
Abstract
Sodium-glucose co-transporter-2 inhibitors are used in the treatment of diabetes but are also emerging as cardioprotective agents in heart diseases even in the absence of type 2 diabetes. In this paper, upon providing a short overview of common pathophysiological features of diabetes, we review the clinically reported cardio- and nephroprotective potential of sodium-glucose co-transporter-2 inhibitors currently available on the market, including Dapagliflozin, Canagliflozin, and Empagliflozin. To that end, we summarize findings of clinical trials that have initially drawn attention to the drugs’ organ-protective potential, before providing an overview of their proposed mechanism of action. Since we particularly expect that their antioxidative properties will broaden the application of gliflozins from therapeutic to preventive care, special emphasis was put on this aspect.
Similar content being viewed by others

Introduction
In 2021, 537 million people worldwide (10.5% of the global population) lived with diabetes and another 541 million adults were at high risk to develop diabetes due to impaired glucose tolerance [1]. More than 90% of diabetic patients suffer from type 2 diabetes, manifested with pancreatic β-cell dysfunction and insulin resistance [1, 2]. Subsequent insulin deficiency and accompanying hyperglycemia adversely affect diverse micro- and macrovascular processes, including diabetic kidney and cardiovascular diseases.
Reduction of blood glucose has been shown to improve microvascular complications such as nephron- and neuropathy, but to be insufficient to prevent the death from cardiovascular events, the most relevant macrovascular complication of type 2 diabetes [2]. Therefore, new drug therapies offering cardioprotective properties are of great interest. One such emerging drug class are sodium-glucose co-transporter-2 (SGLT-2) inhibitors, a relatively novel anti-diabetic medication. Under conditions of functional SGLT and normoglycemia, SGLT-1 and 2 are responsible for about 3% and 97% of renal glucose reabsorption, respectively [3]. The stoichiometry of glucose transport via SGLT is one Na+ ion per glucose molecule for the high-capacity/low-affinity transporter SGLT-2 and two Na+ ions per glucose molecule, for the low-capacity/high-affinity transporter SGLT-1 [4, 5]. While SGLT-2 was so far known to be exclusively expressed in the S1 and S2 segments of the early proximal tubule’s luminal membrane (Fig. 1A), SGLT-1 is mainly located in the S3 segment and the intestine’s brush border membrane but was found in heart, liver and lung as well [6, 7]. In diabetes, the renal transport maximum for glucose can be increased by inducing tubular growth or raising the expression of SGLT-1 and 2, contributing to the state of hyperglycemia [3, 8]. Correspondingly, inhibition of SGLT, especially of SGLT-2, leads to glucose secretion via the urine (glycosuria) [9]. In 2012, the first SGLT-2 inhibitor Dapagliflozin (Fig. 1B) derived from its precursor Phlorizin (a non-specific SGLT inhibitor, Fig. 1B [8]) was approved by the European Medicines Agency (EMA) for the treatment of type 2 diabetes. Dapagliflozin was later followed by Canagliflozin and Empagliflozin (Fig. 1B) among others. Over the past years it has become increasingly recognized that SGLT-2 inhibitors exhibit tissue-protective effects beyond their systematic, glucose-lowering properties. Here (regardless of the patient’s diabetic status) kidney and heart are the organs benefitting the most. In this review, we summarize the clinically reported organ-protective potential of SGLT-2 inhibitors currently available on the market, before specifically focusing on their cardioprotective, antioxidative properties, a trait that will most likely propel this drug class from curative to preventive care.

SGLT-2 expression and inhibitor structure. (A) Localization of SGLT-1 and 2 within the proximal tubule. (B) Chemical structures of Dapa-, Cana- and Empagliflozin in the order of their approval and their precursor Phlorizin.
Renal complications related to diabetes and proposed protective mechanisms of gliflozins
According to a retrospective study by Ling et al. (2020), vascular complications in damaged organs account for 26.8% of all end-stage diabetic related deaths, being one of its leading causes. Of this percentage, 71.1% of death cases resulted from kidney failure, making it the most affected organ [10]. Attempting to cope with hyperglycemia, diabetic patients struggle with renal hyperfiltration (manifested by an increased glomerular filtration rate (GFR)) which results from high glomerular pressure and increased sodium and glucose reabsorption via SGLT-1 and 2. Here, the diabetes-driven boost in Na+ reabsorption in the proximal tubule, causes a decreased Na+ delivery to the macula densa leading to a reduced tubuloglomerular feedback (TGF), accompanied by increasing filtration rates [11]. When inhibiting this process by SGLT-2 inhibitors, sodium delivery to the macula densa is enabled and hyperfiltration rates are reduced [12]. Upon SGLT-2 inhibition, the highly increased sodium cannot be sufficiently cleared by the Na+/K+ ATPases of macula cells, resulting in an osmotic gradient leading to macula cell swelling and leakage of adenosine triphosphate (ATP) from the basolateral membrane, which is extracellularly converted to adenosine. Triggered adenosine signaling leads to Ca2+ mediated vasoconstriction, reducing blood flow and, in consequence, glomerular pressure [13, 14]. On a metabolic level, SGLT-2 inhibitors are proposed to act as stabilizers of hypoxia-inducible factor 1 alpha (HIF-1a), diverting cellular pathways from oxidative metabolism to glycolysis [13, 15]. Hypoxia represents a critical, often detrimental point in end-stage renal diseases but, though counterintuitive, strengthened HIF-1a and 2a signaling has been shown to improve the expression of oxygen-sensitive, reno-protective genes which can aid cell recovery under ischemic conditions [16, 17]. Studies in diabetic mice as well as isolated kidney cells have demonstrated the ability of Empagliflozin to reprogram renal metabolism and to improve mitochondrial function and antioxidative defense [18, 19]. Lastly, glycosuria is recognized as a signal of nutrient-deficiency — especially in kidneys — leading to the activation of autophagy. This in turn rescues the cells from damaged mitochondria and reduces overall oxidative stress burden [20]. For more detailed information on renal effects of gliflozins we direct the reader to other excellent reviews [13, 17, 20, 21].
Cardiovascular complications related to diabetes: focus on oxidative stress
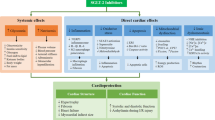
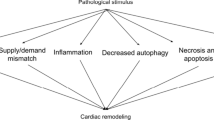
Cardiovascular events such as heart failure (HF) are multicausal, with metabolic syndrome, insulin resistance and diabetes mellitus representing major risk factors [22]. The main pathophysiology of HF is the decreased efficiency of the cardiac muscle to pump blood through the circulatory system [22]. To preserve homeostasis and contractile function in response to stress, the heart offers compensation strategies, including cell growth as well as increase in angiogenesis, energy efficiency, autophagy, and antioxidant generation [23, 24]. Though essential, these adaptive processes might result in fibrosis, altered sarcomere structure, impaired Ca2+ handling, induction of fetal gene programming, mitochondrial dysfunction, disbalance in reactive oxygen species (ROS), metabolic remodeling and cell death, potentially leading to progressive HF in the long term [23,24,25,26,27].
The heart is constantly consuming a variety of energy substrates to fuel life-long contractions. Energy metabolism is governed by multiple layers of crosstalk among metabolic pathways and cellular energy state, as well as substrate and oxygen availability [24, 28]. While glycolysis is the most important route for glucose after cellular uptake, it (and glucose metabolism overall) only contributes little to the total ATP generation in the heart. Oxidative phosphorylation (OXPHOS), utilizing the generated redox equivalents from the metabolization of various energy substrates, is responsible for 95% of ATP production in healthy cardiomyocytes [24, 29, 30]. Thus, high mitochondrial activity and an extensive number of oxidative organelles require non-enzymatic (e.g. glutathione) and enzymatic (e.g., superoxide dismutases, catalase, peroxiredoxins, thioredoxin and glutathione peroxidase systems) antioxidative attenuation of the occurring electron leakage and generated ROS, such as peroxides or free radicals [31, 32]. Under physiological conditions, ROS assert essential signaling functions for the regulation of mitochondrial activity and cellular adaptions to stressors. NADPH oxidases such as NOX4 represent a major source of ROS in cardiac cells, participating in the modulation of the immune response and mitochondrial biogenesis following exercise [31, 33]. However, prolonged exposure to oxidative stress followed by ROS accumulation leads to impaired antioxidative defense, dysregulation of redox signaling and damage to macromolecules, consequently advancing the functional decline of the myocardium [31, 34,35,36].
Heart diseases in various stages can be linked to disrupted and abnormal metabolism in cardiac cells. While healthy cardiomyocytes primarily rely on fatty acids (FA) for ATP production, with beta-oxidation and mitochondrial activity tightly synchronized, impaired cardiomyocytes during pathological remodeling may ultimately show reduced fatty acid oxidation and rely on glucose as fuel instead, as shown in Fig. 2 [24, 37]. This is considered a compensatory mechanism associated with pressure overload and energy depletion, as indicated by elevated levels of intracellular adenosine monophosphate (AMP) in the early pathogenesis of HF [24, 30, 38]. Indeed, failure to increase glucose transport and glycolysis in this AMP-activated protein kinase (AMPK) cascade-dependent way has been shown to advance disease progression in transgenic animal models [39,40,41,42]. The pleiotropic cardioprotective effects of AMPK activation have been suggested not to be fully exclusive to energy repletion [43,44,45,46,47,48]. However, prolonged activation of the glycolytic phenotype may result in decompensation and HF progression. The adaptive increase in glycolysis may not be matched in glucose oxidation and lead to an uncoupling between substrate uptake and oxidation, further impairing cellular metabolic function and redox homeostasis [24, 26, 49, 50].
In the failing heart, oxidative stress is not only linked to dysregulated cardiac metabolism but also to systemic factors such as diabetes, metabolic syndrome and ageing [34, 35]. Hyperglycemia, dyslipidemia, reduced metabolic flexibility and constant low-grade inflammation have been associated not only with increased ROS but also with excess carbonyl stress and the formation of advanced glycation and lipoxidation end-products [51]. Additional alterations of the insulin-resistant heart may result in FA-induced lipotoxicity and further uncoupling of OXPHOS. Namely, disturbance of the mitochondrial membrane structure and Ca2+ homeostasis, as well as increased leak respiration, have all been reported in diabetic heart disease [26, 50, 52]. Restoring metabolic capability, mitochondrial function, and, ultimately, energy output in (diabetic) HF may be essential to ameliorate its progression and facilitate treatment [53, 54].

The progression of heart disease can be associated with profound metabolic remodeling, mitochondrial dysfunction and disturbed redox homeostasis. These interconnected changes may lead to energy depletion, impaired cardiac function and ultimately heart failure.
Cardioprotective effects of SGLT-2 inhibitors from clinical trials
In 2015, the first long-term study on gliflozins, including 7,028 patients, was published. The aim of the EMPA-REG Outcome™ trial [55] was to determine long-term effects of Empagliflozin on cardiovascular safety in type 2 diabetes patients (≥ 18 years) with a high risk of cardiovascular events as well as its cardioprotective effects. Patients suffering from type 2 diabetes, having a glycated hemoglobin value (HbA1c) in a certain range and established cardiovascular diseases were randomly divided into three groups (1:1:1) receiving Empagliflozin (10 mg or 25 mg) or placebo once daily. The background glucose-lowering therapies were kept unchanged during the first 12 weeks but were allowed to be adjusted afterwards [55]. The results showed significant differences between the Empagliflozin and placebo groups regarding lower risk of death from cardiovascular causes, death from any causes and hospitalization due to heart failure. However, there were no significant differences evident between the doses of Empagliflozin (10 mg vs. 25 mg) [56]. Glycated hemoglobin levels were lowered in patients of the Empagliflozin groups after 12 weeks with decreasing impact after adjustment of background therapies and over time. It was further found that drug intake led to slight reduction in weight, waist circumference, uric acid level and blood pressure, systolic as well as diastolic. Levels of LDL and HDL cholesterol were slightly increased [56].
Similar results in terms of positive cardiovascular effects were obtained in a long-term study on Canagliflozin (CANVAS) in which 10,142 patients suffering from type 2 diabetes were included and randomized to receive Canagliflozin (100 mg or 300 mg) or placebo (1:1:1) [57]. However, in CANVAS, patients were either ≥ 30 years and had a history of symptomatic atherosclerotic cardiovascular disease or ≥ 50 years with at least two risk factors for cardiovascular diseases, for instance high systolic blood pressure, a history of diabetes (≥ 10 years), or being a current smoker [57]. One noticeable difference in the adverse side effects (compared to the Empagliflozin trial) was a higher risk of amputations of toe or metatarsal and increased bone fractures. It has to be emphasized though that the highest absolute risk correlated with an already existing history of amputation or peripheral vascular disease in respective patients [57].
A long-term study on Dapagliflozin (DECLARE–TIMI 58) revealed a reduced occurrence of cardiovascular death, myocardial infarction or ischemic stroke in the Dapagliflozin group compared to the placebo. However, contrary to the previous trials these findings were not significant [58]. 17,160 patients were included, suffering from type 2 diabetes, who were ≥ 40 years of age and had an established atherosclerotic cardiovascular disease (40.6%) or multiple risk factors for it (59.4%) [58] The inclusion criteria of all three trials are presented in Table 1.
More recently, in 2019 and 2020, two studies were published, investigating whether the positive cardiovascular effects of Dapagliflozin and Empagliflozin would be restricted to type 2 diabetes patients. The DAPA-HF trial included 4,744 patients (≥ 18 years) suffering from chronic heart failure with a reduced heart function of which 42% had a known diagnosis of type 2 diabetes, 3% were newly diagnosed and 55% did not have a background of diabetes. During the study, all patients received standard heart-failure device therapy as well as standard drugs [59]. Inclusion criteria for the EMPEROR-Reduced (Empagliflozin outcome trial in patients with chronic heart failure with reduced ejection fraction) trial was very similar to DAPA-HF (see Table 2) and included 3,730 participants aged ≥ 18 years, one half each diagnosed or not diagnosed with diabetes, respectively [60]. In both studies a reduced incidence for hospitalization due to HF or death from cardiovascular causes was observed upon treatment with both SGLT-2 inhibitors regardless of diabetic conditions [59, 60]. Subsequently, approvals of both drugs have been extended to an application in heart diseases even in the absence of type 2 diabetes by the EMA [61]. Moreover, renal protective effect of Empa, Dapa- and Canagliflozin was attested in the respective trials and is being further investigated (61).
Potential cardioprotective mechanisms of SGLT-2 inhibitors beyond systemic effects
The trials’ results emphasize that the cardioprotective capability of SGLT inhibitors cannot be solely explained by their glucose-lowering effect [9, 62]. Several experiments have been carried out aiming to provide insights into the underlying key mechanisms.
Treatment of cardiomyocytes from healthy rats and rabbits cultivated in medium containing 5 mM or 10 mM glucose supplemented with Empagliflozin (1 µM) reduced cytoplasmic concentrations of Na+ and Ca2+ and increased mitochondrial Ca2+ concentrations, reversing the negative effects of increased glucose concentrations [63]. Furthermore, Empagliflozin showed properties similar to those of an inhibitor of the cardiac Na+/H+ exchanger (NHE) that seemed to be independent of SGLT-2 expression and glucose [63]. Treatment of these animal cells with Empagliflozin followed by subsequently NHE inhibitor Cariporide only minimally affected the change in cytoplasmic Na+ concentration chosen as indicator for NHE inhibition [63]. The same was true for treatments in reverse order (Cariporide immediately followed by Empagliflozin) [63]. Measuring the alteration of cytoplasmic Na+ concentration and the activity of NHE in healthy mouse cardiomyocytes in the presence of Empa-, Dapa- and Canagliflozin, dosed similarly to the plasma concentrations reached by respective therapies in humans (1 µM, 1 µM and 3 µM, respectively), a decrease in cytosolic concentration of Na+ was found in all three conditions [64]. Furthermore, in silico docking studies on the extracellular Na+ binding site of NHE proved high binding affinity for all three inhibitors (-34.3 kJ/mol Empagliflozin, -32.2 kJ/mol Dapagliflozin, -37.2 kJ/mol Canagliflozin) compared to negative control glucose (-24.3 kJ/mol) [64]. Their glucoside moieties were oriented towards the Na+ binding site, the aglycone part lining the extracellular opening of that site [64]. However, Chung et al. have contradicted the direct inhibition of NHE1 by Empa-, Cana- and Dapagliflozin [65]. The inhibitors’ effects were compared with Cariporide in conditions of intracellular acidosis to evoke large NHE1 fluxes and best show inhibitory effects. While Cariporide showed an inhibited H+ flux in cardiomyocytes, this flux was not affected by the SGLT-2 inhibitors [65].
On metabolic level, the three drugs, Cana-, Empa- and Dapagliflozin, seem to exhibit differential effects. In contrast to Canagliflozin, which significantly activated AMPK in HEK293 cells and murine hepatocytes already at concentrations similar to those in human plasma when treated with the drug, Empa- and Dapagliflozin showed weaker effects even at higher therapeutic doses [66]. Increasing concentrations of Canagliflozin led to an increase in ADP:ATP ratios, suggesting reduced cellular oxygen consumption through inhibition of complex I of the respiratory chain [66]. Similar findings were made in human umbilical vein endothelial cells (HUVEC) and human vascular smooth muscle cells (HaoVSMC) [67]. A study on prediabetic male rats (absent hyperglycaemia), suffering from chronic vascular complications and renal function impairment, has shown that, among other findings, non-esterified fatty acid levels were increased upon Empagliflozin treatment, assumingly leading to the generation of ketone bodies [68]. Beyond that, a conversion from glucose to fatty acid oxidation as energy source was described [68]. Moreover, anti-inflammatory properties of Canagliflozin were observed that might be explained by AMPK activation [67]. AMPK activation accompanied by inhibited mTORC1 activity was also found upon treatment of diabetic and non-diabetic mice with Dapagliflozin, interpreted by the authors as possible explanation for its organ-protective effect [69]. Another effect of this activation was shown to be the inhibition of NFκB signaling in functional studies as well as the involvement in the inhibition of IL-6-stimulated Janus kinase-STAT signaling by directly leading to phosphorylation of Janus kinase-1 in human endothelial cells [70].
SGLT-2 inhibitors and oxidative stress
The first report on the ability of SGLT2 inhibitors to reduce oxidative stress traces back to the initial studies of their effects on hyperglycemia in type 2 diabetes [71]. Experiments in monocytes and endothelial cell lines revealed that the cell’s disfunction in hyperglycemic conditions could be improved by Empagliflozin in a glucose transport-independent mechanism, leading to an attenuated ROS accumulation [72]. The effect of Empa-, Cana- and Dapagliflozin on protease activating receptor 2 (PAR2), whose actions (the widening of blood vessels) are suggested to be compromised due to oxidative stress, were tested [71] in experiments on mouse aorta-derived endothelial cells grown under hyperglycemic (25 mM glucose) and normoglycemic (10 mM glucose) conditions [71]. Treatment with any of the three SGLT-2 inhibitors protected the endothelium from the increased glucose uptake and consequent augmented mitochondrial ROS production, sustaining function of the endothelium [71]. Empagliflozin was found to exert a two-phase effect depending on its concentration (≤ 1 µM: protection of vascular endothelial function; 5 to ≥ 10 µM: no vasodilation or smooth muscle contractile responses anymore) and to improve mitochondrial function, as indicated by a reduced proton leak measured via oxygen consumption rates [71]. Furthermore, treatment with Empagliflozin significantly decreased the amount of H2O2 in mitochondria and cytosol, alleviated oxidative parameters, such as 3-nitrotyrosine, glutathione (GSH), lipid peroxide, and reversed the pathological repression of the protein kinase G (NO-sGC-cGMP-PKG) pathway [73]. eNOS-dependent PKGIα oxidation and polymerization were significantly reduced by Empagliflozin and led to its translocation back to the cytosol [73]. Markers of oxidative stress (lipid hydroperoxide, glutathione peroxidase (GSH-Px), superoxide dismutase (SOD) and malondialdehyde (MDA)) were also investigated in tissue from diabetic mouse hearts treated and not treated with Empagliflozin. Their levels were found to be significantly higher (lipid hydroperoxide, MDA) or lower (SOD, GSH-Px) in diabetic untreated mice compared to Empagliflozin treated ones [74]. The same was observed in the atrial muscle of diabetic and non-diabetic rats [75]. Besides this, the expression of NOX4 — the highest abundant NAD(P)H oxidase isoform in cardiomyocytes — was highly increased in untreated diabetic mice [74]. Among others, the level of the transcription factor Nrf2 (NF-E2-related factor 2), that binds to the antioxidant responsive element (ARE), was markedly increased in the treated mouse heart tissue, hinting that oxidative stress levels were reduced [74]. Furthermore, fibrosis was suppressed by Empagliflozin treatment through inhibition of the transforming growth factor β (TGF-β)/SMAD pathway due to downregulation of TGF-β expression by Empagliflozin and/or elevated levels of Smad7, a negative inhibitor of the aforementioned pathway [74]. The positive effect of Empagliflozin on microvascular morphology incl. fibrosis and membrane thickening was further confirmed by treating diabetic cardiac microvascular endothelial cells (CMEC) with Empagliflozin. The high levels of mitochondrial and intracellular ROS produced by the cells could both be restricted by this treatment [76]. In addition, diabetes induced mitochondrial fission was also prevented by Empagliflozin, which brought the ratio of AMP:ATP closer to the one observed in control cells, leading to the activation of AMPK [76]. Similar findings were also reported for reperfusion injury, where Empagliflozin was able to preserve mitochondrial function through AMPK activation [77]. Markers of microvascular inflammation (ICAM-1, CVAM-1, TNF-α, IL-6) which are markedly increased in heart failure with preserved ejection fraction (HFpEF) were found to be significantly reduced after treatment of fibers from healthy and HFpEF human hearts with Empagliflozin [73] and Canagliflozin [67]. A study in non-diabetic failing pig hearts also showed that Empagliflozin positively influenced cardiac metabolism typically found in failing hearts by diverting ATP production to beta oxidation and ketogenesis, decreasing the consumption of carbohydrates and thereby approaching the conditions found in healthy hearts [9]. Proposed protective mechanisms of SGLT2 inhibitors are also summarized in Fig. 3.

Overview of SGLT2 inhibitors and their proposed mechanisms of oxidative stress relieving effects, including improvement in mitochondrial function, better electron coupling, activation of AMPK and antioxidant production as well as reduction of pro-inflammatory signaling.
Conclusion
SGLT-2 inhibitors show excellent promise for cardio-preventive care and, as demonstrated, cell culture experiments provide a good starting point in understanding how SGLT-2 inhibitors are involved in mechanisms improving cardiovascular conditions. However, most of the studies focused on hyperglycemic conditions [66, 73, 74, 76, 78] and not all were carried out in cardiomyocytes [66, 67, 76, 78]. Some of the clinical studies also searched for protein biomarkers in the blood which could tell us more regarding the underlying effects of the drugs. In case of the EMPEROR study (Empagliflozin), comparing samples taken right before the treatment’s start (baseline), after 12 weeks and 52 weeks of treatment, showed no significant differences in protein expression between the two timepoints during the treatment, hinting that the effect of Empagliflozin is time-independent. However, a small group of intracellular proteins involved in autophagic flux, weakening of oxidative stress and inflammation processes in the heart as well as promotion of repair and regeneration in heart and kidney were found to be differentially expressed upon treatment with Empagliflozin [79]. This finding potentially explains the often-described positive effects of gliflozins on these two organs. The highest increase in protein abundance was found for insulin-like growth factor-binding protein (IGFBP1), transferrin receptor protein 1 (Tfr1) and erythropoietin (EPO), which is functionally related to hemoglobin increase, presumably one of the major benefits of SGLT2 inhibitors for heart and renal function [79].
Overall, both in vitro and in vivo studies agree in one thing: SGLT-2 inhibitors are able to protect our heart independent of their glucose lowering effects. However, more research is ahead of us before we are able to comprehensively decipher their direct and indirect cellular targets and corresponding mechanism of action, in order to maximize their therapeutic potential.
Data Availability
Not applicable.
Abbreviations
- AMP:
-
Adenosine monophosphate
- AMPK:
-
AMP-kinase
- ARE:
-
Antioxidant responsive element
- ATP:
-
Adenosine triphosphate
- CMEC:
-
Cardiac microvascular endothelial cells
- CV:
-
Cardiovascular
- EMA:
-
European Medicines Agency
- EPO:
-
Erythropoietin
- FA:
-
Fatty acids
- GFR:
-
Glomerular filtration rate
- GSH(-Px):
-
Glutathione (peroxidase)
- HaoVSMC:
-
Human vascular smooth muscle cells
- HbA1c :
-
Glycated hemoglobin value
- HDL:
-
High-density lipoprotein
- HF:
-
Heart failure
- HFpEF:
-
Heart failure with preserved ejection fraction
- HIF-1a:
-
Hypoxia-inducible factor 1 alpha
- HUVEC:
-
Human umbilical vein endothelial cells
- IGFBP1:
-
Insulin-like growth factor-binding protein
- LDL:
-
Low density lipoprotein
- MDA:
-
Malondialdehyde
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NHE:
-
Na+/H+ exchanger
- Nrf2:
-
NF-E2-related factor 2
- NYHA:
-
New York Heart Association
- OXPHOS:
-
Oxidative phosphorylation
- PAR2:
-
Protease activating receptor 2
- PKG:
-
Protein kinase G
- ROS:
-
Reactive oxygen species
- SGLT:
-
Sodium-glucose co-transporter
- SOD:
-
Superoxide dismutase
- Tfr1:
-
Transferrin receptor protein 1
- TGF:
-
Tubuloglomerular feedback
- TGF-β:
-
Transforming growth factor β
References
Diabetes Atlas. https://diabetesatlas.org/. Accessed 22 September 2022.
Chatterjee S, Khunti K, Davies MJ. Type 2 diabetes. The Lancet. 2017 Jun;389(10085):2239–51.
Rieg T, Vallon V. Development of SGLT1 and SGLT2 inhibitors.Diabetologia. 2018 Oct22;61(10):2079–86.
Ehrenkranz JRL, Lewis NG, Ronald Kahn C, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005 Jan;21(1):31–8.
Mackenzie B, Loo DDF, Panayotova-Heiermann M, Wright EM. Biophysical characteristics of the pig kidney Na+/Glucose cotransporter SGLT2 reveal a common mechanism for SGLT1 and SGLT2. J Biol Chem. 1996 Dec;271(51):32678–83.
Wright EM. SGLT2 Inhibitors: Physiology and Pharmacology. Kidney360. 2021 Dec 30;2(12):2027–37.
Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012 Jan;14(1):83–90.
Aguillón AR, Mascarello A, Segretti ND, de Azevedo HFZ, Guimaraes CRW, Miranda LSM et al. Synthetic Strategies toward SGLT2 Inhibitors.Org Process Res Dev. 2018 Apr20;22(4):467–88.
Zelniker TA, Braunwald E. Mechanisms of Cardiorenal Effects of Sodium-Glucose cotransporter 2 inhibitors. J Am Coll Cardiol. 2020 Feb;75(4):422–34.
Ling W, Huang Y, Huang YM, Fan RR, Sui Y, Zhao HL. Global trend of diabetes mortality attributed to vascular complications, 2000–2016. Cardiovasc Diabetol. 2020 Dec;20(1):182.
Hallow KM, Gebremichael Y, Helmlinger G, Vallon V. Primary proximal tubule hyperreabsorption and impaired tubular transport counterregulation determine glomerular hyperfiltration in diabetes: a modeling analysis. American Journal of Physiology-Renal Physiology. 2017 May 1;312(5):F819–35.
Wilcox CS. Antihypertensive and renal mechanisms of SGLT2 (sodium-Glucose linked transporter 2) inhibitors. Hypertension. 2020 Apr;75(4):894–901.
Bailey CJ, Day C, Bellary S. Renal protection with SGLT2 inhibitors: Effects in Acute and chronic kidney disease. Curr Diab Rep. 2022 Jan;22(3):39–52.
Thomson SC, Vallon V. Effects of SGLT2 inhibitor and dietary NaCl on glomerular hemodynamics assessed by micropuncture in diabetic rats. American Journal of Physiology-Renal Physiology. 2021 May 1;320(5):F761–71.
Packer M. Mechanisms leading to Differential Hypoxia-Inducible factor signaling in the Diabetic kidney: modulation by SGLT2 inhibitors and Hypoxia Mimetics. Am J Kidney Dis. 2021 Feb;77(2):280–6.
Nangaku M. Chronic hypoxia and Tubulointerstitial Injury: A final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006 Jan;17(1):17–25.
Packer M. Role of Deranged Energy Deprivation Signaling in the pathogenesis of Cardiac and Renal Disease in States of Perceived Nutrient Overabundance. Circulation. 2020 Jun;23(25):2095–105.
Jo HA, Seo JH, Lee S, Yu M, yeon, Bae E, Kim DK, et al. Metabolomic profiling in kidney cells treated with a sodium glucose-cotransporter 2 inhibitor. Sci Rep. 2023 Feb;4(1):2026.
Lu YP, Zhang ZY, Wu HW, Fang LJ, Hu B, Tang C, et al. SGLT2 inhibitors improve kidney function and morphology by regulating renal metabolic reprogramming in mice with diabetic kidney disease. J Transl Med. 2022 Sep;14(1):420.
Sen T, Heerspink HJL. A kidney perspective on the mechanism of action of sodium glucose co-transporter 2 inhibitors.Cell Metab. 2021Apr;33(4):732–9.
Packer M. Critical reanalysis of the Mechanisms underlying the Cardiorenal benefits of SGLT2 inhibitors and reaffirmation of the nutrient Deprivation Signaling/Autophagy hypothesis. Circulation. 2022 Nov;146(18):1383–405.
Davis RC. ABC of heart failure: history and epidemiology. BMJ. 2000 Jan;1(7226):39–42.
Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy.Nat Rev Cardiol. 2018 Jul19;15(7):387–407.
Tran DH, Wang Z. v. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J Am Heart Assoc. 2019 Jun 18;8(12): e012673.
Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y et al. Inducible Overexpression of GLUT1 Prevents Mitochondrial Dysfunction and Attenuates Structural Remodeling in Pressure Overload but Does Not Prevent Left Ventricular Dysfunction. J Am Heart Assoc. 2013 Sep 26;2(5): e000301.
Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, et al. Cardiac insulin-resistance and decreased mitochondrial Energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail. 2013 Sep;6(5):1039–48.
Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE et al. Transient Receptor Potential Channels Contribute to Pathological Structural and Functional Remodeling After Myocardial Infarction.Circ Res. 2014 Aug29;115(6):567–80.
Kolwicz SC, Purohit S, Tian R. Cardiac Metabolism and its Interactions With Contraction, Growth, and Survival of Cardiomyocytes.Circ Res. 2013 Aug16;113(5):603–16.
Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac Energy Metabolism in Heart failure. Circ Res. 2021 May;14(10):1487–513.
Beer M, Seyfarth T, Sandstede J, Landschütz W, Lipke C, Köstler H, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with 31P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002 Oct;40(7):1267–74.
Tomin T, Schittmayer M, Honeder S, Heininger C, Birner-Gruenberger R. Irreversible oxidative post-translational modifications in heart disease.Expert Rev Proteomics. 2019 Aug3;16(8):681–93.
Duarte-Jurado AP, Gopar-Cuevas Y, Saucedo-Cardenas O, Loera-Arias M, de Montes-de-Oca-Luna J, Garcia-Garcia R et al. A,. Antioxidant Therapeutics in Parkinson’s Disease: Current Challenges and Opportunities. Antioxidants. 2021 Mar 15;10(3):453.
Gray SP, Shah AM, Smyrnias I. NADPH oxidase 4 and its role in the cardiovascular system. Vascular Biology. 2019 Aug;12(1):H59–66.
Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure.Journal of Clinical Investigation. 2005 Mar1;115(3):500–8.
Izzo C, Vitillo P, di Pietro P, Visco V, Strianese A, Virtuoso N et al. The Role of Oxidative Stress in Cardiovascular Aging and Cardiovascular Diseases.Life. 2021 Jan15;11(1):60.
Tomin T, Schittmayer M, Sedej S, Bugger H, Gollmer J, Honeder S et al. Mass Spectrometry-Based Redox and Protein Profiling of Failing Human Hearts. Int J Mol Sci. 2021 Feb 11;22(4):1787.
Marsin AS, Bertrand† L, Rider MH, Deprez J, Beauloye C, Vincent‡ MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000 Oct;10(20):1247–55.
Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure.Cardiovasc Res. 2011 May1;90(2):202–9.
Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, et al. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. 2004 Nov;44(5):662–7.
Wang J, Xu J, Wang Q, Brainard RE, Watson LJ, Jones SP, et al. Reduced Cardiac Fructose 2,6 Bisphosphate increases hypertrophy and decreases glycolysis following aortic constriction. PLoS ONE. 2013 Jan;7(1):e53951.
Wang Q, Donthi R, Wang J, Lange AJ, Watson LJ, Jones SP, et al. Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am J Physiol Heart Circ Physiol. 2008 Jun;294(6):H2889–97.
Donthi R, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN. Cardiac expression of kinase-deficient 6-Phosphofructo-2-kinase/Fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem. 2004 Nov;279(46):48085–90.
Kim TT, Dyck JRB. Is AMPK the savior of the failing heart? Trends in Endocrinology & Metabolism. 2015 Jan;26(1):40–8.
Li X, Liu J, Lu Q, Ren D, Sun X, Rousselle T et al. AMPK: a therapeutic target of heart failure—not only metabolism regulation. Biosci Rep. 2019 Jan 31;39(1): BSR20181767.
Gélinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation.Nat Commun. 2018 Jan25;9(1):374.
Zhang F, Liu L, Xie Y, Wang J, Chen X, Zheng S et al. Cardiac contractility modulation ameliorates myocardial metabolic remodeling in a rabbit model of chronic heart failure through activation of AMPK and PPAR-α pathway.Open Medicine. 2022 Feb22;17(1):365–74.
Shu H, Hang W, Peng Y, Nie J, Wu L, Zhang W et al. Trimetazidine Attenuates Heart Failure by Improving Myocardial Metabolism via AMPK.Front Pharmacol. 2021 Sep 15;12: 707399.
Koyani CN, Plastira I, Sourij H, Hallström S, Schmidt A, Rainer PP, et al. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol Res. 2020 Aug;158:104870.
Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters.Cardiovasc Res. 2017 Mar15;113(4):411–21.
Makrecka-Kuka M, Liepinsh E, Murray AJ, Lemieux H, Dambrova M, Tepp K et al. Altered mitochondrial metabolism in the insulin‐resistant heart. Acta Physiologica. 2020 Mar 30;228(3):e13430.
Iacobini C, Vitale M, Haxhi J, Pesce C, Pugliese G, Menini S. Food-Related Carbonyl Stress in Cardiometabolic and Cancer Risk Linked to Unhealthy Modern Diet. Nutrients. 2022 Mar 3;14(5):1061.
Zhao R, Jiang S, Zhang L, Yu Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review).Int J Mol Med. 2019 Jul;44(1):3-15.
Kiyuna LA, Albuquerque RP e, Chen CH, Mochly-Rosen D, Ferreira JCB. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic Biol Med. 2018 Dec;129:155–68.
Noordali H, Loudon BL, Frenneaux MP, Madhani M. Cardiac metabolism — a promising therapeutic target for heart failure. Pharmacol Ther. 2018 Feb;182:95–114.
Zinman B, Inzucchi SE, Lachin JM, Wanner C, Ferrari R, Fitchett D, et al. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME™). Cardiovasc Diabetol. 2014;13(1):102.
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, Cardiovascular Outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015 Nov;26(22):2117–28.
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes.New England Journal of Medicine. 2017 Aug17;377(7):644–57.
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes.New England Journal of Medicine. 2019 Jan24;380(4):347–57.
McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction.New England Journal of Medicine. 2019 Nov21;381(21):1995–2008.
Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure.New England Journal of Medicine. 2020 Oct8;383(15):1413–24.
Rauch-Kröhnert U, Landmesser U. Gliflozine – in Zukunft Kardioprotektiva? Internist (Berl). 2021 Jul 23;62(7):786–95.
Ni L, Yuan C, Chen G, Zhang C, Wu X. SGLT2i: beyond the glucose-lowering effect.Cardiovasc Diabetol. 2020 Dec26;19(1):98.
Baartscheer A, Schumacher CA, Wüst RCI, Fiolet JWT, Stienen GJM, Coronel R et al. Empagliflozin decreases myocardial cytoplasmic Na + through inhibition of the cardiac Na+/H + exchanger in rats and rabbits.Diabetologia. 2017 Mar17;60(3):568–73.
Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na+/H + exchanger, lowering of cytosolic Na + and vasodilation. Diabetologia. 2018 Mar 2;61(3):722–6.
Chung YJ, Park KC, Tokar S, Eykyn TR, Fuller W, Pavlovic D, et al. Off-target effects of sodium-glucose co-transporter 2 blockers: empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+]i in the heart. Cardiovasc Res. 2021 Dec 17;117(14):2794–806.
Hawley SA, Ford RJ, Smith BK, Gowans GJ, Mancini SJ, Pitt RD et al. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes. 2016 Sep 1;65(9):2784–94.
Mancini SJ, Boyd D, Katwan OJ, Strembitska A, Almabrouk TA, Kennedy S, et al. Canagliflozin inhibits interleukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and -independent mechanisms. Sci Rep. 2018 Dec;27(1):5276.
Huttl M, Markova I, Miklankova D, Oliyarnyk O, Trnovska J, Kucera J et al. Metabolic cardio- and reno-protective effects of empagliflozin in a prediabetic rat model.J Physiol Pharmacol.2020 Oct;71(5): 635–645.
Kogot-Levin A, Riahi Y, Abramovich I, Mosenzon O, Agranovich B, Kadosh L et al. Mapping the metabolic reprogramming induced by sodium-glucose cotransporter 2 inhibition.JCI Insight. 2023 Apr 10;8(7):e164296.
Salt IP, Hardie DG. AMP-Activated Protein Kinase.Circ Res. 2017 May26;120(11):1825–41.
El-Daly M, Pulakazhi Venu VK, Saifeddine M, Mihara K, Kang S, Fedak PWM, et al. Hyperglycaemic impairment of PAR2-mediated vasodilation: Prevention by inhibition of aortic endothelial sodium-glucose-co-transporter-2 and minimizing oxidative stress. Vascul Pharmacol. 2018 Oct;109:56–71.
Semo D, Obergassel J, Dorenkamp M, Hemling P, Strutz J, Hiden U et al. The Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Empagliflozin Reverses Hyperglycemia-Induced Monocyte and Endothelial Dysfunction Primarily through Glucose Transport-Independent but Redox-Dependent Mechanisms. J Clin Med. 2023 Feb 8;12(4):1356.
Kolijn D, Pabel S, Tian Y, Lódi M, Herwig M, Carrizzo A et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. 2021 Jan 21;117(2):495–507.
Li C, Zhang J, Xue M, Li X, Han F, Liu X, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019 Dec;2(1):15.
Koizumi T, Watanabe M, Yokota T, Tsuda M, Handa H, Koya J et al. Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats.Front Cardiovasc Med. 2023 Feb 6;10.
Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018 May;15:335–46.
Cai C, Guo Z, Chang X, Li Z, Wu F, He J, et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 2022 Jun;52:102288.
Yaribeygi H, Atkin SL, Butler AE, Sahebkar A. Sodium–glucose cotransporter inhibitors and oxidative stress: An update.J Cell Physiol. 2019 Apr15;234(4):3231–7.
Zannad F, Ferreira JP, Butler J, Filippatos G, Januzzi JL, Sumin M et al. Effect of empagliflozin on circulating proteomics in heart failure: mechanistic insights into the EMPEROR programme. Eur Heart J. 2022 Dec 21;43(48):4991–5002.
Acknowledgements
Some of the schematic illustrations were done in BioRender (purchased license).
Funding
Open access funding provided by Austrian Science Fund (FWF). We thank the TU Wien and the European Union’s Horizon 2020 Marie Sk?odowska-Curie COFUND doctoral program ENROL-Engineering for Life Sciences (grant 101034277), the Austrian Science Fund FWF, grant numbers F73 (SFB “Lipid hydrolysis”) and W1226 (Doctoral School “DK-Metabolic and Cardiovascular Disease”) and the city of Vienna (grant H-867676/2022) for financial support.
Author information
Authors and Affiliations
Contributions
JH wrote the first draft and all authors contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hoehlschen, J., Hofreither, D., Tomin, T. et al. Redox-driven cardioprotective effects of sodium-glucose co-transporter-2 inhibitors: comparative review. Cardiovasc Diabetol 22, 101 (2023). https://doi.org/10.1186/s12933-023-01822-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-023-01822-7