
Abstract
Platelet hyper-reactivity is a crucial cause of accelerated atherosclerosis increasing risk of thrombotic vascular events in diabetic patients. The mechanisms leading to abnormal platelet activity during diabetes are complex and not fully defined. The current study attempted to clarify the role of CTRP9, a novel adiponectin paralog, in enhanced platelet activity and determined whether CTRP9 may inhibit platelet activity. Adult male C57BL/6 J mice were randomized to receive high-fat diet (HFD) or normal diet (ND). 8 weeks after HFD, animals were sacrificed, and both plasma CTRP9 and platelet aggregation were determined. HFD-fed animals increased weight gain significantly, and became hyperglycemic and hyperinsulinemic 8 weeks post-HFD. Compared to ND animals, HFD animals exhibited significantly decreased plasma CTRP9 concentration and increased platelet response to ADP, evidenced by augmented aggregation amplitude, steeper aggregation slope, larger area under the curve, and shorter lag time (P < 0.01). A significant negative correlation between plasma CTRP9 concentration and platelet aggregation amplitude was observed. More importantly, in vitro pre-treatment with CTRP9 significantly inhibited ADP-stimulated platelet activation in platelet samples from both ND and HFD animals. Taken together, our results suggest reduced plasma CTRP9 concentration during diabetes plays a causative role in platelet hyper-activity, contributing to platelet-induced cardiovascular damage during this pathologic condition. Enhancing CTRP9 production and/or exogenous supplementation of CTRP9 may protect against diabetic cardiovascular injury via inhibition of abnormal platelet activity.
Similar content being viewed by others
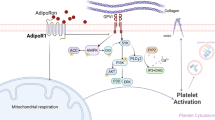

Background
Anti-platelet treatment, such as acetylsalicylic acid and aspirin, reduces cardiovascular morbidity and mortality [1]. It is well known type 2 diabetic patients are at very high cardiovascular risk [2], and should thereby potentially benefit significantly from anti-platelet treatment. Unfortunately, recent large clinical trials have demonstrated acetylsalicylic acid therapy less effectively prevents cardiovascular events in diabetic patients compared to normoglycemic individuals [3]. Moreover, concomitant type 2 diabetes increases the risk of high on-aspirin platelet reactivity (HPR), defined as inadequate inhibition of platelet function [4]. The pathologic mechanisms leading to enhanced platelet activity and reduced platelet response to therapeutic interventions in type 2 diabetes are complex and remain incompletely understood.
An adipokine secreted by adipose tissue [5, 6], adiponectin contains a stalk with 22 collagen repeats and a highly conserved globular domain. Typically present in plasma at concentrations up to 30 µg/ml, adiponectin is markedly down-regulated in association with obesity-linked diseases such as coronary artery disease and type 2 diabetes [7]. Clinical observations have revealed that plasma total adiponectin concentrations are inversely correlated with myocardial infarction risk [8, 9]. Moreover, adiponectin has been reported to have potent anti-platelet action, reducing vascular injury caused by abnormal thrombolysis [10–12]. Adiponectin knockout mice have been generated and studied by many groups. When metabolically challenged (e.g., high-fat diet), adiponectin-null mice develop insulin resistance, endothelial dysfunction, and vascular injury [13]. However, in the absence of dietary or metabolic stress, these animals show a relatively modest phenotype, suggesting potent compensatory mechanisms are in place.
Recently, a highly conserved family of adiponectin paralogs has been discovered, and designated the C1q tumor necrosis factor (TNF) related proteins (CTRPs). Exhibiting a similar structure as APN, each CTRP consists of four distinct domains including a N-terminal signal peptide, a short variable domain, a collagen-like domain, and a C-terminal C1q-like globular domain [14, 15]. Of all CTRPs identified to date, CTRP9 shares the greatest amino acid overlap with APN in its globular C1q domain [16]. We recently demonstrated that CTRP9 is an endothelium-dependent vasodilator and CTRP9 treatment attenuates diabetes-induced endothelial dysfunction [17]. Moreover, others and we have recently demonstrated CTRP9 protects the heart against acute ischemic/reperfusion injury [18, 19]. Most importantly, a recent study demonstrated that, distinctly different from adiponectin-knockout (which lacks phenotypic change under physiologic conditions), CTRP9-knockout mice gain more weight with normal diet and develop spontaneous insulin resistance and type 2 diabetes [20]. In contrast, CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction [21]. These results suggest CTRP9 likely possesses more important metabolic regulatory function than other adipokines. However, the role of CTRP9 in dysregulated platelet hemostasis under diabetic conditions has not been previously investigated.
Therefore, the aims of this study were: (1) to determine the relationship between plasma CTRP9 alteration and platelet aggregation in high-fat diet induced diabetic animals, and (2) to investigate the effect of CTRP9 upon platelet aggregation.
Methods
High-fat diet induced diabetes
We utilized a previously established high-fat diet induced type 2 diabetes model to mimic Western diet-induced obesity/diabetes in human [22]. In brief, adult (8-week old) male C57BL/6 J mice were randomized to receive high-fat diet (HFD, 60 % kcal, Research Diets Inc. D12492i) or normal diet (ND, 10 % kcal control, D12450Bi) containing the same protein content as HFD. All experiments in this study were performed in adherence with the National Institutes of Health Guidelines on the Use of Laboratory Animals, and were approved by the Fourth Military Medical University Committee on Animal Care.
Metabolic characterization
Mice were fasted overnight by removal to a clean cage without food at the end of their dark (feeding) cycle, approximately 6 p.m.. Mice were weighed at 8 a.m. the next morning. 30 µl blood was obtained via tail clip to assess plasma glucose (Accu-Chek Active Blood Glucose Monitoring System, Roche Diagnostics, Indianapolis, IN), plasma insulin (ELISA, Linco, Billerica, MA) and plasma CTRP9 (ELISA, Aviscera Bioscience, Santa Clara, CA). Weight and plasma measurements were recorded initially and every other week thereafter. The Homeostatic Model Assessment-Insulin Resistance (HOMA-IR), a surrogate measure of insulin resistance, was calculated initially and weekly thereafter, via HOMA calculator v2.3 (University of Oxford).
Platelet preparation
Blood was obtained by intracardiac puncture from HFD or ND mice. Blood was drawn into polypropylene syringes containing one-tenth volume of 0.11 M sodium citrate, and centrifuged at 80 g for 10 min to obtain the platelet rich plasma (PRP), or 2400 g for 20 min to obtain platelet poor plasma (PPP).
Platelet aggregation assay
500 µl of reference (PPP) or samples (PRP) was added to P/N 312 cuvettes (Chronolog Corp. Havertown, PA), and inserted into aggregometer wells (Chronolog Corp). 10 µM ADP reagent (Chronolog Corp) was added to the samples to induce platelet aggregation. To determine the effect of CTRP9 on platelet aggregation, recombinant murine CTRP9 (4 µg/ml, Aviscera Bioscience, Santa Clara, CA) was added in PRP for 20 min at 37 °C before ADP addition. CTRP9 is highly conserved throughout evolution. Mouse CTRP9 and its corresponding human ortholog share 100, 85, and 89 % amino acid identity in their short N-terminal variable regions, collagen domains, and C-terminal globular domains, respectively [16]. The amplitude, slope, area under the curve, and lag time of platelet aggregation was calculated automatically by manufacturer-provided software (Chronolog Corp).
Statistical analysis
All values in the text and figures are presented as the mean ± SEM of n independent experiments. Data were analyzed by unpaired t test with GraphPad Prism 6 statistic software (La Jolla, CA). P values less than or equal to 0.05 (2-sided) were considered statistically significant.
Results
Plasma CTRP9 levels were significantly reduced in HFD induced type-2 diabetic mice
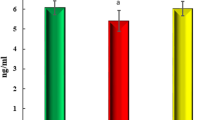
To confirm establishment of type 2 diabetes by HFD, body weight, plasma glucose, and insulin concentration were determined. HFD mice had greater body weight increase compared to normal diet (ND) (Fig. 1). After 2 weeks, fasting plasma glucose increased steadily in HFD mice. 8 weeks post-HFD, fasting plasma insulin was significantly increased (Fig. 2a) and plasma CTRP9 concentration was significantly reduced (Fig. 2b).

High fat diet (HFD) caused significantly greater weight gain (a) and hyperglycemia (b). N = 15 group. *P < 0.05, **P < 0.01 vs. normal diet (ND)

HFD significantly increased plasma insulin concentration (a) and HOMA-IR (b), and reduced plasma CTRP9 concentration (c). N = 15 group. *P < 0.05, **P < 0.01 vs. ND
Platelet aggregation is significantly increased in diabetic animals and negatively correlated with plasma CTRP9
No significant difference was observed in total platelet counts between the two groups (987 ± 36 × 103/µl vs. 966 ± 41 × 103/µl). As illustrated in Fig. 3a and summarized in Fig. 3b, ADP-stimulated platelet aggregation was significantly increased in HFD mice samples. Importantly, a statistically significant negative correlation (P < 0.01) between plasma CTRP9 concentration and platelet aggregation was observed (Fig. 3c). To gain more insight into platelet activity alteration during diabetes, platelet aggregation slope, area under the curve, and lag time (reaction time after ADP addition) were determined. In HFD platelet samples, platelet aggregation slope and area under the curve were significantly increased, whereas the lag time was significantly reduced, compared to ND (Fig. 4), suggesting reduced plasma CTRP9 levels may contribute to abnormal platelet activity in diabetic animals.

ADP-stimulated platelet aggregation is significantly increased in HFD animals. a Typical platelet aggregation tracing. b Bar graph summarizing data from 15 animals/group. c Significant negative correlation between plasma CTRP9 concentration and platelet aggregation was observed. *P < 0.05, **P < 0.01 vs. ND

Both the platelet aggregation slope (a) and area under the curve (b) were significantly increased, but the lag time (c) was significantly reduced in platelet samples isolated from HFD animals. **P < 0.01 vs. ND
Recombinant CTRP9 significantly inhibited diabetic platelet aggregation
To gain more direct evidence that CTRP9 may inhibit platelet function, the effect of in vitro CTRP9 pre-treatment upon ADP-induced platelet aggregation was determined. CTRP9 pre-treatment significantly reduced platelet function as evidenced by reduced platelet aggregation amplitude, decreased platelet aggregation slope, smaller area under the curve, and increased lag time in response to ADP (Fig. 5).

In vitro CTRP9 pre-treatment significantly reduced platelet function as evidenced by reduced platelet aggregation amplitude (a), decreased platelet aggregation slope (b), smaller area under the curve (c), and increased lag time in response to ADP (d). *P < 0.05, **P < 0.01 vs. respective control (without CTRP9 treatment)
Discussion
The current study made several significant observations. First, we demonstrated that plasma concentration of CTRP9, a novel cardioprotective adipokine, is significantly reduced in HFD-induced obesity/diabetic animals. This finding is consistent from a recent study reported by Peterson et al. [21]. Second, we provided the first evidence that platelet aggregation is negatively associated with plasma CTRP9 concentration. Third, we demonstrated for the first time that CTRP9 is a potent anti-platelet adipokine.
The role of platelets in thrombus formation is well known. Previous studies have reported an association between platelet activation and the degree of insulin resistance reflected by HOMA-IR [23]. Platelet hyper-reactivity is one of the most important causes of accelerated atherosclerosis and increased thrombotic risk in diabetic patients [24], contributing to a 2- to 4-fold increased coronary artery disease risk. During platelet activation, arachidonic acid is released from membrane phospholipids, which is oxygenated into thromboxane A2, a potent pro-aggregatory and vaso-constricting compound. Moreover, a previous study has shown that increased platelet aggregation is already detectable in diabetic patients without apparent vascular complications [25], indicating abnormal platelet reactivity may play a causative role in diabetic vascular morbidity and mortality. Therefore, identifying the molecular mechanisms responsible for abnormal platelet activity during diabetes may have great translational benefit.
Adiponectin is the most extensively investigated cardioprotective adipokine [26–29]. Hypoadiponectinemia correlates with increased AMI risk [30–32], and poorer cardiac functional recovery after MI with reperfusion [33, 34]. Exogenous APN supplementation significantly protects the heart against ischemic injury [35, 36]. However, the cardioprotective effects of APN are significantly attenuated in diabetic animals [22]. Moreover, complete APN abrogation results in only mild phenotypic change, unless pathologically challenged (e.g., by high-fat diet or ischemia), suggesting existent overlapping regulators. Efforts to identify such regulators have led to the discovery of a family of APN paralogs, designated the C1q/TNF-related proteins (CTRP1–CTRP15) [14, 37–40]. Although the CTRP family member roster has rapidly grown since their initial discovery 9 years ago, the biological functions of CTRPs have only been realized in recent years [41–44]. Thus far, most published studies focus upon the beneficial metabolic-regulatory functions of CTRPs [15, 16, 45]. Among these studies, CTRP12 is an insulin-sensitizing, anti-inflammatory adipokine, downregulated by obesity [46].
In the current study, we demonstrated plasma CTRP9 concentration is negatively correlated with ADP-induced platelet aggregation. Moreover, we demonstrated that in vitro CTRP9 pre-treatment significantly inhibited platelet activity. It should be indicated that in vitro CTRP9 concentration utilized in this study to inhibit ADP-induced platelet aggregation is much higher than in vivo physiological concentration. This concentration was selected because in vitro platelet aggregation was induced by high concentration of ADP which causes much stronger platelet aggregation than that seen in vivo. As such, higher concentration of CTRP9 is required to achieve a significant inhibition. More importantly, CTRP9 is the closest paralog of adiponectin which circulates in plasma at concentration greater than 10 µg/ml. As adiponectin concentration is significantly reduced in diabetic individuals but biological response to exogenous adiponectin is significantly impaired, supplementation of CTRP9 at super-pharmacological concentration, such as that used in our in vitro platelet inhibition experiment, may represent an effective therapeutic intervention against diabetic pathology.
Taken together, our results suggest that reduced plasma CTRP9 concentration during diabetes plays a causative role in platelet hyper-activity, contributing to platelet-induced cardiovascular damage. Enhancing CTRP9 production and/or exogenous supplementation of CTRP9 may protect against diabetic cardiovascular injury via inhibition of abnormal platelet activity.
Conclusion
Our results show that diabetes induces reduced plasma CTRP9 concentration, which plays a causative role in platelet hyper-activity and subsequent platelet-induced cardiovascular damage during this pathologic condition. Supplementation of exogenous CTRP9 may provide a protection against diabetes induced cardiovascular injury via inhibition of platelet hyper-reactivity.
References
Picker SM. Platelet function in ischemic heart disease. J Cardiovasc Pharmacol. 2013;61(2):166–74.
Soma P, Pretorius E. Interplay between ultrastructural findings and atherothrombotic complications in type 2 diabetes mellitus. Cardiovasc Diabetol. 2015;14:96.
Antithrombotic Trialists Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71–86.
Christensen KH, Grove EL, Wurtz M, Kristensen SD, Hvas AM. Reduced antiplatelet effect of aspirin during 24 hours in patients with coronary artery disease and type 2 diabetes. Platelets. 2015;26(3):230–5.
Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13(2):84–9.
Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care. 2003;26(8):2442–50.
Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, Nishida M, Kihara S, Sakai N, Nakajima T, Hasegawa K, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Hanafusa T, Matsuzawa Y. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20(6):1595–9.
Kumada M, Kihara S, Sumitsuji S, Kawamoto T, Matsumoto S, Ouchi N, Arita Y, Okamoto Y, Shimomura I, Hiraoka H, Nakamura T, Funahashi T, Matsuzawa Y. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol. 2003;23(1):85–9.
Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291(14):1730–7.
Hara K, Omori K, Sumioka Y, Aso Y. Spontaneous platelet aggregation evaluated by laser light scatter in patients with type 2 diabetes: effects of short-term improved glycemic control and adiponectin. Transl Res. 2012;159(1):15–24.
Kato H, Kashiwagi H, Shiraga M, Tadokoro S, Kamae T, Ujiie H, Honda S, Miyata S, Ijiri Y, Yamamoto J, Maeda N, Funahashi T, Kurata Y, Shimomura I, Tomiyama Y, Kanakura Y. Adiponectin acts as an endogenous antithrombotic factor. Arterioscler Thromb Vasc Biol. 2006;26(1):224–30.
Shoji T, Koyama H, Fukumoto S, Maeno T, Yokoyama H, Shinohara K, Emoto M, Shoji T, Yamane T, Hino M, Shioi A, Nishizawa Y. Platelet activation is associated with hypoadiponectinemia and carotid atherosclerosis. Atherosclerosis. 2006;188(1):190–5.
Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8(7):731–7.
Wong GW, Wang J, Hug C, Tsao TS, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci USA. 2004;101(28):10302–7.
Wei Z, Peterson JM, Wong GW. Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): activation OF AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J Biol Chem. 2011;286(18):15652–65.
Wong GW, Krawczyk SA, Kitidis-mitrokostas C, Ge G, Spooner E, Hug C, Gimeno R, Lodish HF. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 2009;23(1):241–58.
Zheng Q, Yuan Y, Yi W, Lau WB, Wang Y, Wang X, Sun Y, Lopez BL, Christopher TA, Peterson JM, Wong GW, Yu S, Yi D, Ma XL. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler Thromb Vasc Biol. 2011;31(11):2616–23.
Kambara T, Ohashi K, Shibata R, Ogura Y, Maruyama S, Enomoto T, Uemura Y, Shimizu Y, Yuasa D, Matsuo K, Miyabe M, Kataoka Y, Murohara T, Ouchi N. CTRP9 protects against myocardial injury following ischemia-reperfusion through AMPK-dependent mechanism. J Biol Chem. 2012;287(23):18965–73.
Yuan YX, Gao E, Wang YJ, Wang XL, Yi W, Lau WB, Wong GW, Koch WJ, Ma XL. CTRPs, a novel therapeutic target against myocardial ischemia/reperfusion injury. Circulation. 2010;122:A14133 (Ref type: generic).
Wei Z, Lei X, Petersen PS, Aja S, Wong GW. Targeted deletion of C1q/TNF-related protein 9 increases food intake, decreases insulin sensitivity, and promotes hepatic steatosis in mice. Am J Physiol Endocrinol Metab. 2014;306(7):E779–90.
Peterson JM, Wei Z, Seldin MM, Byerly MS, Aja S, Wong GW. CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction. Am J Physiol Regul Integr Comp Physiol. 2013;305(5):R522–33.
Yi W, Sun Y, Gao E, Wei X, Lau WB, Zheng Q, Wang Y, Yuan Y, Wang X, Tao L, Li R, Koch W, Ma XL. Reduced cardioprotective action of adiponectin in high-fat diet-induced type ii diabetic mice and its underlying mechanisms. Antioxid Redox Signal. 2011;15(7):1779–88.
Ferroni P, Basili S, Falco A, Davi G. Platelet activation in type 2 diabetes mellitus. J Thromb Haemost. 2004;2(8):1282–91.
Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482–94.
Vericel E, Januel C, Carreras M, Moulin P, Lagarde M. Diabetic patients without vascular complications display enhanced basal platelet activation and decreased antioxidant status. Diabetes. 2004;53(4):1046–51.
Ouchi N, Shibata R, Walsh K. Targeting adiponectin for cardioprotection. Expert Opin Ther Targets. 2006;10(4):573–81.
Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380(1–2):24–30.
Kaplon-Cieslicka A, Postula M, Rosiak M, Peller M, Kondracka A, Serafin A, Trzepla E, Opolski G, Filipiak KJ. Younger age, higher body mass index and lower adiponectin concentration predict higher serum thromboxane B2 level in aspirin-treated patients with type 2 diabetes: an observational study. Cardiovasc Diabetol. 2014;13:112.
Fisman EZ, Tenenbaum A. Adiponectin: a manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc Diabetol. 2014;13:103.
Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med. 2009;6(1):27–35.
Wang Y, Wang X, Jasmin JF, Lau WB, Li R, Yuan Y, Yi W, Chuprun K, Lisanti MP, Koch WJ, Gao E, Ma XL. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol. 2012;32(4):934–42.
Shimano M, Ouchi N, Shibata R, Ohashi K, Pimentel DR, Murohara T, Walsh K. Adiponectin deficiency exacerbates cardiac dysfunction following pressure overload through disruption of an AMPK-dependent angiogenic response. J Mol Cell Cardiol. 2010;4(49):210–20.
Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115(11):1408–16.
Shibata R, Numaguchi Y, Matsushita K, Sone T, Kubota R, Ohashi T, Ishii M, Kihara S, Walsh K, Ouchi N, Murohara T. Usefulness of adiponectin to predict myocardial salvage following successful reperfusion in patients with acute myocardial infarction. Am J Cardiol. 2008;101(12):1712–5.
Kondo K, Shibata R, Unno K, Shimano M, Ishii M, Tetsutaro K, Shintani S, Walsh K, Ouchi N, Murohara T. Impact of a single intracoronary administration of adiponectin on myocardial ischemia/reperfusion injury in a pig model. Circ Cardiovasc Interv. 2010;3(2):166–73.
Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11(10):1096–103.
Davis KE, Scherer PE. Adiponectin: no longer the lone soul in the fight against insulin resistance? Biochem J. 2008;416(2):e7–9.
Peterson JM, Aja S, Wei Z, Wong GW. CTRP1 protein enhances fatty acid oxidation via AMP-activated protein kinase (AMPK) activation and acetyl-CoA carboxylase (ACC) inhibition. J Biol Chem. 2012;287(2):1576–87.
Peterson JM, Seldin MM, Tan SY, Wong GW. CTRP2 overexpression improves insulin and lipid tolerance in diet-induced obese mice. PLoS One. 2014;9(2):e88535.
Peterson JM, Wei Z, Wong GW. C1q/TNF-related Protein-3 (CTRP3), a novel adipokine that regulates hepatic glucose output. J Biol Chem. 2010;285(51):39691–701.
Lei H, Wu D, Wang JY, Li L, Zhang CL, Feng H, Fu FY, Wu LL. C1q/tumor necrosis factor-related protein-6 attenuates post-infarct cardiac fibrosis by targeting RhoA/MRTF—a pathway and inhibiting myofibroblast differentiation. Basic Res Cardiol. 2015;110(4):492.
Yuan Y, Lau WB, Su H, Sun Y, Yi W, Du Y, Christopher T, Lopez B, Wang Y, Ma XL. C1q-TNF-related protein-9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am J Physiol Endocrinol Metab. 2015;308(10):E891–8.
Hong ES, Lim C, Choi HY, Ku EJ, Kim KM, Moon JH, Lim S, Park KS, Jang HC, Choi SH. The amount of C1q-adiponectin complex is higher in the serum and the complex localizes to perivascular areas of fat tissues and the intimal-medial layer of blood vessels of coronary artery disease patients. Cardiovasc Diabetol. 2015;14:50.
Li J, Zhang P, Li T, Liu Y, Zhu Q, Chen T, Liu T, Huang C, Zhang J, Zhang Y, Guo Y. CTRP9 enhances carotid plaque stability by reducing pro-inflammatory cytokines in macrophages. Biochem Biophys Res Commun. 2015;458(4):890–5.
Wong GW, Krawczyk SA, Kitidis-mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR- + ¦ agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J. 2008;416(2):161–77.
Wei Z, Peterson JM, Lei X, Cebotaru L, Wolfgang MJ, Baldeviano GC, Wong GW. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem. 2012;287(13):10301–15.
Acknowledgements
This research was supported by the following grants: National Natural Science Foundation of China (Nos. 81270401, 81470413) and Shaanxi Province Principal Project on Natural Science Research (Nos. 2014JZ010, 2014JM4084).
Authors’ contributions
WW and RL established the diabetes model, carried out the platelet aggregation assay and drafted the manuscript. YW participated in the design of the study and performed the statistical analysis. XM and RL conceived of the study and participated in its design. XM and WBL helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, W., Lau, W.B., Wang, Y. et al. Reduction of CTRP9, a novel anti-platelet adipokine, contributes to abnormal platelet activity in diabetic animals. Cardiovasc Diabetol 15, 6 (2016). https://doi.org/10.1186/s12933-015-0321-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-015-0321-1