SNPs associated with HbA1c) on cardiovascular outcomes, suggesting its possible protective effects against CAD and HF [26]. However, it should be noted that some GKAs have been associated with dyslipidaemia, notably hypertriglyceridemia [16] although this was not reported with Dorzagliatin [23, 24]. This adverse effect is identical to the side effect of GK-GKRP disruptors. Since GKRP is exclusively expressed in the liver, it is reasonable to utilize GCKR SNPs to mimic impaired GK-GKRP interaction in hepatocytes where GK-GKRP disruption might lead to excessive glucose flux and de novo lipogenesis. Since some GKAs are known to interfere GK-GKRP interaction [13], our detailed MR analyses suggested that the risk of hypertriglyceridemia reported with some GKAs might be attributed to enhanced nuclear GK translocation to cytoplasm but not GK activation.
In the two-sample MR analysis, genetic variants located in GCKR gene associated with FPG level in the MAGIC were selected as IVs. Only one SNP, rs1260326, met the stringent selection criteria. This polymorphism (c.1337C > T; p.P446L) is a non-synonymous variant known to impair GK-GKRP interaction in the liver, with enhanced GK translocation from the GK-GKRP complex in the nucleus to the cytoplasm [45], making rs1260326 a reliable proxy. In this study, impaired GK-GKRP interaction instrumented by rs1260326 was associated with decreased risk of T2D without affecting plasma insulin level, further confirming its validity as an IV. Enhanced hepatic de novo lipogenesis due to GK-GKRP disruption may contribute to hepatic lipid accumulation, which plays an important role in the development of MASLD [22]. While cardiovascular diseases share some common cardiometabolic risk factors with MASLD, such as dyslipidaemia, through inflammation and insulin resistance, MASLD can independently promote atherosclerotic plaque formation and progression of cardiovascular disease [21, 46]. In keeping with the shift of glucose to lipid metabolism when the capacity of GK for hepatic glycogen synthesis is exceeded [15, 47], we found genetically-proxied impaired GK-GKRP interaction causally increased plasma TG, LDL-C, ApoB levels, decreased HDL-C level as well as increased risks of MASLD and CAD. Causal associations for HF, PAD and stroke tended toward increased risk, albeit not significant. We validated the effect direction derived from the two-sample MR framework by a one-sample MR analysis in a local, prospective cohort HKDR. The causal estimates in one-sample MR were directionally consistent with those observed in the two-sample MR, supporting the causal impact of GK-GKRP disruption on abnormal lipid metabolism, MASLD and CAD. Colocalization analysis has been demonstrated as an effective method for uncovering the potential pleiotropic effects of specific loci on multiple traits [37]. The PPH4 values derived from our colocalization analyses also suggested that genetically-proxied impaired GK-GKRP interaction shared common causal variants with most outcomes which showed significant association in the MR analyses, proving the absence of pleiotropic effects in IV selection.
We also evaluated impacts of GK activation on these outcomes using GCK SNPs associated with FPG as IVs. The results were overall consistent with our previous analysis using GCK SNPs associated with HbA1c as IVs [26]. In contrast to the impaired GK-GKRP interaction, GK activation did not influence the lipid traits. The slightly decreased TG level might be due to increased insulin secretion with improved clearance of TG from the circulation [48]. Taken together, we inferred that the hypertriglyceridemia risk during GKA development might be better explained by increased cytoplasm GK level in hepatocytes due to enhanced GK translocation, rather than due to up-regulation of hepatic GK activity via conformational change. In mice models fed with normal diet, GK overexpression in the liver lowered blood glucose accompanied by increased hepatic lipogenesis and circulating lipids [49]. In human liver biopsies, increased GCK mRNA expression was associated with increased mRNA expression of hepatic lipogenic enzymes and liver fat [50]. On the other hand, activating GCK mutations in mice and human were associated with increased risk of hypoglycemia but not altered circulating lipid profiles [51,52,53].
For decades, GKAs and GK-GKRP disruptors have been investigated as novel OADs. In this study, utilizing MR framework, we have dissected the different effects of GK activation and GK-GKRP disruption on lipid levels. Whilst both could lower PG, the hyperlipidemic side effects of GK-GKRP disruption might outweigh its glucose-lowering effects with possible adverse long-term outcomes including MASLD and cardiovascular disease. This inference was supported by previous findings that GCKR rs780094 (in high linkage disequilibrium with rs1260326) was associated with a modest decrease in FPG (1.9%) but a proportionately larger increase in plasma TG level (13%) [54]. These adverse effects of GK-GKRP disruption may be particularly relevant to patients with diabetes, who are prone to develop dyslipidemia, notably high TG and low HDL-C, due to insufficient insulin action [55].
We acknowledge certain limitations in our study and the MR results should be interpreted with caution. For MR study, there are three core assumptions: (1) the IV should be associated with the exposure; (2) the IV should not be associated with confounders of the exposure-outcome association; (3) the IV should not directly affect the outcome except through its influence on the exposure. However, the latter two core assumptions can never be entirely proven. Our study is a drug-target MR framework. By employing IVs in specific genes (instead of the whole genome) where their functions and relationships with the exposure are well, we can reduce the risk of confounding and pleiotropy bias.
We acknowledge that GCKR rs1260326 is a pleiotropic SNP associated with multiple traits and dozens of metabolites [19]. The latter are likely due to the downstream effects of impaired GK-GKRP interaction. Although such ‘vertical pleiotropy’ does not interfere with the interpretation of our MR analysis, given its multifaceted associations, it remains possible that rs1260326 might be related to some unknown traits independent of the causal pathway (horizontal pleiotropy).
In addition, most MR studies used GWAS data collected in mid-life, long after the random allocation of genetic variants at conception. Thus, we were unable to account for potential selection bias stemming from competing risks before enrolment, for example, for diseases that typically occur at younger ages which share the same risk factors. Besides, the reported effect sizes cannot be directly utilized to extrapolate the clinical effects of GKA or GK-GKRP disruptor treatment which will depend on the period of treatment exposure. Most randomized controlled trials only investigated the short-term effects of pharmacological treatment as opposed to the impacts of lifelong exposures estimated by MR [56]. We only validated our findings in a local cohort and larger validation studies in other ethnic groups are warranted. Moreover, our study is a biomarker (FPG) weighted drug MR analysis. Using a protein expression weighted drug MR framework to assess whether perturbation of the hepatic GK protein level could also influence the outcomes. would further strengthen our findings [57]. However, high-quality data for GK protein expression are not currently available.
Conclusion
Our genetic evidence suggested that impaired interaction of GK-GKRP complex may increase the risk of liver and cardiovascular diseases, whereas direct GK activation can protect from cardiovascular diseases. Hypertriglyceridemia observed in previous clinical trials with some GKAs are most probably due to unexpected impaired GK-GKRP interaction during GK activation. Future development of GKA needs to avoid interfering GK-GKRP interaction. These findings would need to be confirmed by definitive clinical trials.
Availability of data and materials
The summary statistics used in two-sample MR analyses are publicly available and can be accessed and downloaded through websites listed in Table S1. Due to local laws and regulations, the individual data of the HKDR cannot be shared. Further enquiries can be made to Dr. Juliana CN Chan at jchan@cuhk.edu.hk.
Abbreviations
- ApoB:
-
Apolipoprotein B
- CAD:
-
Coronary artery disease
- CI:
-
Confidence interval
- FPG:
-
Fasting plasma glucose
- GK:
-
Glucokinase
- GKA:
-
Glucokinase activator
- GKRP:
-
Glucokinase regulatory protein
- GWAS:
-
Genome-wide association studies
- HDL-C:
-
High-density lipoprotein cholesterol
- HF:
-
Heart failure
- HKDR:
-
Hong Kong Diabetes Register
- IV:
-
Instrumental variable
- IVW:
-
Inverse variance-weighted
- LDL-C:
-
Low-density lipoprotein cholesterol
- MAGIC:
-
Meta-analyses of glucose and insulin-related traits consortium
- MALSD:
-
Metabolic dysfunction-associated steatotic liver disease
- MODY:
-
Maturity-onset diabetes of the young
- MR:
-
Mendelian randomization
- NAFLD:
-
Non-alcoholic fatty liver disease
- OAD:
-
Oral antidiabetic drug
- OR:
-
Odds ratio
- PAD:
-
Peripheral arterial disease
- PHHI:
-
Persistent hyperinsulinemic hypoglycemia of infancy
- SNP:
-
Single nucleotide polymorphisms
- TG:
-
Triglycerides
- T2D:
-
Type 2 diabetes
References
Khunti K, Ceriello A, Cos X, De Block C. Achievement of guideline targets for blood pressure, lipid, and glycaemic control in type 2 diabetes: a meta-analysis. Diabetes Res Clin Pract. 2018;137:137–48.
Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: a perspective 50 years after demonstrating the presence of the enzyme in islets of langerhans. Front Physiol. 2019;10:148.
Grimsby J, Coffey JW, Dvorozniak MT, Magram J, Li G, Matschinsky FM, et al. Characterization of glucokinase regulatory protein-deficient mice. J Biol Chem. 2000;275(11):7826–31.
Velho G, Froguel P, Clement K, Pueyo ME, Rakotoambinina B, Zouali H, et al. Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. Lancet. 1992;340(8817):444–8.
Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338(4):226–30.
Zečević K, Volčanšek Š, Katsiki N, Rizzo M, Milardović TM, Stoian AP, et al. Maturity-onset diabetes of the young (MODY)—in search of ideal diagnostic criteria and precise treatment. Progress Cardiovasc Dis. 2024. https://doi.org/10.1016/j.pcad.2024.03.004.
Chow E, Wang K, Lim CKP, Tsoi STF, Fan B, Poon E, et al. Dorzagliatin, a dual-acting glucokinase activator, increases insulin secretion and glucose sensitivity in glucokinase maturity-onset diabetes of the young and recent-onset type 2 diabetes. Diabetes. 2023;72(2):299–308.
Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34(12):2560–6.
Kiyosue A, Hayashi N, Komori H, Leonsson-Zachrisson M, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;15(10):923–30.
Katz L, Manamley N, Snyder WJ, Dodds M, Agafonova N, Sierra-Johnson J, et al. AMG 151 (ARRY-403), a novel glucokinase activator, decreases fasting and postprandial glycaemia in patients with type 2 diabetes. Diabetes Obes Metab. 2016;18(2):191–5.
Vella A, Freeman JLR, Dunn I, Keller K, Buse JB, Valcarce C. Targeting hepatic glucokinase to treat diabetes with TTP399, a hepatoselective glucokinase activator. Sci Transl Med. 2019. https://doi.org/10.1126/scitranslmed.aau3441.
Zhu D, Gan S, Liu Y, Ma J, Dong X, Song W, et al. Dorzagliatin monotherapy in Chinese patients with type 2 diabetes: a dose-ranging, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Diabetes Endocrinol. 2018;6(8):627–36.
Futamura M, Hosaka H, Kadotani A, Shimazaki H, Sasaki K, Ohyama S, et al. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem. 2006;281(49):37668–74.
Hale C, Lloyd DJ, Pellacani A, Veniant MM. Molecular targeting of the GK-GKRP pathway in diabetes. Expert Opin Ther Targets. 2015;19(1):129–39.
Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–9.
Toulis KA, Nirantharakumar K, Pourzitaki C, Barnett AH, Tahrani AA. Glucokinase activators for type 2 diabetes: challenges and future developments. Drugs. 2020;80(5):467–75.
Lloyd DJ, St Jean DJ Jr, Kurzeja RJ, Wahl RC, Michelsen K, Cupples R, et al. Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors. Nature. 2013;504(7480):437–40.
Filipski KJ, Pfefferkorn JA. A patent review of glucokinase activators and disruptors of the glucokinase–glucokinase regulatory protein interaction: 2011–2014. Expert Opin Ther Pat. 2014;24(8):875–91.
Brouwers M, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: a double-edged sword? Trends Mol Med. 2015;21(10):583–94.
Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol. 2015;26(2):88–95.
Galiero R, Caturano A, Vetrano E, Cesaro A, Rinaldi L, Salvatore T, et al. Pathophysiological mechanisms and clinical evidence of relationship between Nonalcoholic fatty liver disease (NAFLD) and cardiovascular disease. Rev Cardiovasc Med. 2021;22(3):755–68.
Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci. 2016;61(5):1282–93.
Zhu D, Li X, Ma J, Zeng J, Gan S, Dong X, et al. Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2022;28(5):965–73.
Yang W, Zhu D, Gan S, Dong X, Su J, Li W, et al. Dorzagliatin add-on therapy to metformin in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2022;28(5):974–81.
Klein KR, Freeman JLR, Dunn I, Dvergsten C, Kirkman MS, Buse JB, et al. The SimpliciT1 study: a randomized, double-blind, placebo-controlled phase 1b/2 adaptive study of TTP399, a hepatoselective glucokinase activator, for adjunctive treatment of type 1 diabetes. Diabetes Care. 2021;44(4):960–8.
Wang K, Shi M, Huang C, Fan B, Luk AOY, Kong APS, et al. Evaluating the impact of glucokinase activation on risk of cardiovascular disease: a Mendelian randomisation analysis. Cardiovasc Diabetol. 2022;21(1):192.
Chen J, Spracklen CN, Marenne G, Varshney A, Corbin LJ, Luan J, et al. The trans-ancestral genomic architecture of glycemic traits. Nat Genet. 2021;53(6):840–60.
Richardson TG, Sanderson E, Palmer TM, Ala-Korpela M, Ference BA, Davey Smith G, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med. 2020;17(3): e1003062.
Ghodsian N, Abner E, Emdin CA, Gobeil É, Taba N, Haas ME, et al. Electronic health record-based genome-wide meta-analysis provides insights on the genetic architecture of non-alcoholic fatty liver disease. Cell Rep Med. 2021;2(11): 100437.
van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122(3):433–43.
Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524–37.
Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11(1):163.
Bonàs-Guarch S, Guindo-Martínez M, Miguel-Escalada I, Grarup N, Sebastian D, Rodriguez-Fos E, et al. Re-analysis of public genetic data reveals a rare X-chromosomal variant associated with type 2 diabetes. Nat Commun. 2018;9(1):321.
Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–9.
Chan JCN, Lim LL, Luk AOY, Ozaki R, Kong APS, Ma RCW, et al. From Hong Kong Diabetes Register to JADE program to RAMP-DM for data-driven actions. Diabetes Care. 2019;42(11):2022–31.
Wang K, Shi M, Yang A, Fan B, Tam CHT, Lau E, et al. GCKR and GCK polymorphisms are associated with increased risk of end-stage kidney disease in Chinese patients with type 2 diabetes: The Hong Kong Diabetes Register (1995–2019). Diabetes Res Clin Pract. 2022;193: 110118.
Zuber V, Grinberg NF, Gill D, Manipur I, Slob EAW, Patel A, et al. Combining evidence from Mendelian randomization and colocalization: review and comparison of approaches. Am J Hum Genet. 2022;109(5):767–82.
Pierce BL, Ahsan H, VanderWeele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2010;40(3):740–52.
Wald A. The fitting of straight lines if both variables are subject to error. Ann Math Stat. 1940;11(3):284–300.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–74.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2019;4:186.
Hughes RA, Davies NM, Davey Smith G, Tilling K. Selection bias when estimating average treatment effects using one-sample instrumental variable analysis. Epidemiology. 2019;30(3):350–7.
Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–906.
Rees MG, Wincovitch S, Schultz J, Waterstradt R, Beer NL, Baltrusch S, et al. Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk. Diabetologia. 2012;55(1):114–22.
Targher G, Bertolini L, Rodella S, Tessari R, Zenari L, Lippi G, et al. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care. 2007;30(8):2119–21.
Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998;14(4):263–83.
Dimitriadis G, Mitrou P, Lambadiari V, Maratou E, Raptis SA. Insulin effects in muscle and adipose tissue. Diabetes Res Clin Pract. 2011;93(Suppl 1):S52–9.
O’Doherty RM, Lehman DL, Télémaque-Potts S, Newgard CB. Metabolic impact of glucokinase overexpression in liver: lowering of blood glucose in fed rats is accompanied by hyperlipidemia. Diabetes. 1999;48(10):2022–7.
Peter A, Stefan N, Cegan A, Walenta M, Wagner S, Königsrainer A, et al. Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J Clin Endocrinol Metab. 2011;96(7):E1126–30.
Gloyn AL, Noordam K, Willemsen MA, Ellard S, Lam WW, Campbell IW, et al. Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes. 2003;52(9):2433–40.
Pino MF, Kim KA, Shelton KD, Lindner J, Odili S, Li C, et al. Glucokinase thermolability and hepatic regulatory protein binding are essential factors for predicting the blood glucose phenotype of missense mutations. J Biol Chem. 2007;282(18):13906–16.
Wabitsch M, Lahr G, Van de Bunt M, Marchant C, Lindner M, von Puttkamer J, et al. Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycaemia of infancy. Diabet Med. 2007;24(12):1393–9.
Orho-Melander M, Melander O, Guiducci C, Perez-Martinez P, Corella D, Roos C, et al. Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes. 2008;57(11):3112–21.
Adiels M, Olofsson SO, Taskinen MR, Borén J. Diabetic dyslipidaemia. Curr Opin Lipidol. 2006;17(3):238–46.
Ference BA. How to use Mendelian randomization to anticipate the results of randomized trials. Eur Heart J. 2018;39(5):360–2.
Schmidt AF, Finan C, Gordillo-Marañón M, Asselbergs FW, Freitag DF, Patel RS, et al. Genetic drug target validation using Mendelian randomisation. Nat Commun. 2020;11(1):3255.
Acknowledgements
We sincerely acknowledge the participants of each study and the researchers who made the GWAS data openly available.
Funding
No funding has been received for preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
K.W., M.S., C.L., L.C., E.C. and J.C.N.C. conceived and designed the study. R.C.W.M., A.L. and A.P.S.K. contributed to data collection of the Hong Kong Diabetes Register. K.W. and M.S. undertook the analyses. K.W. wrote the manuscript with important intellectual input from all authors. All authors gave final approval of the version to be published. E.C. and J.C.N.C. are the guarantors of this work and take responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The data used in the two-sample MR analyses are publicly available and anonymized. All contributing studies had received appropriate ethical approval and patient consent at each original GWAS study site, and can be found in the original studies. The individual-level data used in one-sample MR analyses received ethical approval from the Clinical Research Ethics Committee of the Chinese University of Hong Kong (CREC 2009-421 CREC 2019-080).
Consent for publication
Not applicable.
Competing interests
E.C. has received institutional research support from Sanofi, Medtronic, Powder Pharmaceuticals Inc. and Hua Medicine and speaker fees from Sanofi and Novartis. J.C.N.C has received research grants and/or honoraria for consultancy and/or giving lectures from Applied Therapeutics, AstraZeneca, Bayer, Boehringer Ingelheim, Celltrion, Eli-Lilly, Hua Medicine, Powder Pharmaceuticals, Merck Serono, Merck Sharp & Dohme, Novo Nordisk, Pfizer, Servier, Sanofi, Viatris and ZP Therapeutics. A.P.S.K. has received honoraria for consultancy or giving lectures from Abbott, Astra Zeneca, Bayer, Boehringer Ingelheim, Dexcom, Eli-Lilly, Kyowa Kirin, Merck Serono, Merck Sharp & Dohme, Nestle, Novo-Nordisk, Pfizer and Sanofi. R.C.W.M. has received research grants and/or honoraria for consultancy or giving lectures, from AstraZeneca, Boehringer Ingelheim, Bayer, Kyowa Kirin, Merck, Novo Nordisk, Pfizer, Roche Diagnostics and Tricida Inc. A.O.Y.L. has served as an advisory committee member for AstraZeneca, Boehringer Ingelheim, Sanofi, and Amgen, and has received research grants and travel grants from AstraZeneca, Boehringer Ingelheim, MSD, Novartis, Novo Nordisk, Sanofi, and Amgen. J.C.N.C. and R.C.W.M. hold patents for using biomarkers to predict risks of diabetes and its complications and are co-founders of GemVCare, a technology start-up initiated with support from the Hong Kong Government Innovation and Technology Commission and its Technology Start-up Support Scheme for Universities (TSSSU). K.W., C.L., L.C. are employees of Hua Medicine (Shanghai) Co., Ltd. Other authors have no relevant conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, K., Shi, M., Luk, A.O.Y. et al. Impaired GK-GKRP interaction rather than direct GK activation worsens lipid profiles and contributes to long-term complications: a Mendelian randomization study. Cardiovasc Diabetol 23, 228 (2024). https://doi.org/10.1186/s12933-024-02321-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-024-02321-z