
Abstract
Background
Glucokinase activators (GKAs) are an emerging class of glucose lowering drugs that activate the glucose-sensing enzyme glucokinase (GK). Pending formal cardiovascular outcome trials, we applied two-sample Mendelian randomisation (MR) to investigate the impact of GK activation on risk of cardiovascular diseases.
Methods
We used independent genetic variants in or around the glucokinase gene meanwhile associated with HbA1c at genome-wide significance (P < 5 × 10−8) in the Meta-Analyses of Glucose and Insulin-related traits Consortium study (N = 146,806; European ancestry) as instrumental variables (IVs) to mimic the effects of GK activation. We assessed the association between genetically proxied GK activation and the risk of coronary artery disease (CAD; 122,733 cases and 424,528 controls), peripheral arterial disease (PAD; 7098 cases and 206,541 controls), stroke (40,585 cases and 406,111 controls) and heart failure (HF; 47,309 cases and 930,014 controls), using genome-wide association study summary statistics of these outcomes in Europeans. We compared the effect estimates of genetically proxied GK activation with estimates of genetically proxied lower HbA1c on the same outcomes. We repeated our MR analyses in East Asians as validation.
Results
Genetically proxied GK activation was associated with reduced risk of CAD (OR 0.38 per 1% lower HbA1c, 95% CI 0.29–0.51, P = 8.77 × 10−11) and HF (OR 0.54 per 1% lower HbA1c, 95% CI 0.41–0.73, P = 3.55 × 10−5). The genetically proxied protective effects of GKA on CAD and HF exceeded those due to non-targeted HbA1c lowering. There was no causal relationship between genetically proxied GK activation and risk of PAD or stroke. The estimates in sensitivity analyses and in East Asians were generally consistent.
Conclusions
GKAs may protect against CAD and HF which needs confirmation by long-term clinical trials.
Similar content being viewed by others

Background
The increasing burden of diabetes is a major healthcare concern globally. The large proportion of patients with suboptimal glycaemic control [1] who are at high risk for multiple complications calls for novel treatment strategies. Glucokinase (GK) serves as a glucose sensor which converts glucose to glucose-6-phosphate, the first step towards ATP production [2]. The latter is essential for insulin secretion which promotes peripheral glucose uptake for cellular metabolism or storage as glycogen. Alpha-cell GK also suppresses glucose-regulated glucagon secretion [3]. The regulatory importance of GK is evidenced by maturity onset diabetes of the young type 2 (MODY2) due to inactivating mutations in this enzyme [4].
Many glucokinase activators (GKAs) have been designed and tested as new target-specific glucose-lowering drugs since 2003 [5]. These small molecules can bind to an allosteric site in the enzyme and facilitate GK activation by stabilizing a high-affinity conformation of GK to glucose. This allosteric site harbours most of the activating mutations implicated in genetic conditions such as congenital hyperinsulinism [5]. The binding of GKA to GK can improve the enzymatic kinetics and alter glucose sensitivity. Depending on the site of action, GKAs are further divided into dual-acting pancreatic and hepatic GKA and liver-selective GKA [6]. Within pancreatic β-cells, GKA facilitates glucokinase activation and enhances pancreatic glucose-stimulated insulin secretion. In the liver, GKA could activate glucokinase both directly and by dissociating the GK-GKRP (GK regulatory protein) complex to promote hepatic glucose uptake and glycogen synthesis [7].
Several GKAs have shown promising glucose-lowering effect in patients with type 2 diabetes. For example, dorzagliatin, a dual pancreatic and hepatic allosteric GKA lowered HbA1c by 1.07% (95% confidence interval [CI] − 1.19 to − 0.95) at a dose of 75 mg twice daily versus placebo which reduced HbA1c by –0.50% (95% CI –0.68 to –0.32) during a 24-week of double-blind treatment in 463 drug-naive patients with type 2 diabetes in a phase 3 study [8]. Meanwhile, a liver-selective GKA, TTP-339, produced a placebo-subtracted 0.9% (95% CI − 1.5 to − 0.3%) reduction in HbA1c over 6 months in 42 patients with type 2 diabetes at a dose of 800 mg once daily [9].
Cardiovascular diseases (CVDs) are major complications amongst patients with type 2 diabetes [10]. Early intensive blood-glucose lowering can protect diabetic populations from cardiovascular complications [11]. Nevertheless, glucose-lowering drugs may also act via other pleiotropic pathways that affect the cardiovascular safety, including those that are associated with an increased (e.g., rosiglitazone) or reduced (e.g., glucagon-like peptide-1 receptor agonists and sodium-glucose co-transporter-2 inhibitors) risk of CVD, as well as those that have neutral effects (e.g., dipeptidyl peptidase-4 inhibitors) [12].
Given the colossal cost in drug development and clinical trials, Mendelian randomisation (MR) provides a tool, where researchers can leverage genetic variants that are randomly allocated at conception as proxies to investigate the causal effect of an exposure on an outcome [13]. Moreover, MR studies are now increasingly applied to infer health effects of medications by proxying drug effects using variants located in the target genes [14]. Some examples include the use of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) variants to mimic long-term treatment effects of HMGCR inhibitors (statins) [15] and the use of glucagon like peptide 1 receptor (GLP1R) variants to mimic that of GLP1R agonists [16].
With several GKAs in late phase clinical trials, in this study we utilized the MR framework to investigate the potential effects of GK activation by GKA on CVDs.
Methods
Study design
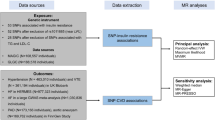
Figure 1 and Additional file 1: Figure S1 illustrate the conceptual framework of our study. MR study is based on the three core assumptions: (1) instrumental variables (IVs) must be strongly associated with the exposure (typically P < 5 × 10−8); (2) IVs affect the outcome only through exposure and are not directly associated with the outcome; (3) IVs are not correlated with known exposure–outcome confounders. In the main analysis, we employed genetic variants located in or near the GCK gene as IVs of GK activation and evaluated the causal effects of genetically proxied GK activation on risk of CVDs using two-sample MR. We further performed MR analysis of genetically predicted lower HbA1c as a proxy of non-targeted HbA1c lowering, to assess whether the causal relevance of GKA and outcomes was contributed solely by its glucose-lowering effect. Throughout the study, we utilized the summary-level genetic data from published genome-wide association studies (GWAS) in MR analyses (Additional file 1: Table S1). All data used in the present work are publicly available and anonymized. All contributing studies had received appropriate ethical approval and patient consent.

Directed acyclic graph of MR. MR is based on the three core assumptions: (1) IVs must be strongly associated with the exposure (typically P < 5 × 10−8); (2) IVs affect the outcome only through exposure and are not directly associated with the outcome; (3) IVs are not correlated with known exposure–outcome confounders. MR, Mendelian randomisation; IV, instrumental variable; SNP, single-nucleotide polymorphism; GK, glucokinase
Identifying IVs for GK activation
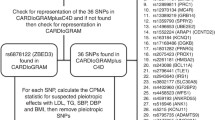
The GCK gene encodes GK which is the protein target of GKA. For selection of IVs, we used single-nucleotide polymorphisms (SNPs) in or around GCK that were significantly associated with HbA1c in the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) study [17]. In the main analysis, we selected genetic variants within 100 kb downstream and upstream of GCK (genomic position on build GRCh37.p13: chromosome 7: 44182812–44229038) gene associated with HbA1c (%, per 1% equals to per 11 mmol/mol) at a genome-wide level of statistical significance (P ≤ 5 × 10−8) and in low linkage disequilibrium (r2 < 0.3) in MAGIC. SNPs were selected by linkage disequilibrium clumping algorithm in PLINK (r2 threshold = 0.3, window size = 1000 kb) retaining the one with the smallest P-value. The 1000 Genomes European data were used as the reference for linkage disequilibrium estimation. The GWAS data used in the main analysis were restricted to those derived from subjects of European ancestry (N = 146,806) to avoid biases from population stratification.
Outcome data
In this study, we focused on CVDs including coronary artery disease (CAD), peripheral arterial disease (PAD), stroke and heart failure (HF) as defined by the respective Consortiums in the original GWAS studies. We used the plasma insulin level and incident type 2 diabetes as positive controls given their expected associations with GKA use.
For CAD, we used summary statistics including 122,733 cases and 424,528 controls from a meta-analysis of CAD GWAS based on the CARDIOGRAMplusC4D Consortium and the UK BioBank [18]. Summary statistics for PAD (7098 cases and 206,541 controls) were obtained from the FinnGen Consortium. Summary statistics for stroke (40,585 cases and 406,111 controls) were obtained from the MEGASTROKE Consortium [19]. Summary statistics for HF (47,309 cases and 930,014 controls) were obtained from the Heart Failure Molecular Epidemiology for Therapeutic Targets (HERMES) Consortium [20]. Summary statistics for type 2 diabetes (12,931 cases and 57,196 controls) were obtained from the 70KforT2D study [21]. Summary statistics for plasma insulin level (3301 subjects) were obtained from a GWAS published by Sun et al. [22]. All participants were European ancestry and non-overlapping with those in the MAGIC. The detailed information of disease diagnoses and definitions can be found in the original publications or Consortium websites and were also briefly summarized in Additional file 1: Supplementary method.
Sensitivity analysis
To assess the robustness of associations detected in the main analysis, we applied more stringent genetic variant selection criteria to exclude potential invalid instruments as sensitivity analyses. Specifically, we selected uncorrelated genetic variants (r2 threshold = 0.01, window size = 1000 kb, European 1000 Genome Project as reference panel) located completely in the gene body of GCK (genomic position on build GRCh37.p13: chromosome 7: 44182812–44229038) and were significantly associated with HbA1c (P ≤ 5 × 10−8) in MAGIC (restricted to European ancestry). This was followed by two-sample MR analyses on the CVDs as previously defined.
Replication in Asian population
We assessed the robustness of effect direction and statistical significance of the causal relationship between genetically proxied GK activation and CVD by repeating the same MR analyses in East Asians. We selected the IVs based on the associations derived from East Asian population (N = 33,307) in the MAGIC. For replication, we extracted SNPs within 100 kb downstream and upstream of GCK gene significantly associated (P ≤ 5 × 10−8) with HbA1c (%) and in low linkage disequilibrium (r2 threshold = 0.3, window size = 1000 kb, East Asian 1000 Genome Project as reference panel). We collected the East-Asian-specific summary statistics of CVDs from Biobank Japan [23, 24].
Comparing effects of genetically proxied GK activation with genetically predicted lower HbA1c (non-targeted HbA1c lowering)
To assess whether the cardiovascular effects of GK activation were different from those of non-targeted HbA1c lowering, we also estimated the causal associations of non-targeted HbA1c lowering with the outcomes. We used genetic variants associated with HbA1c throughout the genome in addition to the GCK gene region in MAGIC (European ancestry, P ≤ 5 × 10−8, r2 threshold = 0.001, window size = 10000 kb, European 1000 Genome Project as reference panel) to mimic non-targeted HbA1c lowering. The two-sample MR analyses were implemented on the outcomes with significant causal associations in the main analyses. We then estimated the magnitude of difference (βdiff) by taking the difference between the MR beta coefficients for genetically proxied GK activation and genetically predicted lower HbA1c (βGK activation − βLower HbA1c) [16]. The standard error for βdiff (SEdiff) was derived using the propagation of error method as follows:
where SEGK activation and SELower HbA1c are the standard errors of the MR estimates for the associations of genetically proxied GK activation and genetically predicted lower HbA1c with the respective outcomes.
Statistical analysis
To validate the instrumental strength for MR analyses, we calculated the F-statistic to check if it exceeded the empirical threshold 10 [25]. We switched the effect allele of each variant to the HbA1c-decreasing allele to align with the expected effect of GKA. We harmonized the genetic associations of IVs with the exposures and outcomes by aligning effect alleles. We excluded palindromic variants with intermediate frequencies due to uncertainty in identifying the effect allele on the same strand in the two datasets [26]. The Wald ratio (the ratio of the genetic association with outcome to the genetic association of exposure) for each SNP was calculated.
We assessed the causal relationships by combining the Wald ratio using the random-effects inverse variance-weighted (IVW) method [27]. For the results indicating possible causal relationships in the IVW model, we performed additional analyses using sensitivity MR methods. These included Cochran’s Q statistic to measure heterogeneity among IVs [28], the weighted median method to provide consistent estimates if more than half of the genetic variants were valid IVs [29], the MR-Egger regression to detect horizontal pleiotropy [30], the MR Pleiotropy RESidual Sum and Outlier (MR-PRESSO) method to remove horizontal pleiotropy by detecting and correcting for potential outliers [31].
We carried out colocalization analysis to assess the validity of the instrumental variable assumptions, in case that the exposure and outcome might be causally influenced by distinct variants that happen to be in linkage disequilibrium [32]. We used the “coloc” package to quantify the probability of shared causal variants across exposure and outcomes that showed significant causality. This package uses approximate Bayes factor (ABF) computation to generate posterior probabilities (PP) with 5 exclusive hypotheses: (i) neither trait has a genetic association (PPH0); (ii) only trait 1 has a genetic association (PPH1); (iii) only trait 2 has a genetic association (PPH2); (iv) both traits are associated but with different causal variants (PPH3); (v) both traits are associated and share a single causal variant (PPH4). Colocalization analysis was performed by generating ± 100 kb windows from the GCK gene region.
All analyses were performed using R 4.1.2 software with the R packages “TwoSampleMR”, “MRPRESSO” and “coloc”. A P-value of < 0.05 was deemed statistically significant.
Results
Main analyses
We identified seventeen SNPs (F-statistic = 94) in or around GCK gene associated with HbA1c (%) from MAGIC (European ancestry) as IVs for GK activation (Additional file 1: Table S2). As positive controls, we confirmed the genetically proxied GK activation were associated with higher plasma insulin level (beta 1.52 per 1% lower HbA1c, 95% CI 0.65–2.38, P = 0.0006) and lower risk of type 2 diabetes (odds ratio [OR] 0.09 per 1% lower HbA1c, 95% CI 0.05–0.17, P = 2.76 × 10−15; Table 1).
Genetically proxied GK activation were associated with decreased risk of CAD (OR 0.38 per 1% lower HbA1c, 95% CI 0.29–0.51, P = 8.77 × 10−11) and HF (OR 0.54 per 1% lower HbA1c, 95% CI 0.41–0.73, P = 3.55 × 10−5; Fig. 2). There was no significant heterogeneity in the IVW model (PHeterogeneity = 0.292 for CAD, and 0.752 for HF) nor horizontal pleiotropy in MR-Egger regression (PEgger-intercept = 0.067 for CAD, and 0.498 for HF; Table 2). No horizontal pleiotropy and outliers were detected in the MR-PRESSO model (PGlobal-test = 0.266 for CAD, and 0.777 for HF). Results from weighted median model indicated that more than half of the genetic variants were valid IVs and the estimates were similar as those estimated by the IVW method (ORweighted-median for CAD 0.39, 95% CI 0.27–0.57, Pweighted-median = 9.56 × 10−7; ORweighted-median for HF 0.51, 95% CI 0.34–0.76, Pweighted-median = 8.62 × 10−4).

Associations of genetically proxied GK activation with risks of CAD, HF, PAD and stroke. The population was restricted to European ancestry. All estimations were based on the inverse variance weighted method. 1% lower HbA1c equals to 11 mmol/mol lower. OR, odds ratio; CI, confidence interval; GK, glucokinase; SNP, single-nucleotide polymorphism; CAD, coronary artery disease; HF, heart failure; PAD, peripheral arterial disease
We performed colocalization analysis to assess potential confounding due to linkage disequilibrium (Additional file 1: Table S3). The posterior probability that genetically proxied GK activation and CAD or HF shared different causal variants was low (PPH3 = 0.052 for CAD; PPH3 = 0.017 for HF), though we also did not have enough evidence to support colocalization (PPH4 = 0.190 for CAD; PPH4 = 0.048 for HF). Of note, the probability of [PPH4/(PPH3 + PPH4)] which represents the probability of colocalization conditional on the presence of a causal variant for outcomes provided some evidence for colocalization (0.785 for CAD; 0.738 for HF).
We did not observe causal relationship between genetically proxied GK activation and PAD (OR 1.17, 95% CI 0.59–2.33, P = 0.659) or stroke (OR 0.96, 95% CI 0.68–1.35, P = 0.810; Fig. 2).
Sensitivity analyses
As sensitivity analyses, we identified two uncorrelated GCK variants (F-statistic = 207) significantly associated with HbA1c (%) in MAGIC (European ancestry) as IVs for GK activation (Additional file 1: Table S4). The results of sensitivity analyses were overall consistent with those of main analyses. GK activation proxied by the two variants remained significantly associated with reduced risk of CAD (OR 0.43, 95% CI 0.26–0.71, P = 0.001) and the association was slightly attenuated for HF (OR 0.56, 95% CI 0.31–1.02, P = 0.056; Fig. 2). There was no heterogeneity in the IVW model (PHeterogeneity = 0.915 for CAD, and 0.466 for HF). We did not observe causal relationship in sensitivity analyses between genetically proxied GK activation and PAD (OR 0.89, 95% CI 0.14–5.79, P = 0.901) or stroke (OR 0.92, 95% CI 0.38–2.23, P = 0.857; Fig. 2).
Replication analyses in Asian population
For replication in Asian population, we identified three SNPs (F-statistic = 56) associated with HbA1c (%) as the IVs (Additional file 1: Table S5). GK activation proxied by the three SNPs were significantly associated with reduced risk of CAD (OR 0.47, 95% CI 0.28–0.80, P = 0.005). The estimates of HF risk (OR 0.67, 95% CI 0.22–2.08, P = 0.493) was directionally consistent with the results in European population, albeit not significant due to fewer cases in Biobank Japan (Additional file 1: Table S6). No causal relationship was observed between genetically proxied GK activation and PAD (OR 1.01, 95% CI 0.27–3.83, P = 0.987) or stroke (OR 0.84, 95% CI 0.44–1.59, P = 0.594; Additional file 1: Table S6).
Comparisons of genetically proxied GK activation with genetically predicted lower HbA1c (non-targeted HbA1c lowering)
We identified 75 genome-wide SNPs (Additional file 1: Table S7) associated with HbA1c in MAGIC (European ancestry, P ≤ 5 × 10−8, r2 < 0.001) as IVs for non-targeted HbA1c lowering (Fig. 3). Genetically predicted lower HbA1c was associated with a decreased risk of type 2 diabetes (OR 0.28 per 1% lower HbA1c, 95% CI 0.11–0.71, P = 0.008) and the effect size was smaller in magnitude than the estimate of genetically proxied GK activation, albeit not significant (Pdifference = 0.061). There was no causal relationship between genetically predicted lower HbA1c and insulin level (beta 1.52 [0.65, 2.38] versus − 0.12 [− 0.58, 0.34] per 1% lower HbA1c, Pdifference = 0.001), confirming the validity of the IVs selected for GK activation. Though genetically predicted lower HbA1c also reduced risk of CAD (OR 0.76 per 1% lower HbA1c, 95% CI 0.58–0.98, P = 0.035), the risk-reducing effect size for CAD by genetically proxied GK activation was two-fold larger than that of genetically predicted lower HbA1c (Pdifference = 0.0006). We did not observe causal relationship between genetically predicted lower HbA1c and HF (OR 0.97 per 1% lower HbA1c, 95% CI 0.79–1.21, P = 0.810). We observed similar results after excluding the GCK variants (rs2908277 and rs2971670) from the 75 SNPs that mimicked non-targeted HbA1c lowering (Additional file 1: Table S8).

Comparisons of effects of genetically proxied GK activation and genetically predicted lower HbA1c. All estimations were based on the inverse variance weighted method. The population was restricted to European ancestry. 1% lower HbA1c equals to 11 mmol/mol lower. OR, odds ratio; CI, confidence interval; GK, glucokinase; SNP, single-nucleotide polymorphism; T2D, type 2 diabetes; CAD, coronary artery disease; HF, heart failure
Discussion
In this study, we investigated the causal relationships between GK activation and CVDs using MR approach. We provided genetic evidence inferring that GK activation by GKA treatment may reduce risks of CAD and HF. Upon comparison, the effect of GK activation exceeded that proxied by non-targeted HbA1c lowering. Taken together, our findings suggested that GK-targeted glucose-lowering medications may have stronger protective effects on CAD and HF than non-targeted glucose-lowering regimens. Our results provide the premise for designing RCTs of GKA in cardiovascular protection.
Since GKAs are not yet available in the market, there are no observational studies to support the potential long-term effects of GKA treatment. Compared with observational studies, MR design is less susceptible to confounding and time-related biases by leveraging the random allocation of genetic variants at conception by nature. Although we found genetically predicted lower HbA1c was also associated with reduced risk of CAD which was in line with previous MR analysis [33], there is heterogeneity regarding the cardiovascular effects of glucose-lowering drugs in clinical trials. Not all glucose-lowering drugs prevented these complications and some thiazolidinediones were associated with increased cardiovascular risk [12].
Of note, some of GKAs had shown adverse effects in clinical trials. One is higher incidence of hypoglycaemia events due to overstimulation of pancreatic GK, which affect hepato-selective GKAs to a lesser extent. Hypoglycemia has been associated with an increased risk of cardiovascular events [34] via a number of potential mechanisms [35]. In 52-week trials of the dual-acting GKA dorzagliatin in drug-naïve and metformin treated Chinese patients with type 2 diabetes versus placebo, the reported incidence of clinically significant hypoglycaemia (< 3.0 mmol/L) was low (0.3% and 0.8% respectively) with no severe hypoglycaemic events [8, 36]. Overstimulation of hepatic GK may lead to hypertriglyceridemia [37, 38], which could increase the risk of CVDs. In phase 3 trials of dorzagliatin among metformin-treated T2D patients, hypertriglyceridemia related to the study drug was higher in the intervention group versus placebo (2 vs. 0.5%) [36]. Overall, the potential adverse effects of GKA on hypoglycemia and lipids may depend on the degree and balance of pancreatic and hepatic GK activation.
Our study had several strengths. We applied different MR sensitivity methods to test the robustness of our findings against different MR assumptions and replicated our findings in European and East Asian populations. However, we acknowledge certain limitations and recommend that our results should be interpreted with appropriate caution. First, although our work provided a robust indication of significance and effect direction, the effect estimates reported in this study do not directly reflect the clinical effect sizes or health impacts of GKA treatment on cardiovascular events. The magnitude by which the reported effect estimates approximated true effects would depend on the degree by which genetically-proxied GK activation mimicked the true GKA effect on HbA1c reduction. Moreover, compared to RCTs that investigate short-term pharmacological treatment.
GKAs include dual-acting pancreatic and hepatic GKA and liver-selective GKA. In the current study, we were not able to select organ-specific GCK variants associated with HbA1c to distinguish the effects of pancreatic and hepatic GK activation. Therefore, our results might better proxy dual-acting GKA than the liver-selective GKA. In response to rising blood glucose, hepatic GK can be released from GK-GKRP complex for glucose uptake and glycogen synthesis in the liver [39]. It has been reported that GKA can influence the interaction of GK and GKRP and enhance GK translocation [7]. To this end, analysis of genetically predicted HbA1c/glucose reduction instrumented by GCKR SNPs might provide better approximation to mimic liver-selective GKA. However, we were not able to identify any GCKR SNPs that were associated with HbA1c at a genome-wide level of statistical significance in the MAGIC. Of note, studies have shown a common functional variant in GCKR (rs1260326) associated with increased translocation of GK from GK-GKRP complex, lower fasting plasma glucose, but with increased de novo lipogenesis, non-alcoholic fatty liver disease (NAFLD) and higher serum triglycerides [40], as well as an increased risk of CAD [41]. Therefore, GCKR SNPs should be also considered in some way in the future analyses.
We did not observe causal relationships between genetically proxied GK activation and PAD or stroke, which could be due to few number of events in the original GWAS, an absence of true causal associations or the bias in our IV selection. We only proxied the glycaemic effects of GKAs using the HbA1c-associated SNPs in GCK, but GKAs might exert effects on CVD through other mechanisms which were not fully captured by our selected IVs. In addition, MR studies of possible drug effects may be subject to selection bias due to inclusion of survivors with certain genetic make-up of the outcomes under investigation. MR studies usually utilise GWAS recruited in mid-life well after the random allocation of genetic variants at conception. Therefore, we were not able to exclude potential selection bias from competing risk before recruitment for diseases which share the same risk factors that typically occur at younger ages, which may attenuate the effect estimates in the analyses.
Finally, our study was different from a drug-target MR analysis, where the primary aim was to assess whether perturbation of the protein (or functional protein) level of certain drug target could affect the outcomes [42]. This is mainly because GKAs activate GK by changing the conformation of the enzyme rather than regulating its protein expression. Nevertheless, a drug-target MR on GCK or GKRP warrants future investigation to explore the alternative druggable potentials of the GK-related pathways for treating both diabetes and cardiovascular complications.
Conclusions
In conclusion, we provided genetic evidence suggesting that GK activation by GKA treatment might reduce risks of CAD and HF. Patients with type 2 diabetes at high risk of cardiovascular complications may benefit from this novel glucose-lowering drug although RCTs are required to confirm these findings.
Availability of data and materials
All summary statistics used in the present work are publicly available, and can be accessed and downloaded through websites listed in Table S1.
Abbreviations
- CAD:
-
Coronary artery disease
- CVD:
-
Cardiovascular disease
- GK:
-
Glucokinase
- GKA:
-
Glucokinase activator
- GKRP:
-
Glucokinase regulatory protein
- GLP1R:
-
Glucagon like peptide 1 receptor
- GWAS:
-
Genome-wide association studies
- HF:
-
Heart failure
- HMGCR:
-
3-Hydroxy-3-methylglutaryl-CoA reductase
- IV:
-
Instrumental variable
- IVW:
-
Inverse variance-weighted
- MAGIC:
-
Meta-Analyses of Glucose and Insulin-related traits Consortium
- MR:
-
Mendelian randomisation
- MR-PRESSO:
-
MR Pleiotropy RESidual Sum and Outlier
- PAD:
-
Peripheral arterial disease
References
Khunti K, Ceriello A, Cos X, De Block C. Achievement of guideline targets for blood pressure, lipid, and glycaemic control in type 2 diabetes: a meta-analysis. Diabetes Res Clin Pract. 2018;137:137–48.
Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: a perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front Physiol. 2019;10:148.
Basco D, Zhang Q, Salehi A, Tarasov A, Dolci W, Herrera P, et al. α-Cell glucokinase suppresses glucose-regulated glucagon secretion. Nat Commun. 2018;9(1):546.
Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanne-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30(11):1512–26.
Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301(5631):370–3.
Sarabu R, Berthel SJ, Kester RF, Tilley JW. Glucokinase activators as new type 2 diabetes therapeutic agents. Expert Opin Ther Pat. 2008;18(7):759–68.
Futamura M, Hosaka H, Kadotani A, Shimazaki H, Sasaki K, Ohyama S, et al. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem. 2006;281(49):37668–74.
Zhu D, Li X, Ma J, Zeng J, Gan S, Dong X, et al. Dorzagliatin in drug-naive patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2022.
Vella A, Freeman JLR, Dunn I, Keller K, Buse JB, Valcarce C. Targeting hepatic glucokinase to treat diabetes with TTP399, a hepatoselective glucokinase activator. Science translational medicine. 2019;11(475).
Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88–98.
Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321(7258):405–12.
Zhu J, Yu X, Zheng Y, Li J, Wang Y, Lin Y, et al. Association of glucose-lowering medications with cardiovascular outcomes: an umbrella review and evidence map. Lancet Diabetes Endocrinol. 2020;8(3):192–205.
Smith GD, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Evans DM, Davey SG. Mendelian randomization: new applications in the coming age of hypothesis-free causality. Annu Rev Genomics Hum Genet. 2015;16:327–50.
Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med. 2016;375(22):2144–53.
Daghlas I, Karhunen V, Ray D, Zuber V, Burgess S, Tsao PS, et al. Genetic evidence for repurposing of GLP1R (Glucagon-like peptide-1 receptor) agonists to prevent heart failure. J Am Heart Assoc. 2021;10(13): e020331.
Chen J, Spracklen CN, Marenne G, Varshney A, Corbin LJ, Luan J, et al. The trans-ancestral genomic architecture of glycemic traits. Nat Genet. 2021;53(6):840–60.
van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122(3):433–43.
Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524–37.
Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11(1):163.
Bonàs-Guarch S, Guindo-Martínez M, Miguel-Escalada I, Grarup N, Sebastian D, Rodriguez-Fos E, et al. Re-analysis of public genetic data reveals a rare X-chromosomal variant associated with type 2 diabetes. Nat Commun. 2018;9(1):321.
Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–9.
Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53(10):1415–24.
Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat Genet. 2020;52(7):669–79.
Pierce BL, Ahsan H, VanderWeele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2010;40(3):740–52.
Hartwig FP, Davies NM, Hemani G, Davey SG. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol. 2016;45(6):1717–26.
Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–906.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–74.
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28(1):30–42.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Zuber V, Grinberg NF, Gill D, Manipur I, Slob EAW, Patel A, et al. Combining evidence from Mendelian randomization and colocalization: review and comparison of approaches. Am J Hum Genet. 2022;109(5):767–82.
Au Yeung SL, Luo S, Schooling CM. The impact of glycated hemoglobin (HbA(1c)) on cardiovascular disease risk: a Mendelian randomization study using UK Biobank. Diabetes Care. 2018;41(9):1991–7.
Amiel SA, Aschner P, Childs B, Cryer PE, De Galan BE, Frier BM, et al. Hypoglycaemia, cardiovascular disease, and mortality in diabetes: epidemiology, pathogenesis, and management. Lancet Diabetes Endocrinol. 2019;7(5):385–96.
Chow E, Iqbal A, Walkinshaw E, Phoenix F, Macdonald IA, Storey RF, et al. Prolonged prothrombotic effects of antecedent hypoglycemia in individuals with type 2 diabetes. Diabetes Care. 2018;41(12):2625–33.
Yang W, Zhu D, Gan S, Dong X, Su J, Li W, et al. Dorzagliatin add-on therapy to metformin in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2022;28(5):974–81.
Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34(12):2560–6.
Katz L, Manamley N, Snyder WJ, Dodds M, Agafonova N, Sierra-Johnson J, et al. AMG 151 (ARRY-403), a novel glucokinase activator, decreases fasting and postprandial glycaemia in patients with type 2 diabetes. Diabetes Obes Metab. 2016;18(2):191–5.
Van Schaftingen E, Detheux M, Veiga da Cunha M. Short-term control of glucokinase activity: role of a regulatory protein. FASEB J. 1994;8(6):414–9.
Brouwers M, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: a double-edged sword? Trends Mol Med. 2015;21(10):583–94.
Simons P, Simons N, Stehouwer CDA, Schalkwijk CG, Schaper NC, Brouwers M. Association of common gene variants in glucokinase regulatory protein with cardiorenal disease: a systematic review and meta-analysis. PLoS ONE. 2018;13(10): e0206174.
Schmidt AF, Finan C, Gordillo-Marañón M, Asselbergs FW, Freitag DF, Patel RS, et al. Genetic drug target validation using Mendelian randomisation. Nat Commun. 2020;11(1):3255.
Acknowledgements
Summary statistics were contributed by MAGIC, CARDIOGRAMplusC4D Consortium, FinnGen Consortium, MEGASTROKE consortium, HERMES Consortium, 70KforT2D study, International Consortium of Blood Pressure, UK BioBank and BioBank Japan. We truly acknowledge the participants of each study and the researchers who made the GWAS data openly available.
Funding
No funding has been received for preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
KW, MS, EC and JCNC conceived and designed the study. KW, CH and BF collected data. KW, MS, CH and BF undertook analyses with feedback from EC and JCNC. KW, EC and JCNC interpreted the results. KW wrote the manuscript with critical comments and revise by EC, JCNC, MS, RCWM, AOYL, APSK, CH and BF. All authors gave final approval of the version to be published. EC is the guarantor of this work and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All data used in the present work are publicly available and anonymized. All contributing studies had received appropriate ethical approval and patient consent at each original GWAS study site, and can be found in the original studies.
Consent for publication
Not applicable.
Competing interests
EC has received institutional research support from Sanofi, Medtronic, Powder Pharmaceuticals Inc. and Hua Medicine and speaker fees for Sanofi and Novartis. JCNC has received research grants and/or honoraria for consultancy and/or giving lectures from AstraZeneca, Bayer, Boehringer Ingelheim, Celltrion, Eli-Lilly, Hua Medicine, Lee Powder, Merck Serono, Merck Sharp & Dohme, Pfizer, Servier, Sanofi and Viatris, holds patents for using biomarkers to predict risks of diabetes and its complications and is a co-founder of GemVCare, a biotechnology company partially supported by the Hong Kong Government startup fund. RCWM is a co-founder of GemVCare, a technology start-up initiated with support from the Hong Kong Government Innovation and Technology Commission and its Technology Start-up Support Scheme for Universities (TSSSU). Other authors have no relevant conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Table S1. Information of included summary-level statistics. Table S2. Associations of instrumental variables for GK activation in main analyses with exposure and outcomes. Table S3. Colocalization analysis of genetically proxied GK activation and outcomes. Table S4. Associations of instrumental variables for GK activation in sensitivity analyses with exposure and outcomes. Table S5. Instrumental variables for GK activation in East Asian population and their associations with HbA1c. Table S6. Associations of genetically proxied GK activation with risks of cardiovascular outcomes in East Asian population. Table S7. Instrumental variables for non-targeted HbA1c lowering and their associations with HbA1c. Table S8. Associations of genetically predicted lower HbA1c with outcomes after removing GCK variants. Figure S1. Conceptual framework of study design. Supplementary Method. Brief summary of outcome definition.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, K., Shi, M., Huang, C. et al. Evaluating the impact of glucokinase activation on risk of cardiovascular disease: a Mendelian randomisation analysis. Cardiovasc Diabetol 21, 192 (2022). https://doi.org/10.1186/s12933-022-01613-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-022-01613-6