
Abstract
The participation of sunlight in the natural chemistry of the earth is presented as a unique field of study, from historical observations to prospects for future inquiry. A compilation of known reactions shows the extent of light-driven interactions between naturally occurring components of land, air, and water, and provides the backdrop for an outline of the mechanisms of these phenomena. Catalyzed reactions, uncatalyzed reactions, direct processes, and indirect processes all operate in natural photochemical transformations, many of which are analogous to well-known biological reactions. By overlaying photochemistry and surface geochemistry, complementary approaches can be adopted to identify natural photochemical reactions and discern their significance in the environment.
Similar content being viewed by others


Background
Photogeochemistry has been defined as the photochemistry of Earth-abundant minerals in shaping biogeochemistry [1], and this can be extended to the entire interface between photochemistry and geochemistry to include any chemical reaction induced by sunlight among naturally occurring substances. The term has been used previously on only several other isolated occasions [2, 3], but if existing research is surveyed for studies that fit this definition, an appreciable body of knowledge emerges.
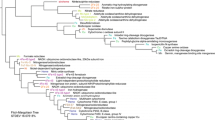
The context of a photogeochemical reaction is implicitly the surface of the earth, since that is where sunlight is available (ignoring other sources of light such as bioluminescence). Reactions may occur among constituents of land such as minerals, plant residue, and the organic and inorganic components of soil; constituents of surface water such as sediment and dissolved organic matter; and constituents of the atmospheric boundary layer directly influenced by contact with land or water, such as organic aerosols, mineral aerosols, and gases. Figure 1 shows some examples of photochemical reactions among these substances. Sunlight penetrates up to approximately 0.3 mm in soils and particulate minerals, depending on the wavelength of light and the nature of the particles [4], and many meters in clear water, depending on the concentration of light-absorbing molecules [5, 6]. Light of wavelengths less than about 290 nm is completely absorbed by the present atmosphere and therefore does not reach Earth’s surface [7, 8].

Photogeochemistry is the study of sunlight-induced chemical reactions among substances that are found naturally on Earth’s surface and intermingle across its domains. Examples of photochemical reactions are shown that occur in the basic domains of land, air, and water. Reaction details and references can be found in Table 1
Photogeochemistry describes photochemical reactions on Earth that are not facilitated by living organisms. The reactions that comprise photosynthesis in plants and other organisms, for example, are not included, since the physiochemical context for these reactions is installed by the organism, and must be maintained in order for the reactions to continue (the photoreactions cease if the organism dies). However, if a certain substance is produced by an organism, and the organism dies but the substance remains (e.g., plant residue or biogenic mineral precipitates), photoreactions involving this substance still contribute to photogeochemistry.
History
The most famous example of a photochemical reaction involving natural compounds is the production of indigoid dyes from the secretions of marine mollusks, known since antiquity [9]; the role of sunlight was emphasized in a study by William Cole in 1685 [10]. The development of modern photochemistry in general was fostered by similar adventitious observations of the effect of sunlight on natural compounds. For example, Hyde Wollaston in 1811 [11] observed that guaiac, a tree resin, rapidly turned green in the air when exposed to sunlight (due to photooxidation). Natural photodegradation was also known, as described by Berzelius in 1829 [12]: “Light fades and destroys the majority of plant colorants. Every day we see that of the sun weakening the dyes of our fabrics”. This phenomenon was also mentioned by John William Draper in 1845 [13]. Georges Witz in 1883 described the degradation of cellulose by sunlight, remarking on the influence of air and moisture, and further noted that degradation was greatly accelerated by ferric oxide [14]. By the end of the 19th century, photodegradation of organic matter in natural waters was recognized as a universal phenomenon [15]. In addition to degradation, other light-induced transformations were also recorded. Louis Pasteur described how a dark-colored material is produced in cinchona bark under the influence of sunlight, an observation that he confirmed in the laboratory with specific compounds [16], and Hermann Trommsdorff [17] and Karl Fritzsche [18] were also among those who observed changes in natural organic substances when they were illuminated. Many inorganic substances were also known to change (e.g., in color or crystal structure) upon exposure to light [13]. For example, since 1881 it has been known that zinc sulfide, normally white, becomes dark when exposed to sunlight [19]; John Cawley remarked that “I have prepared pigments so sensitive as to be turned almost black when exposed to bright sunlight for one or two minutes” [20]. Investigation of the light-induced reactions of this compound [21], which occurs as a natural mineral, provided some additional empirical contributions to photochemistry and the “photochemical metallurgy” of zinc, and its photocatalytic properties are still studied at present [22, 23]. Many natural inorganic compounds used throughout the ages as pigments in painting also slowly degrade by exposure to sunlight; artists like Van Gogh were aware of this [24]. Some of these compounds, such as mercury(II) sulfide, undergo a number of light-mediated reactions [25] which are environmentally relevant.
Around the time of these and other observations, experiments increased in an effort to reproduce natural processes. The hypothesis of von Baeyer in 1870 [26], in which formaldehyde was proposed to be the initial product of plant photosynthesis followed by polymerization into sugars, inspired numerous attempts to obtain formaldehyde from carbon dioxide and water. For example, the formation of lower uranium oxides was observed upon irradiation of a solution of uranium acetate and carbon dioxide, implying the formation of a reducing agent assumed to be formaldehyde [27]. Some experiments included reducing agents such as hydrogen gas [28], and others reportedly detected formaldehyde and other products in the absence of additives [29, 30], suggesting that reducing power was produced from the decomposition of water during exposure to light. In addition to this main focus on the synthesis of formaldehyde and simple sugars, other light-driven reactions were occasionally noted, such as the decomposition of formaldehyde and subsequent release of methane [28]. Many experiments explored the effect of a catalyst in converting light energy into chemical energy; some effective “transformers” (as they were sometimes called) were similar to naturally occurring minerals, including iron(III) oxide or colloidal iron(III) hydroxide [30,31,32], zinc oxide [33], and cobalt, copper, nickel, and iron carbonates [30, 33]. By this time, interest had spread to other light-induced reactions involving naturally occurring materials. These studies sometimes reported photoreactions analogous to biological processes, such as oxidation of simple carbon compounds [34] or nitrification in soil [35].
Overview of photogeochemical reactions
Table 1 presents a selection of documented photochemical reactions (with light >290 nm) among naturally occurring substances, ranging from general reactions such as mineralization of organic matter to specific reactions such as methylation and demethylation of mercury. This compilation is by no means exhaustive, either in reactions or references, but illustrates the general scope and diversity of abiotic photochemical reactions that may occur at the surface of the earth.
Classification of photogeochemical reactions
The same principles that form the foundation of photochemistry can also be used to describe and explain photogeochemical reactions. If specific reactions are known, they may be distinguished as either photosynthetic reactions, photocatalytic reactions, or uncatalyzed reactions. In the most general sense, photosynthesis refers to any photochemical reaction for which the change in energy (ΔG) is positive. The energy of the products is greater than that of the reactants, and therefore the reaction is thermodynamically unfavorable, except through the action of light in conjunction with a catalyst [36] or a chromophoric system, for example, that mimics what occurs in plants [37]. Examples of photosynthetic reactions include the production of H2 and O2 from water and the reaction of CO2 and water to form O2 and reduced carbon compounds such as methane and methanol. Photocatalysis refers to photochemical reactions, accelerated by the presence of a catalyst, that have a negative change in energy and are therefore thermodynamically favored [36], such as the reaction of organic compounds with O2 to form CO2 and water. Finally, uncatalyzed photoreactions proceed through the action of light alone. For example, many organic compounds absorb light and suffer decomposition as a result. Figure 2 depicts a simple scheme for classifying photoreactions based on the requirement for a catalyst and whether a reaction proceeds by a direct or indirect mechanism, as further described below. Figure 3 shows some of the processes that operate in these reactions, also discussed below.

Photogeochemical reactions, if enough information is known, can be classified using general principles of photochemistry. Examples are given for each of four categories in a simple scheme of classification based on the mechanism of reaction. Light-absorbing materials are shaded and catalysts are shown in italics. Intermediate processes in indirect reactions are indicated as separate reactions below the main reaction arrow. For additional explanation of these mechanisms, see the text and the references for specific reactions listed in Table 1

Simplified representations and some examples of processes that occur in photochemical reactions of natural substances: a promotion of electrons (e −) and generation of electron vacancies (holes, h +) upon irradiation of a semiconductor, which may then reduce and oxidize other substances; b excitement of organic compounds by sunlight which then directly react with other substances or are themselves altered, with examples of photochemical acidification, dissolution, and crosslinking; c photocatalysis via surface adsorption, which makes a species, here N2O, susceptible to the effect of light; d indirect generation, via a photosensitizer, of electrons and holes in a semiconductor: the difference between the highest occupied molecular orbital (HOMO) of the sensitizer and its lowest unoccupied molecular orbital (LUMO) is smaller than the band gap of the semiconductor, and therefore less energy is required to excite the sensitizer; e cooperative generation of transient reactive species by compounds that do not individually absorb sunlight; f generation of transient reactive species by light-absorbing compounds. Arrows with shadows indicate reactions induced by light (hν), asterisks (*) indicate excited species (electrons promoted to higher energy levels), single brackets (]) indicate mineral surfaces, and dotted lines (…) indicate surface adsorption. The references cited in the text offer additional, detailed explanations of these processes
Catalysis
A catalyst is a substance that increases the rate of a chemical reaction due to a change in mechanism, but does not experience any net change itself during the course of the reaction [37, 38]. A photocatalyst does this by absorbing light, but as described below, other substances that do not absorb light may nevertheless catalyze light-induced reactions. Strictly speaking, the term catalysis should not be used unless it can be shown that the number of product molecules produced per number of active sites on a substance (the turnover number) is greater than one [39]; this is difficult to do in practice, although it is often assumed to be true if there is no loss in the activity of the substance for an extended period of time [36]. Reactions which are not definitively catalytic may be designated as assisted photoreactions [36, 38] or photosensitized reactions. Photosensitized reactions involve transfer of energy from a light-absorbing species (photosensitizer) to another, nonabsorbing species, and therefore facilitate reaction of this nonabsorbing species [40]. If the photosensitizer remains intact it is effectively a photocatalyst. Furthermore, a substance may initially act as a photocatalyst in a reaction even if it eventually suffers light-induced decomposition. Descriptors such as those given here are most applicable when all of the participants in a specific reaction can be identified, not just individual reactants or products. In contrast, it is hard to classify observations in complex matrices such as soil if the complete reactions responsible for the observations are not first discerned.
Direct reactions
Photochemical reactions can be further categorized as either direct or indirect. Direct reactions involve the substance that initially absorbs light [41,42,43] which reacts with other substances or is itself changed. Many photochemical reactions on Earth may be directly mediated by naturally occurring semiconductors that absorb ultraviolet and visible radiation. These are mostly transition metal oxides and sulfides and include abundant, widely distributed minerals such as hematite (Fe2O3), magnetite (Fe3O4), goethite and lepidocrocite (FeOOH), anatase and rutile (TiO2), pyrolusite (MnO2), pyrite (FeS2) chalcopyrite (CuFeS2), and sphalerite (ZnS) [44, 45]. Other types of minerals are also known to absorb light and directly participate in photoreactions, including silicates such as Ag6Si2O7 [46] and phosphates such as Cu2(OH)PO4 [47]. Light of energy equal to or greater than the band gap of a semiconductor is sufficient to promote electrons from the valence band to a higher energy level in the conduction band, leaving behind electron vacancies or holes (Fig. 3a). The excited electron and hole in the semiconductor can then, respectively, reduce and oxidize other compounds having appropriate redox potentials relative to the potentials of the valence and conduction bands [48]. The band gaps and absolute energy levels of many minerals are suitable, in theory, for a diverse array of photoreactions at interfaces with water, gases, and other solids. Naturally occurring semiconductors are almost exclusively inorganic compounds, with notable exceptions (notable because they occur widely) being melanin [49] and possibly cellulose [50, 51] and peptides [52,53,54].
Natural semiconducting minerals, like most minerals, are rarely pure; additional metals are almost always present [44], and these substitutional impurities can cause changes in energy levels and conductivity [44, 55]. Such alterations are manifested in photocatalytic activity. For example, the band gap of TiO2 decreases due to Fe impurities [56, 57], which extends its response to a wider range of solar radiation compared to pure TiO2; the efficiencies of photochemical oxidation and reduction reactions of TiO2 are also greater if Fe impurities are present [57, 58]. Similarly, the presence of Ti or V in magnetite enhances its photocatalytic activity relative to pure magnetite [59]. In addition to atoms of foreign elements, another common “defect” in minerals is deviation from stoichiometry due to vacancies (missing atoms), and this can also affect photochemical properties. For example, sulfur deficiencies in ZnS crystals impart increased photocatalytic activity under visible light to a material that normally absorbs little or no visible light [23]. In addition to chemical alterations, the photocatalytic activity of materials like these is also influenced by physical properties such as crystal structure and specific surface area [23, 56, 60].
Like inorganic minerals, many natural organic compounds also absorb sunlight and can react directly with other compounds or undergo reactions themselves (Fig. 3b); these include dissolved organic matter [61,62,63], “bioorganic” substances [64], chlorophyll [65], atmospheric humic-like substances [42], and soot or black carbon [42, 66]. Moreover, two species may combine to form a new species with even greater propensity to undergo direct photoreactions, as is often the case with intramolecular or intermolecular charge-transfer complexes among components of organic matter [67] or between transition metals and organic matter [68]. Sometimes this even leads to catalytic or autocatalytic cycles [69,70,71].
Finally, materials that do not absorb sunlight, such as silica, may nonetheless enable direct photoreactions. These materials are usually catalysts and act primarily via surface adsorption, which can alter the bond lengths and energies of a substance when it is bound to the catalyst [72, 73] and consequently alter the amount or wavelengths of sunlight absorbed by this substance [74, 75]. The bound substance then becomes susceptible to photolysis and other photoreactions (Fig. 3c). Depending on the nature of a substance, however, adsorption onto materials such as clay and ash can sometimes impede rather than facilitate photoreactions [76,77,78].
Indirect reactions
Indirect photochemical reactions are initiated by substances that absorb radiation and subsequently facilitate other reactions that do not involve the original light-absorbing substance [42]. For example, excited electrons and holes can be indirectly generated in semiconductors by light of lower energy than the band gap: the semiconductor itself does not absorb this light, but another substance (possibly even another semiconductor) that does absorb this light may be excited, and if this substance is in contact with the semiconductor and has appropriate energy levels, electrons can then be transferred between the excited substance and the semiconductor [48, 68, 79,80,81] (Fig. 3d). The semiconductor, now carrying additional electrons or holes, can participate in redox reactions that would not otherwise occur. For example, TiO2 has a large band gap and is not normally excited by visible light; however, organic matter and natural chlorophyll derivatives are excited upon absorption of visible light, and in proximity to TiO2 can transfer electrons to TiO2 [82, 83]. This process is called charge injection, and is an example of photosensitization—reactions of TiO2 with additional substances are facilitated by the initial presence of organic matter or chlorophyll derivatives.
A substance may also participate indirectly in photochemical reactions by generating reactive species upon irradiation; these reactive species then engage in other reactions that do not involve the original light-absorbing substance [42]. For example, some aluminosilicates (e.g., zeolites) and non-transition-metal oxides (e.g., SiO2, Al2O3, MgO) can react with the oxygen in air upon irradiation to produce reactive oxygen species (ROS) such as singlet oxygen and superoxide [84, 85]. Photodegradation of an organic compound was observed in the presence of kaolinite and montmorillonite, for example, and was attributed to the formation of ROS on the surface of these minerals in the presence of molecular oxygen and water [86]. Since the organic compound in question does not absorb sunlight and the ROS are produced in a separate reaction, this is an indirect photoreaction, facilitated by the clay minerals which presumably act as catalysts by generating ROS from O2 upon exposure to light (Fig. 3e).
Along with minerals [87], other substances can indirectly facilitate photoreactions by generating reactive species in sunlight: dissolved and particulate organic matter [88,89,90,91,92,93,94,95], dissolved organic matter and silicate minerals in synergy [63], cellulose [50, 96, 97], lignin [98, 99], leaves of phototoxic plants [100], chlorophyll [101], nitrite and nitrate [102,103,104], flavins [41, 105], tryptophan and tyrosine [99, 106, 107], and aqueous iron(III) species [108,109,110]. In contrast to the typically strong oxidizing action of ROS, a strongly reducing species can also be generated which is usually represented as e − (aq), a hydrated electron, although its true nature and features are not completely understood. Hydrated electrons are evident upon irradiation of dissolved organic matter, for example [94, 95]. As might be expected, reactive species are formed on exposed soil surfaces [111, 112]; both the mineral and organic components of soil contribute to this process [113]. Indirect photolysis of organic compounds in soil has been observed to occur at depths of up to 2 mm due to migration of reactive species; in contrast, direct photolysis (in which the degraded compound itself absorbs light) is restricted to a photic depth of about ten times less [114, 115]. Both light penetration and transport processes such as diffusion influence the extent to which compounds are degraded by light in soil and similar media [116]. Indirect processes may operate during photodegradation of plant material as well [117]. In certain instances, however, the same substances listed above may also inhibit the formation of reactive species and therefore retard indirect photoreactions, as observed for chlorophyll [118], carotenoids [119], and organic matter in soil and water [76, 120].
Experimental approaches
Studies in photogeochemistry may take several different paths, depending on the source of inspiration for identifying and investigating natural photochemical reactions (Fig. 4). Oftentimes photogeochemistry distinctly parallels biogeochemistry. As mentioned above, early research sometimes intentionally used biological phenomena as a starting point to search for analogous photochemical reactions. Other studies simply explored the effect of light on different materials, and as a result also discovered photochemical reactions analogous to biological processes. Photochemical counterparts have since been confirmed for many well-known biochemical reactions. These include photochemical disproportionation of acetic acid [121, 122] which is analogous to acetoclastic methanogenesis, and light-induced depletion of O2 via a catalytic cycle involving iron and organic matter [123], analogous to consumption of O2 by microorganisms. Estimates of the environmental significance of photochemical reactions relative to biological reactions have been offered on occasion, as for photochemical production of gases from plant litter [124, 125], and the photofixation of N2 in deserts, estimated as 20 kg N ha−1 year−1, which is about one third of that fixed by lightning and about 10% of that fixed biologically on Earth [126]. In contrast to these processes, in which biological reactions predominate (at least on a global level), the rate of degradation of dissolved lignin in rivers by photochemical mechanisms was found to be several times larger than by biological mechanisms [127]. Witz, based on his (nonbiological) studies with cellulose and other plant fibers [14], concluded that light is indeed an integral participant in natural decomposition: “In nature, once the life of plants is extinguished, cellulosic matter and other structured matter must no doubt pass progressively under the influence of light, air, and humidity … and are eventually transformed into gaseous compounds and colored humic materials.”

The study of photogeochemistry reflects the overlap between surface geochemistry and photochemistry. The curved arrows represent three different but complementary approaches which can lead to the discovery of natural photoreactions: observing natural phenomena, extending known natural photoreactions, and contextualizing photoreactions that are not known to occur naturally
Extension of known photoreactions
The most obvious experimental precedent in photogeochemistry is a natural photoreaction that has already been ascertained. Known reactions may be further investigated as to their context, mechanisms, and environmental significance. For example, the greenhouse gases CO2, CH4, and N2O are the subject of a large amount of ongoing interdisciplinary research. Natural production and consumption of these gases at the earth’s surface are ascribed largely to biological activity [128,129,130,131], which remains the focus of most research, in spite of studies that have demonstrated photochemical production and consumption (see Table 1). Similarly, mineralization of organic carbon, nitrogen, and phosphorus in soil and water, the biological drivers of which are extensively studied, may also proceed photochemically. It is interesting to note that biologically recalcitrant portions of organic matter can be quite susceptible to photodegradation [132, 133]; the consequent release of labile organic and inorganic compounds can stimulate biological activity [134,135,136].
Sometimes a particular reaction, when placed in a certain environmental context, may even affect existing paradigms. For example, it is generally (and logically) assumed that in water classified as anoxic there can be no reactions involving molecular oxygen, including aerobic metabolism. However, some naturally occurring minerals are known to facilitate the photochemical oxidation of water to molecular oxygen; such “photochemical sources of oxidizing power in low-oxygen environments” [137] may be active alongside or in place of other sources of oxygen such as air or photosynthetic organisms. Similarly, organic acids known to be produced during the photodecomposition of organic matter may form a connection between light exposure and soil acidity, a simple but unestablished possibility next to the usual factors that determine soil pH.
While investigation of known natural photoreactions can be extended by pursuing additional work with the same substances, knowledge of natural photoreactions may also support inquiry into photoreactions of distinct but related substances. For example, the susceptibility to photodegradation of polycyclic aromatic hydrocarbons and related condensed aromatic compounds has been reported [e.g., 78, 138–140]. These studies focus on relatively simple molecules which are either regarded as naturally occurring pollutants or are components of dissolved organic matter. At the same time, the incomplete combustion of natural organic materials leaves solid residues (“charcoal”, “biochar”, or “pyrogenic black carbon”) that contain analogous extended aromatic structure [141,142,143]. It may therefore be suggested that this ubiquitous material, commonly deemed environmentally persistent [63, 140, 143, 144] and therefore paradoxical (since it does not accumulate in the environment) [145, 146], is also degraded upon exposure to sunlight.
The study of photogeochemistry, while purely chemical in nature, may even venture into the domain of biology and identify more of the ways in which compounds derived from living organisms can influence abiotic photochemistry [e.g., 81], as well as more of the unique relationships between photochemical reactions and biological metabolism known as photobiocatalysis [147,148,149].
Observation of natural phenomena
Specific photoreactions are often planned and conveniently observed in the laboratory, using artificial light sources or sunlight itself, where it is easy to confirm the identity of the substances involved, design reaction vessels, characterize the light, and adjust the reaction environment. However, observations of natural phenomena can offer opportunities to consider unknown photochemical reactions possibly associated with these phenomena. For example, by the 1970s it was generally agreed that nitrous oxide (N2O) has a short residence time in the troposphere, although the explanation for its removal was incomplete. Since N2O does not absorb light of wavelengths greater than 290 nm, direct photolysis had been discarded as a possible explanation. It was then observed that light would decompose chloromethanes when they were adsorbed on silica sand [150], and this occurred at lower energies (longer wavelengths) than the absorption spectra for the free compounds. The same phenomenon was observed for N2O on natural sand, leading to the conclusion that particulate matter in the atmosphere is responsible for the destruction of N2O via surface-sensitized photolysis [151]. Indeed, the idea of such a sink for atmospheric N2O was supported by reports of low concentrations of N2O in the air above deserts, where there is a large amount of suspended particulate matter [152]. In general, simple atmospheric gases (e.g., CO2, CO, CH4, N2O, N2, H2O, H2, O2) do not absorb ultraviolet and visible sunlight at the earth’s surface, and the cooperation of particulate matter is necessary for photoreactions involving these gases; such reactions are therefore heterogeneous. Other gases, however, such as some of the volatile compounds emitted from living plants [153, 154], burning plants [155] and soils [156], do absorb sunlight and can undergo homogeneous as well as heterogeneous reactions.
As another example, the observation that the amount of nitrous acid in the atmosphere greatly increases during the day led to insight into the surface photochemistry of humic acids and soils and an explanation for the original observation [157]. Fluctuations such as this are often a clue to the existence of photochemical reactions, which operate only during the day. Diurnal photogeochemical cycles often have a significant influence on the amounts of redox-sensitive elements in aqueous environments [70, 158,159,160]. Furthermore, multiple elemental cycles can be linked via photoreactions that directly affect both elements, as occurs during the concurrent oxidation of organic matter and reduction of iron [92]. The effect of light on one element can also indirectly affect other elements: a daily cycle of photoreduction, reoxidation, and precipitation of iron(III) species affects dissolved As, Cu, and P, which adsorb to iron(III) oxides as they reappear at night and may be subsequently released the next day upon photoreduction of the same iron oxides [158, 159, 161].
Contextualization of nonnatural photoreactions
Although photogeochemistry describes reactions among substances known to occur naturally, studies of similar substances may nonetheless point towards greater understanding of natural processes. A general example demonstrates this: it has been shown that samples of clay minerals found in soils can accelerate the photodegradation of synthetic chemicals via production of reactive oxygen species [e.g., 86]; it may therefore be assumed that many naturally occurring compounds are similarly affected. The conversion of N2 to NH3 and NO3 − has been observed upon irradiation with visible light in the presence of Fe2Ti2O7 [162, 163]. While such a compound is not known to occur naturally, it is related to known minerals like ilmenite (FeTiO3), ulvospinel (Fe2TiO4), pseudorutile (Fe2Ti3O9), and various titanium-substituted iron oxides, and can form when ilmenite is heated [162, 164]; these naturally occurring minerals might therefore also react with N2 under certain conditions.
Outlook
Principles of photochemistry can be readily merged with geochemistry in investigation as well as education. Given the broad response of natural substances to light, recognizing photochemical reactions in the environment is part of understanding its fabric of interconnected processes, particularly on land, where this has not been explored as much as in water or the atmosphere. As remarked by Formenti and Teichner [40] concerning heterogeneous photochemistry, “there are so many different possibilities”, an outlook reiterated by Cooper and Herr [165] for aqueous photochemistry which is easily extended to photogeochemistry: “there are a seemingly endless number of combinations and permutations to study.” This does not enjoin an unattainable research agenda, but rather affirms ample opportunity for geoscientists to incline their curiosity toward what happens on Earth when the sun appears.
References
Kim JD, Yee N, Nanda V, Falkowski PG (2011) Anoxic photochemical oxidation of siderite generates molecular hydrogen and iron oxides. Proc Nat Acad Sci USA 110:10073–10077
Schrauzer GN, Strampach N, Hui LN, Palmer NR, Salehi J (1983) Nitrogen photoreduction on desert sands under sterile conditions. Proc Nat Acad Sci USA 80:3873–3876
Falkowski PG (2015) From light to life. Origins Life Evol B 45:347–350
Ciani A, Goss KU, Schwarzenbach RP (2005) Light penetration in soils and particulate minerals. Eur J Soil Sci 56:561–574
Kirk JTO (1977) Attenuation of light in natural waters. Aust J Mar Fresh Res 28:497–508
Piazena H, Perez-Rodriguez E, Hader DP, Lopez-Figueroa F (2002) Penetration of solar radiation into the water column of the central subtropical Atlantic Ocean—optical properties and possible biological consequences. Deep Sea Res Pt II Top Stud Oceanogr 49:3513–3528
Seinfield JH, Pandis SN (1998) Atmospheric Chemistry and Physics: from Air Pollution to Climate Change. Wiley, New York
Madronich S, Flocke S (1999) The role of solar radiation in atmospheric chemistry. In: Boule P (ed) Handbook of environmental chemistry, volume 2 part L: environmental photochemistry. Springer, Berlin
McGovern PE, Michel RH (1990) Royal purple dye: the chemical reconstruction of the ancient Mediterranean industry. Acc Chem Res 23:152–158
Cole W (1685) A letter from Mr. Willam Cole of Bristol, to the Philosophical Society of Oxford; containing his observations on the purple fish. Phil Trans 15:1278–1286
Wollaston H (1811) Ueber gewisse chemische Wirkungen des Lichts [On certain chemical effects of light]. Ann Phys Gilbert 39:291–299
Berzelius JJ (1829) Traité de Chimie, Première Partie—Chimie Minérale. Firmin Didot Frères, Paris, p 52
Draper JW (1845) A treatise on the forces which produce the organization of plants, with an appendix, containing several memoirs on capillary action, electricity, and the chemical action of light, Chap 10, 2nd edn. Harper and Brothers, New York
Witz G (1883) Recherches sur certaines alterations du coton [Studies on certain changes in cotton]. Bulletin de la Société Industrielle de Rouen 11:169–232
Whipple GC, Hazen A, Soper GA, Fuller GW, Maignen JPA, Chester JN, Stearns FP, Fitzgerald D, Dunham HF (1901) The decolorization of water. Am Soc Civ Eng 46:141–181
Pasteur L (1853) Alcaloïdes des quinquinas. L’Institut Journal Universel 21:249–250
Trommsdorff H (1834) Ueber Santonin. Ann Pharm 11:190–207
Fritzsche KJ (1867) Ueber die festen Kohlenwasserstoffe des Steinkohlentheers [On the solid hydrocarbons of coal tar]. J Prakt Chem 101:333–343
O’Brien WJ (1915) A study of lithopone. J Phys Chem 19:113–144
Cawley J (1891) On the curious behaviour of certain zinc sulphide compounds. Chem News J Ind Sci 63:88–89
Job A, Emschwiller G (1922) Sur la reduction photochimique du sulfure de zinc [On the photochemical reduction of zinc sulfide]. CR Hebd Acad Sci 177:313–316
Reber JF, Meier K (1984) Photochemical production of hydrogen with zinc sulfide suspensions. J Phys Chem 88:5903–5913
Wang G, Huang BB, Li ZJ, Lou ZZ, Wang ZY, Dai Y, Whangbo MH (2015) Synthesis and characterization of ZnS with controlled amount of S vacancies for photocatalytic H2 production under visible light. Sci Rep 2015:8544
Everts S (2016) Van Gogh’s fading colors inspire scientific inquiry—lessons learned from the chemical breakdown of pigments in the Post-Impressionist’s masterpieces. Chem Eng News 94:32–33
Keune K, Boon JJ (2005) Analytical imaging studies clarifying the process of the darkening of vermilion in paintings. Anal Chem 77:4742–4750
von Baeyer A (1870) Ueber die Wasserentziehung und ihre Bedeutung fur das Pflanzenleben unt die Gahrung [On the removal of water and its importance for plant life and fermentation]. Ber Dtsch Chem Ges 3:63–75
Bach A (1893) Contribution à l’étude des phénomènes chimiques de l’assimilation de l’acide carbonique par les plantes à chlorophylle [Contribution to the study of the chemical phenomena of carbonic acid assimilation by plants with chlorophyll]. CR Acad Sci 116:1145–1148
Berthelot D, Gaudechon H (1910) Synthèse photochimique des hydrates de carbone aux dépens des éléments de l’anhydride carbonique et de la vapeur d’eau, en l’absence de chlorophylle; synthèse photochimique des composés quaternaires [Photochemical synthesis of carbohydrates from the elements of carbonic anhydride and water vapor, in the absence of chlorophyll; photochemical synthesis of quaternary compounds]. CR Acad Sci 150:1690–1693
Usher FL, Priestly JH (1911) The mechanism of carbon assimilation: part III. Proc R Soc Lond B 84:101–112
Rajvansi AR, Dhar NR (1932) Photosynthesis in tropical sunlight. Part III: synthesis of formaldehyde. J Phys Chem 36:568–574
Moore B, Webster TA (1913) Synthesis by sunlight in relationship to the origin of life: synthesis of formaldehyde from carbon dioxide and water by inorganic colloids acting as transformers of light energy. Proc R Soc Lond B 87:163–176
Baly ECC (1930) Photosynthesis of carbohydrates. Nature 126:666–667
Dhar NR, Ram A (1932) Photoreduction of carbonic acid, bicarbonates, and carbonates to formaldehyde. Nature 129:205
Baur E, Neuweiler C (1927) Über photolytische Bildung von Hydroperoxyd [On the photolytic formation of hydrogen peroxide]. Helv Chim Acta 10:901–907
Dhar NR, Bhattacharya AK, Biswas NN (1932) Photonitrification in soil. Soil Sci 35:281–284
Mills A, LeHunte S (1997) An overview of semiconductor photocatalysis. J Photochem Photobiol A Chem 108:1–35
Braslavsky SE, Braun AM, Cassano AE, Emeline AV, Litter MI, Palmisano L, Parmon VN, Serpone N (2011) Glossary of terms used in photocatalysis and radiation catalysis (IUPAC recommendations). Pure Appl Chem 83:931–1014
Emeline AV, Otroshchenki VA, Ryabchuk VK, Serpone N (2003) Abiogenesis and photostimulated heterogeneous reactions in the interstellar medium and on primitive earth. Relevance to the genesis of life. J Photochem Photobiol C Photochem Rev 3:203–224
Emeline AV, Ryabchuk VK, Serpone N (2007) Photoreactions occurring on metal oxide surfaces are not all photocatalytic. Description of criteria and conditions for processes to be photocatalytic. Catal Today 122:91–100
Formenti M, Teichner SJ (1978) Heterogeneous photo-catalysis. In: Kemball C, Dowden DA (eds) Specialist periodical reports, catalysis, vol 2. The Chemical Society (Royal Society of Chemistry), London
Larson RA, Weber EJ (1994) Reaction mechanisms in environmental organic chemistry. Lewis, Boca Raton
George C, Ammann M, D’Anna B, Donaldson DJ, Nizkorodov SA (2015) Heterogeneous photochemistry in the atmosphere. Chem Rev 115:4218–4258
Kisch H (2015) Semiconductor photocatalysis. Wiley, Weinheim
Shuey RT (1975) Semiconducting ore minerals. Developments in economic geology, vol 4. Elsevier, Amsterdam
Xu Y, Schoonen MAA (2000) The absolute energy positions of conduction and valence bands of selected semiconducting minerals. Am Miner 85:543–556
Lou ZZ, Huang BB, Wang ZY, Ma XC, Zhang R, Zhang XY, Qin XY, Dai Y, Whangbo MH (2014) Ag6Si2O7: a silicate photocatalyst for the visible region. Chem Mater 26:3873–3875
Wang G, Huang BB, Ma XC, Wang ZY, Qin XY, Zhang XY, Dai Y, Whangbo MH (2013) Cu2(OH)PO4, a near-infrared-activated photocatalyst. Angew Chem Int Ed 52:4810–4813
Schoonen MAA, Xu Y, Strongin DR (1998) An introduction to geocatalysis. J Geochem Explor 62:201–215
Mostert AB, Powell BJ, Pratt FL, Hanson GR, Sarna T, Gentle IR, Meredith P (2012) Role of semiconductivity and ion transport in the electrical conduction of melanin. Proc Nat Acad Sci USA 109:8943–8947
Phillips GO, Hinojosa O, Arthur JC Jr, Mares T (1966) Photochemical initiation of free radicals in cotton cellulose. Text Res J 36:822–827
Simão CD, Reparaz JS, Wagner MR, Graczykowski B, Kreuzer M, Ruiz-Blanco YB, García Y, Malho JM, Goñi AR, Ahopelto J, Sotomayor-Torres CM (2015) Optical and mechanical properties of nanofibrillated cellulose: toward a robust platform for next-generation green technologies. Carbohyd Polym 126:40–46
Evans MG, Gergely J (1949) A discussion of the possibility of bands of energy levels in proteins. Biochim Biophys Acta 3:188–197
Cardew MH, Eley DD (1959) The semiconductivity of organic substances. Part 3. Haemoglobin and some amino acids. Discuss Faraday Soc 27:115–128
Hauser CAE, Zhang SG (2010) Peptides as biological semiconductors. Nature 468:516–517
Hu C (2010) Modern semiconductor devices for integrated circuits. Prentice Hall, Upper Saddle
Liu K, Rykov AI, Wang JH, Zhang T (2015) Recent advances in the application of Mossbauer spectroscopy in heterogeneous catalysis. Adv Catal 58:1–142
Choi WY, Termin A, Hoffmann MR (1994) The role of metal ion dopants in quantum-sized TiO2: correlation between photoreactivity and charge carrier recombination dynamics. J Phys Chem 98:13669–13679
Soria J, Conesa JC, Augugliaro V, Palmisano L, Schiavello M, Sclafani A (1991) Dinitrogen photoreduction to ammonia over titanium-dioxide powders doped with ferric ions. J Phys Chem 95:274–282
Liang XL, Zhong YH, Zhu SY, Ma LY, Yuan P, Zhu JX, He HP, Jiang Z (2012) The contribution of vanadium and titanium on improving methylene blue decolorization through heterogeneous UV-Fenton reaction catalyzed by their co-doped magnetite. J Hazard Mater 199:247–254
Osterloh FE (2008) Inorganic materials as catalysts for photochemical splitting of water. Chem Mater 20:35–54
Zepp RG, Baughman GL, Schlotzhauer PF (1981) Comparison of photochemical behavior of various humic substances in water. 1. Sunlight induced reactions of aquatic pollutants photosensitized by humic substances. Chemosphere 10:109–117
Timko SA, Romera-Castillo C, Jaffe R, Cooper WJ (2014) Photo-reactivity of natural dissolved organic matter from fresh to marine waters in the Florida Everglades, USA. Environ Sci Process Impacts 16:866–878
Fu HY, Liu HT, Mao JD, Chu WY, Li QL, Alvarez PJJ, Qu XL, Zhu DQ (2016) Photochemistry of dissolved black carbon released from biochar: reactive oxygen species generation and phototransformation. Environ Sci Technol 50:1218–1226
Gomis J, Vercher RF, Amat AM, Martire DO, Gonzales MC, Bianco-Prevot A, Montoneri E, Arques A, Carlos L (2013) Application of soluble bio-organic substances (SBO) as photocatalysts for wastewater treatment: sensitizing effect and photo-Fenton-like process. Catal Today 209:176–180
Reeser DI, George C, Donaldson DJ (2009) Photooxidation of halides by chlorophyll at the air-salt water interface. J Phys Chem A 113:8591–8595
George C, D’Anna B, Herrmann H, Weller C, Vaida V, Donaldson DJ, Bartels-Rausch T, Ammann M (2014) Emerging areas in atmospheric photochemistry. Atmos Aerosol Chem 399:1–53
Del Vecchio R, Blough NV (2004) On the origin of the optical properties of humic substances. Environ Sci Technol 38:3885–3891
Waite TD (1990) Photo-redox processes at the mineral-water interface. In: Hochella MF, White AF (eds) Mineral-water interface geochemistry (Reviews in Mineralogy vol. 23). Mineralogical Society of America, Washington DC
Ciesla P, Kocot P, Mytych P, Stasicka Z (2004) Homogeneous photocatalysis by transition metal complexes in the environment. J Mol Catal A 224:17–33
Waite TD (2005) Role of iron in light-induced environmental processes. In: Boule P, Bahnemann DW, Robertson P (eds) Handbook of environmental chemistry, vol. 2 Part M: environmental photochemistry part II. Springer, Berlin
Siffert C, Sulzberger B (1991) Light-induced dissolution of hematite in the presence of oxalate: a case study. Langmuir 7:1627–1634
Parida SK, Dash S, Patel S, Mishra BK (2006) Adsorption of organic molecules on silica surface. Adv Colloid Interface 121:77–110
Al-abadleh HA, Grassian VH (2003) Oxide surfaces as environmental interfaces. Surf Sci Rep 52:63–161
Leermakers PA, Thomas HT, Weis LD, James FC (1966) Spectra and photochemistry of molecules adsorbed on silica gel. IV. J Am Chem Soc 88:5075–5083
Schiavello M (1988) Basic concepts in photocatalysis. In: Schiavello M (ed) Photocatalysis and environment: trends and applications. Kluwer, Dordrecht
Katagi T (2004) Photodegradation of pesticides on plant and soil surfaces. Rev Environ Contam Toxicol 182:1–189
Thomas JK (1993) Physical aspects of photochemistry and radiation chemistry of molecules adsorbed on SiO2, γ-Al2O3, zeolites, and clays. Chem Rev 93:301–320
Korfmacher WA, Wehry EL, Mamantov G, Natusch DFS (1980) Resistance to photochemical decomposition of polycyclic aromatic hydrocarbons vapor-adsorbed on coal fly ash. Environ Sci Technol 14:1094–1099
Gratzel M (1989) Heterogeneous photochemical electron transfer. CRC Press, Boca Raton
Hagfeldt A, Boschloo G, Sun LC, Kloo L, Pettersson H (2010) Dye-sensitized solar cells. Chem Rev 110:6595–6663
Braun A, Boudoire F, Bora DK, Faccio G, Hu YL, Kroll A, Mun BS, Wilson ST (2015) Biological components and bioelectronics interfaces of water splitting photoelectrodes for solar hydrogen production. Chem Eur J 21:4188–4199
Vinodgopal K (1994) Environmental photochemistry: electron transfer from excited humic acid to TiO2 colloids and semiconductor mediated reduction of oxazine dyes by humic acid. Res Chem Intermed 20:825–833
Kay A, Grätzel M (1993) Artificial photosynthesis. 1. Photosensitization of TiO2 solar cells with chlorophyll derivatives and related natural porphyrins. J Phys Chem 97:6272–6277
Che M, Tench AJ (1983) Characterization and reactivity of molecular oxygen species on oxide surfaces. Adv Catal 32:1–148
Gohre K, Miller GC (1985) Photochemical generation of singlet oxygen on non-transition-metal oxide surfaces. J Chem Soc, Faraday Trans 81:793–800
Katagi T (1990) Photoinduced oxidation of the organophosphorus fungicide tolclofos-methyl on clay minerals. J Agr Food Chem 38:1595–1600
Schoonen MAA, Cohn CA, Roemer E, Laffers R, Simon SR, O’Riordan T (2006) Mineral-induced formation of reactive oxygen species. Rev Mineral Geochem 64:179–221
Coelho C, Guyot G, ter Halle A, Cavani L, Ciavatta C, Richard C (2011) Photoreactivity of humic substances: relationship between fluorescence and singlet oxygen production. Environ Chem Lett 9:447–451
Sharpless CM, Blough NV (2014) The importance of charge-transfer interactions in determining chromophoric dissolved organic matter (CDOM) optical and photochemical properties. Environ Sci Process Impacts 16:654–671
Appiani E, McNeill K (2015) Photochemical production of singlet oxygen from particulate organic matter. Environ Sci Technol 49:3514–3522
Vione D, Minella M, Maurino V, Minero C (2014) Indirect photochemistry in sunlit surface waters: photoinduced production of reactive transient species. Chem Eur J 20:10590–10606
Voelker BM, Morel FMM, Sulzberger B (1997) Iron redox cycling in surface waters: effects of humic substances and light. Environ Sci Technol 31:1004–1011
Baxter RM, Carey JH (1983) Evidence for photochemical generation of superoxide ion in humic waters. Nature 306:575–576
Thomas-Smith TE, Blough NV (2001) Photoproduction of hydrated electron from constituents of natural waters. Environ Sci Technol 35:2721–2726
Zepp RG, Braun AM, Hoigne J, Leenheer JA (1987) Photoproduction of hydrated electrons from natural organic solutes in aquatic environments. Environ Sci Technol 21:485–490
Malesic J, Kolar J, Strlic M, Kocar D, Fromageot D, Lemaire J, Haillant O (2005) Photo-induced degradation of cellulose. Polym Degrad Stabil 89:64–69
Hon NS (1976) Fundamental degradation processes relevant to solar irradiation of cellulose: ESR studies. J Macromol Sci Chem 10:1175–1192
Gellerstedt G, Kringstad K, Lindfors EL (1978) Singlet oxygen oxidation of lignin structures. In: Ranby B, Rabek JF (eds) Singlet oxygen: reactions with organic compounds and polymers. Wiley, Chichester
Davidson RS (1996) The photodegradation of some naturally occurring polymers. J Photochem Photobiol B Biol 33:3–25
Berenbaum MR, Larson RA (1988) Flux of singlet oxygen from leaves of phototoxic plants. Experientia 44:1030–1032
Rontani JF (1999) Photodegradation of lipidic compounds during the senescence of phytoplankton. In: Boule P (ed) Handbook of environmental chemistry, vol 2 part L: environmental photochemistry. Springer, Berlin
Boule P, Bolte M, Richard C (1999) Phototransformations induced in aqueous media by NO3 −/NO2 −, Fe(III) and humic substances. In: Boule P (ed) Handbook of environmental chemistry, vol 2 part L: environmental photochemistry. Springer, Berlin
Mack J, Bolton JR (1999) Photochemistry of nitrite and nitrate in aqueous solution: a review. J Photochem Photobiol, A 128:1–13
Minero C, Chiron S, Falletti G, Maurino V, Pelizzetti E, Ajassa R, Carlotti ME, Vione D (2007) Photochemical processes involving nitrite in surface water samples. Aquat Sci 69:71–85
Mopper K, Zika RG (1987) Natural photosensitizers in sea water: riboflavin and its breakdown products. In: Zika RG, Cooper WJ (eds) Photochemistry of environmental aquatic systems. American Chemical Society, Washington DC
McCormick JP, Thomason T (1978) Near-ultraviolet photooxidation of tryptophan—proof of formation of superoxide ion. J Am Chem Soc 100:312–313
Draper WM, Crosby DG (1983) Photochemical generation of superoxide radical anion in water. J Agr Food Chem 31:734–737
Stumm W, Sulzberger B (1992) The cycling of iron in natural environments—considerations based on laboratory studies of heterogeneous redox processes. Geochim Cosmochim Acta 56:32333
Feng W, Nansheng D (2000) Photochemistry of hydrolytic iron(III) species and photoinduced degradation of organic compounds: a minireview. Chemosphere 41:1137–1147
Deguillaume L, Leriche M, Desboeufs K, Mailhot G, George C, Chaumerliac N (2005) Transition metals in atmospheric liquid phases: sources, reactivity, and sensitive parameters. Chem Rev 105:3388–3431
Gohre K, Miller GC (1983) Singlet oxygen generation on soil surfaces. J Agr Food Chem 31:1104–1108
Georgiou CD, Sun HJ, McKay CP, Grintzalis K, Papapostolou I, Zisimopoulos D, Panagiotidis K, Zhang GS, Koutsopoulou E, Christidis GE, Margiolaki I (2015) Evidence for photochemical production of reactive oxygen species in desert soils. Nature Commun 6:7100
Gohre K, Scholl R, Miller GC (1986) Singlet oxygen reactions on irradiated soil surfaces. Environ Sci Technol 20:934–938
Herbert VR, Miller GC (1990) Depth dependence of direct and indirect photolysis on soil surfaces. J Agr Food Chem 38:913–918
Mayer LM, Thornton KR, Schick LL, Jastrow JD, Harden JW (2012) Photodissolution of soil organic matter. Geoderma 170:314–321
Balmer ME, Goss KU, Schwarzenbach RP (2000) Photolytic transformation of organic pollutants on soil surfaces—an experimental approach. Environ Sci Technol 34:1240–1245
McNally AM, Moody EC, McNeill K (2005) Kinetics and mechanism of the sensitized photodegradation of lignin model compounds. Photochem Photobiol Sci 4:268–274
Krasnovsky AA, Lopez J, Cheng P, Liddell PA, Blankenship RE, Moore TA, Gust D (1994) Generation and quenching of singlet molecular oxygen by aggregated bacteriochlorophyll d in model systems and chlorosomes. Photosynth Res 40:191–198
Foote CS, Chang YC, Denny RW (1970) Chemistry of singlet oxygen X. carotenoid quenching parallels biological protection. J Am Chem Soc 92:5216–5218
McKay G, Rosario-Ortiz FL (2016) Photochemical reactivity of organic matter and its size fractions. In: Calza P, Vione D (eds) Surface water photochemistry. Royal Society of Chemistry, Cambridge
Kraeutler B, Bard AJ (1977) Heterogeneous photocatalytic synthesis of methane from acetic acid—new Kolbe reaction pathway. J Am Chem Soc 100:2239–2240
Miyoshi H, Mori H, Yoneyama H (1991) Light-induced decomposition of saturated carboxylic acids on iron oxide incorporated clay suspended in aqueous solutions. Langmuir 7:503–507
Miles CJ, Brezonik PL (1981) Oxygen consumption in humic-colored waters by a photochemical ferrous-ferric catalytic cycle. Environ Sci Technol 15:1089–1095
Rutledge S, Campbell DI, Baldocchi D, Schipper LA (2010) Photodegradation leads to increased carbon dioxide losses from terrestrial organic matter. Glob Change Bio 16:3065–3074
Lee H, Rahn T, Throop HL (2012) An accounting of C-based trace gas release during abiotic plant litter degradation. Glob Change Bio 18:1185–1195
Schrauzer GN (2011) Photoreduction of nitrogen on TiO2 and TiO2-containing minerals. In: Zang L (ed) Energy efficiency and renewable energy through nanotechnology. Springer, London
Hernes PJ, Benner R (2003) Photochemical and microbial degradation of dissolved lignin phenols: implications for the fate of terrigenous dissolved organic matter in marine environments. J Geophys Res 108:3291
Schoell M (1988) Origins of methane in the earth. Chem Geol 71:1–10
Denman KL, Brasseur G, Chidthaisong A, Ciais P, Cox PM, Dickinson RE, Hauglustaine D, Heinze C, Holland E, Jacob D, Lohmann U, Ramachandran S, da Silva Dias PL, Wofsy SC, Zhang X (2007) Couplings between changes in the climate system and biogeochemistry. In: Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL (eds) Climate change 2007: the physical science basis. Contribution of working group i to the fourth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge
Wuebbles DJ (2009) Nitrous oxide: no laughing matter. Science 326:56–57
Singh BK, Bardgett RD, Smith P, Reay DS (2010) Microorganisms and climate change: terrestrial feedbacks and mitigation options. Nat Rev Microbiol 8:779–790
Vahatalo AV, Zepp RG (2005) Photochemical mineralization of dissolved organic nitrogen to ammonium in the Baltic Sea. Environ Sci Technol 39:6985–6992
Francko DA, Heath RT (1979) Functionally distinct classes of complex phosphorus compounds in lake water. Limnol Oceanogr 24:463–473
Moran MA, Zepp RG (1997) Role of photoreactions in the formation of biologically labile compounds from dissolved organic matter. Limnol Oceanogr 42:1307–1316
Wetzel RG, Hatcher PG, Bianchi TS (1995) Natural photolysis by ultraviolet irradiance of recalcitrant dissolved organic matter to simple substrates for rapid bacterial metabolism. Limnol Oceanogr 40:1369–1380
Austin AM, Mendez SM, Ballare CL (2016) Photodegradation alleviates the lignin bottleneck for carbon turnover in terrestrial ecosystems. Proc Nat Acad Sci USA 113:4392–4397
Eggleston CM, Stern JR, Strellis TM, Parkinson BA (2012) A natural photoelectrochemical cell for water splitting: implications for early Earth and Mars. Am Miner 97:1804–1807
Fasnacht MP, Blough NV (2003) Mechanisms of the aqueous photodegradation of polycyclic aromatic hydrocarbons. Environ Sci Technol 37:5767–5772
Zhang LH, Li PJ, Gong ZQ, Li XM (2008) Photocatalytic degradation of polycyclic aromatic hydrocarbons on soil surfaces using TiO2 under UV light. J Hazard Mater 158:478–484
Marques M, Mari M, Audi-Miro C, Sierra J, Soler A, Nadal M, Domingo JL (2016) Photodegradation of polycyclic aromatic hydrocarbons in soils under a climate change base scenario. Chemosphere 148:495
Keiluweit M, Nico PS, Johnson MG, Kleber M (2010) Dynamic molecular structure of plant biomass-derived black carbon (biochar). Environ Sci Technol 44:1247–1253
Preston CM, Schmidt MWI (2006) Black (pyrogenic) carbon: a synthesis of current knowledge and uncertainties with special consideration of boreal regions. Biogeosciences 3:397–420
Schmidt MWI, Noack AG (2000) Black carbon in soils and sediments: analysis, distribution, implications, and current challenges. Glob Biogeochem Cycle 14:777–793
Lehmann J (2007) A handful of carbon. Nature 447:143–144
Druffel ERM (2004) Comments on the importance of black carbon in the global carbon cycle. Mar Chem 92:197–200
Czimczik CI, Masiello CA (2007) Controls on black carbon storage in soils. Global Biogeochem Cycle 21:GB3005
Nikandrov VV (1998) Inorganic semiconductors as photosensitizers in biochemical redox reactions. Biochem Moscow Suppl Ser A 12:755–769
Lu AH, Li Y, Jin S, Wang X, Wu XL, Zeng CP, Li Y, Ding HR, Hao RX, Lv M, Wang CQ, Tang YQ, Dong HL (2012) Growth of non-phototrophic microorganisms using solar energy through mineral photocatalysis. Nature Commun 3:768
Macia-Agullo JA, Corma A, Garcia H (2015) Photobiocatalysis: the power of combining photocatalysis and enzymes. Chem Eur J 21:10940–10959
Ausloos P, Rebbert RE, Glasgow L (1977) Photodecomposition of chloromethanes adsorbed on silica surfaces. J Res Nat Bur Stand 82:1–8
Rebbert RE, Ausloos P (1978) Decomposition of N2O over particulate matter. Geophys Res Lett 5:761–764
Pierotti D, Rasmussen LE, Rasmussen RA (1978) The Sahara as a possible sink for trace gases. Geophys Res Lett 5:1001–1004
Misztal PK, Hewitt CN, Wildt J, Blande JD, Eller ASD, Fares S, Gentner DR, Gilman JB, Graus M, Greenberg J, Guenther AB, Hansel A, Harley P, Huang M, Jardine K, Karl T, Kaser L, Keutsch FN, Kiendler-Scharr Kleist E, Lerner BM, Li T, Mak J, Nölscher AC, Schnitzhofer R, Sinha V, Thornton B, Warneke C, Wegener F, Werner C, Williams J, Worton DR, Yassaa N, Goldstein AH (2015) Atmospheric benzenoid emissions from plants rival those from fossil fuels. Sci Rep 2015:12064
Richards-Henderson NK, Pham AT, Kirk BB, Anastasio C (2015) Secondary organic aerosol from aqueous reactions of green leaf volatiles with organic triplet excited states and singlet molecular oxygen. Environ Sci Technol 49:268–276
Smith JD, Kinney H, Anastasio C (2016) Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products. Atmos Environ 126:36–44
Insam H, Seewald MSA (2010) Volatile organic compounds (VOCs) in soils. Biol Fert Soils 46:199–213
Stemmler K, Ammann M, Donders C, Kleffmann J, George C (2006) Photosensitized reduction of nitrogen dioxide on humic acid as a source of nitrous acid. Nature 440:195–198
Sarmiento AM, Oliveira V, Gómez-Ariza JL, Nieto JM, Sánchez-Rodas D (2007) Diel cycles of arsenic speciation due to photooxidation in acid mine drainage from the Iberian Pyrite Belt (SW Spain). Chemosphere 66:677–683
Gammons CH, Nimick DA, Parker SR, Cleasby TE, McCleskey RB (2005) Diel behavior of iron and other heavy metals in a mountain stream with acidic to neutral pH: Fisher Creek, Montana, USA. Geochim Cosmochim Acta 69:2505–2516
Dill C, Kuiken T, Zhang H, Ensor M (2006) Diurnal variation of dissolved gaseous mercury (DGM) levels in a southern reservoir lake (Tennessee, USA) in relation to solar radiation. Sci Total Env 357:176–193
Tate CM, Broshears RE, McKnight DM (1995) Phosphate dynamics in an acidic mountain stream: interactions involving algal uptake, sorption by iron oxide, and photoreduction. Limnol Oceanogr 40:938–946
Rusina O, Linnik O, Eremenko A, Kisch H (2003) Nitrogen photofixation on nanostructured iron titanate films. Chem Eur J 9:561–565
Linnik O, Kisch H (2006) On the mechanism of nitrogen photofixation at nanostructured iron titanate films. Photochem Photobiol Sci 5:938–942
Gupta SK, Rajakumar V, Grieveson P (1991) Phase transformations during heating of ilmenite concentrates. Metall Trans B 22:711–716
Cooper WJ, Herr FL (1987) Introduction and overview. In: Zika RG, Cooper WJ (eds) Photochemistry of environmental aquatic systems. American Chemical Society, Washington DC
Austin AT, Vivanco L (2006) Plant litter decomposition in a semi-arid ecosystem controlled by photodegradation. Nature 442:555–558
Brandt LA, Bohnet C, King JY (2009) Photochemically induced carbon dioxide production as a mechanism for carbon loss from plant litter in arid ecosystems. J Geophys Res 114:G02004
Tarr MA, Miller WL, Zepp RG (1995) Direct carbon monoxide photoproduction from plant matter. J Geophys Res 100:11403–11413
Schade GW, Hofmann RM, Crutzen PJ (1999) CO emissions from degrading plant matter (I). Measurements. Tellus B 51:889–908
Bruhn D, Albert KR, Mikkelsen TN, Ambus P (2013) UV-induced carbon monoxide emission from living vegetation. Biogeosciences 10:7877–7882
Fraser WT, Blei E, Fry SC, Newman MF, Reay DS, Smith KA, McLeod AR (2015) Emission of methane, carbon monoxide, carbon dioxide and short-chain hydrocarbons from vegetation foliage under ultraviolet radiation. Plant, Cell Environ 38:980–989
Egerton GS (1949) The mechanism of the photochemical degradation of textile materials. J Soc Dyers Colour 65:764–780 (comment by Cunliffe)
Vigano I, van Weelden H, Holziner R, Keppler F, McLeod A, Rockmann T (2008) Effect of UV radiation and temperature on the emission of methane from plant biomass and structural components. Biogeosciences 5:937–947
Bruhn D, Mikkelsen TN, Rolsted MMM, Egsgaard H, Ambus P (2014) Leaf surface wax is a source of plant methane formation under UV radiation and in the presence of oxygen. Plant Biol 16:512–516
McLeod AR, Fry SC, Loake GJ, Messenger DJ, Reay DS, Smith KA, Yun B (2008) Ultraviolet radiation drives methane emissions from terrestrial plant pectins. New Phytol 180:124–132
Bloom AA, Lee-Taylor J, Madronich S, Messenger DJ, Palmer PI, Reay DS, McLeod AR (2010) Global methane emission estimates from ultraviolet irradiation of terrestrial plant foliage. New Phytol 183:417–425
Derendorp L, Holzinger R, Rockmann T (2011) UV-induced emissions of C2-C5 hydrocarbons from leaf litter. Environ Chem 8:602–611
Gallo ME, Sinsabaugh RL, Cabaniss SE (2006) The role of ultraviolet radiation in litter composition in arid ecosystems. Appl Soil Ecol 34:82–91
Foereid B, Bellarby J, Meier-Augenstein W, Kemp H (2010) Does light exposure make plant litter more degradable? Plant Soil 333:275–285
Sancier KM, Wise H (1981) Photoassisted oxidation of organic matter catalyzed by sand. Atmos Environ 15:639–640
Jugold A, Althoff F, Hurkuck M, Greule M, Lelieveld J, Keppler F (2012) Non-microbial methane formation in oxic soils. Biogeosciences 9:11961–11987
Helms JR, Glinski DA, Mead RN, Southwell MW, Avery GB, Kieber RJ, Skrabal SA (2014) Photochemical dissolution of organic matter from resuspended sediments: impact of source and diagenetic state on photorelease. Org Geochem 73:83–89
Schiebel HN, Wang XC, Chen RF, Peri F (2015) Photochemical release of dissolved organic matter from resuspended salt marsh sediments. Estuar Coast 38:1692–1705
Tarr MA, Wang WW, Bianchi TS, Engelhaupt E (2001) Mechanisms of ammonia and amino acid photoproduction from aquatic humic and colloidal material. Water Res 35:3688–3696
Schmitt-Kopplin P, Hertkorn N, Schulten HR, Kettrup A (1998) Structural changes in a dissolved soil humic acid during photochemical degradation processes under O2 and N2 atmosphere. Environ Sci Technol 32:2531–2541
Brinkmann T, Horsch P, Sartorius D, Frimmel FH (2003) Photoformation of low-molecular-weight organic acids from brown water dissolved organic matter. Environ Sci Technol 37:4190–4198
Noorjahan A, Kumari VD, Subrahmanyam A, Panda L (2005) Immobilized Fe(III)-HY: an efficient and stable photo-Fenton catalyst. Appl Catal B 57:291–298
Conrad R, Seiler W, Bunse G, Giehl H (1982) Carbon monoxide in sea water (Atlantic Ocean). J Geophys Res 87:8839–8852
Mopper K, Zhou XL, Kieber RJ, Sikorski RJ, Jones RD (1991) Photochemical degradation of dissolved organic carbon and its impact on the oceanic carbon cycle. Nature 353:60–62
Miller WL, Zepp RG (1995) Photochemical production of dissolved inorganic carbon from terrestrial organic matter—significance to the oceanic organic carbon cycle. Geophys Res Lett 22:417–420
Valentine RL, Zepp RG (1993) Formation of carbon monoxide from the photodegradation of terrestrial dissolved organic carbon in natural waters. Environ Sci Technol 27:409–412
Matthews RW (1986) Photooxidation of organic material in aqueous suspensions of titanium dioxide. Water Res 20:569–578
Chen HM, Abdulla HAN, Sanders RL, Myneni SCB, Mopper K, Hatcher PG (2014) Production of black carbon-like and aliphatic molecules from terrestrial dissolved organic matter in the presence of sunlight and iron. Environ Sci Technol Lett 1:399–404
Aarnos H, Ylöstalo P, Vähätalo AV (2012) Seasonal phototransformation of dissolved organic matter to ammonium, dissolved inorganic carbon, and labile substrates supporting bacterial biomass across the Baltic Sea. J Geophys Res 117:G01004
Zhang Y, Xie H (2015) Photomineralization and photomethanification of dissolved organic matter in Saguenay River surface water. Biogeosciences 12:6823–6836
Kieber DJ, McDaniel J, Mopper K (1989) Photochemical source of biological substrates in sea water—implications for carbon cycling. Nature 341:637–639
Kieber RJ, Zhou XL, Mopper K (1990) Formation of carbonyl compounds from UV-induced photodegradation of humic substances in natural waters: fate of riverine carbon in the sea. Limnol Oceanogr 35:1503–1515
Palit CC, Dhar NR (1928) Oxidation of carbohydrates, fats, and nitrogenous products by air in presence of sunlight. J Phys Chem 32:1263–1268
Egerton GS (1949) The mechanism of the photochemical degradation of textile materials. J Soc Dyers Colour 65:764–780
Reine AH, Arthur JC Jr (1970) Photochemistry of cotton cellulose. The direct action of near-ultraviolet light on purified fibrous cotton cellulose. Text Res J 40:90–92
Andrady AL, Torikai A, Kobatake T (1996) Spectral sensitivity of chitosan photodegradation. J Appl Polym Sci 62:1465–1471
Dahm A, Lucia LA (2004) Titanium dioxide catalyzed photodegradation of lignin in industrial effluents. Ind Eng Chem Res 43:7996–8000
Castellan A, Colombo N, Vanucci C, Fornier de Violet P, Bouas-Laurent H (1990) A photochemical study of an o-methylated α-carbonyl β-1 lignin model dimer: 1,2-di(3′,4′-dimethoxyphenyl)ethanone (deoxyveratroin). J Photochem Photobiol, A 51:451–467
Weir NA, Arct J, Ceccarelli A, Puumala D (1996) Long-wave photochemistry of di-methoxylated compounds related to lignin. Polym Degrad Stabil 52:119–129
Neumann MG, Machado AEH (1989) The role of oxygen in the photodegradation of lignin solution. J Photochem Photobiol, B 3:473–481
Gellerstedt G, Pettersson E (1975) Light-induced oxidation of lignin. The behaviour of structural units containing a ring-conjugated double bond. Acta Chem Scand B 29:1005–1010
Pattison DI, Rahmanto AS, Davies MJ (2012) Photo-oxidation of proteins. Photochem Photobiol Sci 11:38–53
Wheeler J (1972) Some effects of solar levels of ultraviolet radiation on lipids in artificial sea water. J Geophys Res 77:5302–5306
Harvey GR, Boran DA, Chesal LA, Tokar JM (1983) The structure of marine fulvic and humic acids. Mar Chem 12:119–132
Rossignol S, Tinel L, Bianco A, Passananti M, Brigante M, Donaldson DJ, George C (2016) Atmospheric photochemistry at a fatty acid-coated air-water interface. Science 353:699–702
Mills A, Davies RH, Worsley D (1993) Water purification by semiconductor photocatalysis. Chem Soc Rev 22:417–425
Bridgen CT, Poulston S, Twigg MV, Walker AP, Wilkins AJJ (2001) Photo-oxidation of short-chain hydrocarbons over titania. Appl Catal B 32:63–71
O’Brien RJ, Holmes JR, Bocklan AH (1975) Formation of photochemical aerosol from hydrocarbons. Environ Sci Technol 9:568–576
Borghesi D, Vione D, Maurino V, Minero C (2005) Transformations of benzene photoinduced by nitrate salts and iron oxide. J Atmos Chem 52:259–281
Minero C, Bono F, Rubertelli F, Pavino D, Maurino V, Pelizzetti E, Vione D (2007) On the effect of pH in aromatic photonitration upon nitrate photolysis. Chemosphere 66:650–656
Vione D, Maurino V, Minero C, Vincenti M, Pelizzetti E (2001) Aromatic photonitration in homogeneous and heterogeneous aqueous systems. Environ Sci Pollut Res 10:321–324
Niessen R, Lenoir D, Boule P (1988) Phototransformation of phenol induced by excitation of nitrate ions. Chemosphere 17:1977–1984
Grosjean D (1984) Atmospheric reactions of ortho cresol: gas phase and aerosol products. Atmos Environ 18:1641–1652
Zepp RG, Schlotzhauer PF (1983) Influence of algae on photolysis rates of chemicals in water. Environ Sci Technol 17:462–468
Stubbins A, Spencer RGM, Chen H, Hatcher PG, Mopper K, Hernes PJ, Mwamba VL, Mangangu AM, Wabakanghanzi JN, Six J (2010) Illuminated darkness: molecular signatures of Congo River dissolved organic matter and its photochemical alteration as revealed by ultrahigh precision mass spectrometry. Limnol Oceanogr 55:1467–1477
Ward CP, Sleighter RL, Hatcher PG, Cory RM (2014) Insights into the complete and partial photooxidation of black carbon in surface waters. Environ Sci Process Impacts 16:721–731
Han C, Liu YC, Ma JZ, He H (2012) Key role of organic carbon in the sunlight-enhanced atmospheric aging of soot by O2. Proc Nat Acad Sci USA 109:21250–21255
Wise H, Sancier KM (1991) Photocatalyzed oxidation of crude oil residue by beach sand. Catal Lett 11:277–284
Moffett JW, Zika RG (1987) Photochemistry of copper complexes in sea water. In: Zika RG, Cooper WJ (eds) Photochemistry of Environmental Aquatic Systems. American Chemical Society, Washington DC
Natarajan P, Ferraudi G (1981) Photochemical properties of copper(II)-amino acid complexes. Inorg Chem 20:3708–3712
Milne PJ, Zika RG (1993) Amino acid nitrogen in atmospheric aerosols: occurrence, sources, and photochemical modification. J Atmos Chem 16:361–398
Ohtani B, Tsuru S, Nishimoto S, Kagiya T, Izawa K (1990) Photocatalytic one-step syntheses of cyclic imino acids by aqueous semiconductor suspensions. J Org Chem 55:5551–5553
Pal B, Ikeda S, Ohtani B (2003) Photoinduced chemical reactions on natural single crystals and synthesized crystallites of mercury(II) sulfide in aqueous solution containing naturally occurring amino acids. Inorg Chem 42:1518–1524
Sclafani A, Palmisano L, Schiavello M (1993) N2 photoreduction and phenol and nitrophenol isomers photooxidation as examples of heterogeneous photocatalytic reactions. Res Chem Intermed 18:211–226
Chatterjee S, Sarkar S, Bhattacharyya SN (1994) Photodegradation of phenol by visible light in the presence of colloidal Fe2O3. J Photochem Photobiol, A 81:199–203
Larson RA, Ellis DD, Ju HL, Marley KA (1989) Flavin-sensitized photodecomposition of anilines and phenols. Environ Toxicol Chem 8:1165–1170
Canonica S, Jans U, Stemmler K, Hoigne J (1995) Transformation kinetics of phenols in water—photosensitization by dissolved natural organic material and aromatic ketones. Environ Sci Technol 29:1822–1831
Gibbs HD (1912) The action of sunlight upon phenolic compounds and aniline. J Am Chem Soc 34:1190–1207
Haggiage E, Coyle EE, Joyce K, Oelgemoller M (2009) Green photochemistry: solarchemical synthesis of 5-amido-1,4-naphthoquinones. Green Chem 11:318–321
Schonberg A, Mustafa A, Barakat MZ (1947) Dimerization reactions in sunlight. Nature 160:401–402
Bruce JM (1962) The preparation of some 2,3:6,7-dibenzobiphenylenes. J Chem Soc 1962:2782–2785
Krauch CH, Farid S (1966) Competition between cyclobutane and oxetane formation in the photoaddition of 1,4-naphthoquinone to benzocyclic olefins. Tetrahedron Lett 39:4783–4788
Ciamician G, Silber P (1907) Chemische Lichtwirkungen. XI. Mitteilung [Chemical effects of light. XI. Communication]. Ber Dtsch Chem Ges 40:2415–2424
Quinkert G (1965) Light-induced formation of acids from cyclic ketones. Angew Chem Int Ed 4:211–222
Haas Y (2004) Photochemical α-cleavage of ketones: revisiting acetone. Photoch Photobio Sci 3:6–16
Schonberg A, Mustafa A (1947) Reactions of non-enolizable ketones in sunlight. Chem Rev 40:181–200
Ito Y, Kunimoto K, Miyachi S, Kako T (1991) Photocatalytic cleavage of 1,2-diols by a cofacially hindered water-soluble iron(III) porphyrin. Tetrahedron Lett 32:4007–4010
Hein SM (2006) An exploration of a photochemical pericyclic reaction using NMR data. J Chem Educ 83:940–942
Sakata T, Kawai T, Hashimoto K (1984) Heterogeneous photocatalytic reactions of organic acids and water—new reaction paths besides the photo-Kolbe reaction. J Phys Chem 88:2344–2350
Schenk GO, Ziegler K (1944) Die synthese des ascaridols. Naturwissenschaften 32:157
Kormann C, Bahnemann DW, Hoffmann MR (1988) Photocatalytic production of H2O2 and organic peroxides in aqueous suspensions of TiO2, ZnO, and desert sand. Environ Sci Technol 22:798–806
Chiron F, Chalchat JC, Garry RP, Pilichowski JF, Lacoste J (1997) Photochemical hydroperoxidation of terpenes I. synthesis and characterization of α-pinene, β-pinene and limonene hydroperoxides. J Photochem Photobiol, A 111:75–86
Paillous N, Fery-Forgues S (1994) Interest of photochemical methods for induction of lipid peroxidation. Biochimie 76:355–368
Khan NA, Lundberg WO, Holman RT (1954) Displacement analysis of lipids. IX. Products of the oxidation of methyl linoleate. J Am Chem Soc 76:1779–1784
Wang WH, Beyerle-Pfnür R, Lay JP (1988) Photoreaction of salicylic acid in aquatic systems. Chemosphere 17:1197–1204
Harada H, Ueda T, Sakata T (1989) Semiconductor effect on the selective photocatalytic reaction of α-hydroxycarboxylic acids. J Phys Chem 93:1542–1548
Matsushima R, Ichikawa Y, Kuwabara K (1980) Photooxidation of 2-hydroxy acids by copper(II) species in aqueous solution. B Chem Soc Jpn 52:1902–1907
Styler SA, Donaldson DJ (2012) Heterogeneous photochemistry of oxalic acid on Mauritanian sand and Icelandic volcanic ash. Environ Sci Technol 46:8756–8763
Leland JK, Bard AJ (1987) Photochemistry of colloidal semiconducting iron oxide polymorphs. J Phys Chem 91:5076–5083
Nikandrov VV, Gratzel CK, Moser JE, Gratzel M (1997) Light induced redox reactions involving mammalian ferritin as photocatalyst. J Photochem Photobiol, B 41:82–89
Guzman MI, Colussi AJ, Hoffmann MR (2006) Photoinduced oligomerization of aqueous pyruvic acid. J Phys Chem A 110:3619–3626
Dallin E, Wan P, Krogh E, Gill C, Moore RM (2009) New pH-dependent photosubstitution pathways of syringic acid in aqueous solution: relevance in environmental photochemistry. J Photochem Photobiol, A 207:297–305
Yanagida S, Azume T, Kawakami H, Kizumoto H, Sakurai H (1984) Photocatalytic carbon-carbon bond formation with concurrent hydrogen evolution on colloidal zinc sulfide. J Chem Soc, Chem Commun 1:21–22
Claeys M, Graham B, Vas G, Wang W, Vermeylen R, Pashynska V, Cafmeyer J, Guyon P, Andreae MO, Artaxo P, Maenhaut W (2004) Formation of secondary organic aerosols through photooxidation of isoprene. Science 303:1173–1176
Wat CK, Johns T, Towers GHN (1980) Phototoxic and antibiotic activities of plants of the Asteraceae used in folk medicine. J Ethnopharmacol 2:279–290
Larson RA, Berenbaum MR (1988) Environmental phototoxicity. Environ Sci Technol 22:354–360
Tennakone K, Jayatissa AH, Punchihewa S (1989) Selective photoreduction of carbon dioxide to methanol with hydrous cuprous oxide. J Photochem Photobiol, A 49:369–375
Matsumoto Y, Obata M, Hombo J (1994) Photocatalytic reduction of carbon dioxide on p-type CaFe2O4 powder. J Phys Chem 98:2950–2951
Yamashita H, Nishiguchi N, Kamada N, Anpo M, Teraoka Y, Hatano H, Ehara S, Kikui K, Palmisano L, Sclafani A, Schiavello M, Fox MA (1994) Photocatalytic reduction of CO2 with H2O on TiO2 and Cu/TiO2 catalysts. Res Chem Intermed 20:815–823
Matsuoka M, Anpo M (2003) Local structures, excited states, and photocatalytic reactivities of highly dispersed catalysts constructed within zeolites. J Photochem Photobiol, C 3:225–252
Roy SC, Varghese OK, Paulose M, Grimes CA (2010) Toward solar fuels: photocatalytic conversion of carbon dioxide to hydrocarbons. ACS Nano 4:1259–1278
Habisretinger SN, Schmidt-Mende L, Stolarczyk JK (2013) Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew Chem Int Ed 52:7372–7408
Li KF, An XQ, Park KH, Khraisheh M, Tang JW (2014) A critical review of CO2 photoconversion: catalysts and reactors. Catal Today 223:3–12
Lichtin NN, Vijayakumar KM, Rubio BI (1987) Photoassisted reduction of CO2 by H2 over metal oxides in the absence and presence of water vapor. J Catal 104:246–251
Sastre F, Puga AV, Liu LC, Corma A, Garcia H (2014) Complete photocatalytic reduction of CO2 to methane by H2 under solar light irradiation. J Am Chem Soc 136:6798–6801
Premkumar J, Ramaraj R (1997) Photocatalytic reduction of carbon dioxide to formic acid at porphyrin and phthalocyanine adsorbed Nafion membranes. J Photochem Photobiol, A 110:53–58
Ohta K, Ueda Y, Nakaguchi S, Mizuno T (1998) Photochemical reduction of CO2 using Cu-loaded silcate rock powder suspended in water. Can J Chem 76:228–233
Yamamura S, Kojima H, Iyoda J, Kawai W (1987) Formation of ethyl alcohol in the photocatalytic reduction of carbon dioxide by SiC and ZnSe/metal powders. J Electroanal Chem 225:287–290
Cook RL, MacDuff RC, Sammells AF (1988) Photoelectrochemical carbon dioxide reduction to hydrocarbons at ambient temperature and pressure. J Electrochem Soc 135:3069–3070
Eggins BR, Robertson PKJ, Stewart JH, Woods E (1993) Photoreduction of carbon dioxide on zinc sulfide to give four-carbon and two-carbon acids. J Chem Soc, Chem Commun 4:349–350
Liu YY, Huang BB, Dai Y, Zhang XY, Qin XY, Jiang MH, Whangbo MH (2009) Selective ethanol formation from photocatalytic reduction of carbon dioxide in water with BiVO4 photocatalyst. Catal Commun 11:210–213
Tahir M, Amin NS (2013) Photocatalytic reduction of carbon dioxide with water vapors over montmorillonite modified TiO2 nanocomposites. Appl Catal B 142:512–522
Lien CF, Chen MT, Lin YF, Lin JL (2004) Photooxidation of methane over TiO2. J Chin Chem Soc 51:37–42
Yoshida H, Matsushita N, Kato Y, Hattori T (2003) Synergistic active sites on SiO2-Al2O3-TiO2 photocatalysts for direct methane coupling. J Phys Chem B 106:8355–8362
Hari P, Raivonen M, Vesala T, Munger JW, Pilegaard K, Kulmana M (2003) Atmospheric science: ultraviolet light and leaf emission of NOx. Nature 422:134
Bruhn D, Albert KR, Mikkelsen TN, Ambus P (2014) UV-induced N2O emission from plants. Atmos Environ 99:206–214
Bushaw KL, Zepp RG, Tarr MA, Schulz-Jander D, Bourbonniere RA, Hodson RE, Miller WL, Bronk DA, Moran MA (1996) Photochemical release of biologically available nitrogen from aquatic dissolved organic matter. Nature 381:404–407
Rao GG, Varadanam CI (1938) Photo-ammonification of organic nitrogenous compounds in the soil. Nature 142:618
Jeff S, Hunter K, Vandergucht D, Hudson J (2012) Photochemical mineralization of dissolved organic nitrogen to ammonia in prairie lakes. Hydrobiologia 693:71–80
Lundeen RA, Janssen EML, Chu CH, McNeill K (2014) Environmental photochemistry of amino acids, peptides, and proteins. Chimia 68:812–817
Porcal P, Kopacek J, Tomkova I (2014) Seasonal photochemical transformations of nitrogen species in a forest stream and lake. PLoS ONE 9:e116364
Kieber RJ, Li A, Seaton PJ (1999) Production of nitrite from the photodegradation of dissolved organic matter in natural waters. Environ Sci Technol 33:993–998
Rao GG, Dhar NR (1930) Photosensitized oxidation of ammonia and ammonium salts and the problem of nitrification in soils. Soil Sci 31:379–384
ZoBell CE (1933) Photochemical nitrification in sea water. Science 77:27–28
McLean WR, Ritchie M (1965) Reactions on titanium dioxide: the photo-oxidation of ammonia. J Appl Chem 15:452–460
Mozzanega H, Herrmann JM, Pichat P (1979) NH3 oxidation over UV-irradiated TiO2 at room temperature. J Phys Chem 83:2251–2255
Dhar NR (1934) Denitrification in sunlight. Nature 134:572–573
Dhar NR, Pant NN (1944) Nitrogen loss from soils and oxide surfaces. Nature 153:115–116
Rubasinghege G, Spak SN, Stainer CO, Carmichael GR, Grassian VH (2011) Abiotic mechanism for the formation of atmospheric nitrous oxide from ammonium nitrate. Environ Sci Technol 45:2691–2697
Dalton JS, Janes PA, Jones NG, Nicholson JA, Hallam KR, Allen GC (2002) Photocatalytic oxidation of NOx gases using TiO2: a surface spectroscopic approach. Environ Poll 120:415–422
Bedjanian Y, El Zein A (2012) Interaction of NO2 with TiO2 surface under UV irradiation: products study. J Phys Chem A 116:1758–1764
Milis A, Domenech X (1993) Photoassisted oxidation of nitrite to nitrate over different semiconducting oxides. J Photochem Photobiol, A 72:55–59
Ohtani B, Kakimoto M, Miyadzu H, Nishimoto S, Kagiya T (1988) Effect of surface-adsorbed 2-propanol on the photocatalytic reduction of silver and/or nitrate ions in acidic TiO2 suspension. J Phys Chem 92:5773–5777
Warneck P, Wurzinger C (1988) Product quantum yields for the 305-nm photodecomposition of NO3 − in aqueous solution. J Phys Chem 92:6278–6283
Grannas AM, Jones AE, Dibb J, Ammann M, Anastasio C, Beine HJ, Bergon M, Bottenheim J, Boxe CS, Carver G, Chen G, Crawford JH, Domine F, Frey MM, Guzman MI, Heard DE, Helmig D, Hoffmann MR, Honrath RE, Huey LG, Hutterli M, Jacobi HW, Klan P, Lefer B, McConnell J, Plane J, Sander R, Savarino J, Shepson PB, Simpson WE, Sodeau JR, von Glasow R, Weller R, Wolff EW, Zhu T (2007) An overview of snow photochemistry: evidence, mechanisms, and impacts. Atmos Chem Phys 7:4329–4373
Ketir W, Bouguelia A, Trari M (2009) NO3 − removal with a new delafossite CuCrO2 photocatalyst. Desalination 244:144–152
Ndour M, Conchon P, D’Anna B (2009) Photochemistry of mineral dust surfaces as a potential atmospheric renoxification process. Geophys Res Lett 36:L05816
Rubasinghege G, Grassian VH (2009) Photochemistry of adsorbed nitrate on aluminum oxide particle surfaces. J Phys Chem A 113:7818–7825
Gankanda A, Grassian VH (2014) Nitrate photochemistry on laboratory proxies of mineral dust aerosol: wavelength dependence and action spectra. J Phys Chem C 118:29117–29125
Lesko DMB, Coddens EM, Swomely HD, Welch RM, Borgatta J, Navea JG (2015) Photochemistry of nitrate chemisorbed on various metal oxide surfaces. Phys Chem Chem Phys 17:20775–20785
Moore B (1918) The formation of nitrites from nitrates in aqueous solution by the action of sunlight, and the assimilation of the nitrites by green leaves in sunlight. Proc R Soc Lond B 90:158–167
Rao GG, Murty KS (1937) Photocatalytic reduction of nitrate and the simultaneous oxidation of ammonia to nitrite. Proc Natl Inst Sci India 3:133–137
Spokes LJ, Liss PS (1996) Photochemically induced redox reactions in seawater. II. Nitrogen and iodine. Mar Chem 54:1–10
Bems B, Jentoft FC, Schlogl R (1998) Photoinduced decomposition of nitrate in drinking water in the presence of titania and humic acids. Appl Catal B Environ 20:155–163
Bartels-Rausch T, Brigante M, Elshorbany YF, Ammann M, D’Anna B, George C, Stemmler K, Ndour M, Kleffmann J (2010) Humic acid in ice: photo-enhanced conversion of nitrogen dioxide into nitrous acid. Atmos Environ 44:5443–5450
Monge ME, D’Anna B, Mazri L, Giroir-Fendler A, Ammann M, Donaldson DJ, George C (2010) Light changes the atmospheric reactivity of soot. Proc Natl Acad Sci USA 107:6605–6609
Cunningham J, Kelly JJ, Penny AL (1971) Reactions involving electron transfer at semiconductor surfaces. 2. Photoassisted dissociation of nitrous oxide over illuminated ferric oxide and zinc oxides. J Phys Chem 75:617–625
Tanaka K, Blyholder G (1971) Photocatalytic reactions on semiconductor surfaces. I. Decomposition of nitrous oxide on zinc oxide. J Phys Chem 75:1037–1043
Anpo M, Matsuoka M, Hano K, Mishima H, Ono T, Yamashita H (1997) Photocatalytic decomposition of N2O on Cu+/Y-zeolite catalysts prepared by ion exchange. Korean J Chem Eng 14:498–501
Khan F, Yue PL, Ruzzuti L, Augugliaro V, Schiavello M (1981) Photochemical synthesis of ammonia over zeolites. J Chem Soc, Chem Commun 20:1049–1050
Yue PL, Khan F, Rizzuti L (1983) Photocatalytic ammonia synthesis in a fluidized bed reactor. Chem Eng Sci 38:1893–1900
Dhar NR, Francis AM (1951) Fixation of atmospheric nitrogen under sterile conditions, in sand, using different energy materials. Proc Natl A Sci India 20:112–117
Dhar NR (1958) Influence de la lumière sur la fixation de l’azote [Influence of light on the fixation of nitrogen]. J Chim Phys PCB 55:980–984
Miyama H, Fujii N, Nagae Y (1980) Heterogeneous photocatalytic synthesis of ammonia from water and nitrogen. Chem Phys Lett 74:523–524
Tennakone K, Ileperuma OA, Bandara JMS, Thaminimulla CTK, Ketipearachchi US (1991) Simultaneous reductive and oxidative photocatalytic nitrogen fixation in hydrous iron(III) oxide loaded nafion films in aerated water. J Chem Soc Chem Comm 8:579–580
Khader MM, Lichtin NN, Vurens GH, Salmeron M, Somorjai GA (1987) Photoassisted catalytic dissociation of H2O and reduction of N2 to NH3 on partially reduced Fe2O3. Langmuir 3:303–304
Schrauzer GN, Guth TD (1977) Photolysis of water and photoreduction of nitrogen on titanium dioxide. J Amer Chem Soc 99:7189–7190
Bickley RI, Vishwanathan V (1979) Photocatalytically induced fixation of molecular nitrogen by near UV radiation. Nature 280:306–308
Ileperuma OA, Weerasinghe FNS, Bandara TSL (1989) Photoinduced oxidative nitrogen fixation reactions on semiconductor suspensions. Sol Energy Mater 19:409–414
Al-Taani AA (2008) Non-biological fixation of atmospheric nitrogen to nitrate on titanium dioxide and desert soil surfaces. Thesis (Ph.D.), University of Nevada, Reno
Tennakone K, Ileperuma OA, Thaminimulla CTK, Bandara JMS (1992) Photo-oxidation of nitrogen to nitrite using a composite ZnO-Fe2O3 catalyst. J Photochem Photobiol, A 66:375–378
Porcal P, Amirbahman A, Kopacek J, Novak F, Norton SA (2009) Photochemical release of humic and fulvic acid-bound metals from simulated soil and streamwater. J Environ Monit 11:1064–1071
Shiller AM, Duan S, van Erp P, Bianchi TS (2006) Photo-oxidation of dissolved organic matter in river water and its effect on trace element speciation. Limnol Oceanogr 51:1716–1728
Helms JR, Mao JD, Schmidt-Rohr K, Abdulla H, Mopper K (2013) Photochemical flocculation of terrestrial dissolved organic matter and iron. Geochim Cosmochim Acta 121:398–413
Braterman PS, Cairns-Smith AG, Sloper RW (1983) Photo-oxidation of hydrated Fe2+ - significance for banded iron formations. Nature 303:163–164
Goldberg MC, Cunningham KM, Weiner ER (1993) Aquatic photolysis: photolytic redox reactions between goethite and adsorbed organic acids in aqueous solutions. J Photochem Photobiol, A 73:105–120
Finden DAS, Tipping E, Jaworski GHM, Reynolds CS (1984) Light-induced reduction of natural iron(III) oxide and its relevance to phytoplankton. Nature 309:783–784
Waite TD, Morel FMM (1984) Photoreductive dissolution of colloidal iron oxides in natural waters. Environ Sci Technol 18:860–868
Faust BC, Hoffmann MR (1986) Photoinduced reductive dissolution of α-Fe2O3 by bisulfite. Environ Sci Technol 20:943–948
Pehkonen SO, Siefert R, Erel Y, Webb S, Hoffmann MR (1993) Photoreduction of iron oxyhydroxides in the presence of important atmospheric organic compounds. Environ Sci Technol 27:2056–2062
Zhu XR, Prospero JM, Savoie DL, Millero FJ, Zika RG, Saltzman ES (1993) Photoreduction of iron(III) in marine mineral aerosol solutions. J Geophys Res 98:9039–9046
Sherman DM (2005) Electronic structures of iron(III) and manganese(IV) (hydr)oxide minerals: thermodynamics of photochemical reductive dissolution in aquatic environments. Geochim Cosmochim Acta 69:3249–3255
Kim K, Choi W, Hoffmann MR, Yoon HI, Park BK (2010) Photoreductive dissolution of iron oxides trapped in ice and its environmental implications. Environ Sci Technol 44:4142–4148
Weiss J (1935) Photochemical reactions connected with the quenching of fluorescence of dyestuffs by ferrous ions. Nature 136:794–795
Borowska ZK, Mauzerall DC (1987) Efficient near ultraviolet light induced formation of hydrogen by ferrous hydroxide. Origins Life Evol B 17:251–259
Balzani V, Carassiti V (1970) Photochemistry of Coordination Compounds. Academic Press, London
Bienfait HF, Scheffers MR (1992) Some properties of ferric citrate relevant to the iron nutrition of plants. Plant Soil 143:141–144
Sunda WG, Huntsman SA, Harvey GR (1983) Photo-reduction of manganese oxides in seawater and its geochemical and biological implications. Nature 301:234–236
Waite TD, Wrigley IC, Szymczak R (1988) Photoassisted dissolution of a colloidal manganese oxide in the presence of fulvic acid. Environ Sci Technol 22:778–785
Bertino DJ, Zepp RG (1991) Effects of solar radiation on manganese oxide reactions with selected organic compounds. Environ Sci Technol 25:1267–1273
Spokes LJ, Liss PS (1995) Photochemically induced redox reactions in seawater. I. Cations. Mar Chem 49:201–213
Kim K, Yoon HI, Choi W (2012) Enhanced dissolution of manganese oxide in ice compared to aqueous phase under illuminated and dark conditions. Environ Sci Technol 46:13160–13166
Nico PS, Anastasio C, Zasoski RJ (2002) Rapid photo-oxidation of Mn(II) mediated by humic substances. Geochim Cosmochim Acta 66:4047–4056
Lozano A, García J, Domènech X, Casado J (1992) Heterogeneous photocatalytic oxidation of manganese(II) over TiO2. J Photoch Photobiol A 69:237–240
Kim I, Hosein H, Strongin DR, Douglas T (2002) Photochemical reactivity of ferritin for Cr(VI) reduction. Chem Mater 14:4874–4879
Mytych P, Stasicka Z (2004) Photochemical reduction of chromium(VI) by phenol and its halogen derivatives. Appl Catal B 52:167–172
Zhang H (2006) Photochemical redox reactions of mercury. Struct Bond 120:37–79
Lalonde JD, Amyot M, Kraepiel AML, Morel FMM (2001) Photooxidation of Hg(0) in artificial and natural waters. Environ Sci Technol 35:1367–1372
Lalonde JD, Poulain AJ, Amyot M (2002) The role of mercury redox reactions in snow on snow-to-air mercury transfer. Environ Sci Technol 36:174–178
O’Driscoll NJ, Lean DRS, Loseto LL, Carignan R, Siciliano SD (2004) Effect of dissolved organic carbon on the photoproduction of dissolved gaseous mercury in lakes: potential impacts of forestry. Environ Sci Technol 38:2664–2672
Bartels-Rausch T, Krysztofiak G, Bernhard A, Schlappi M, Schwikowski M, Ammann M (2011) Photoinduced reduction of divalent mercury in ice by organic matter. Chemosphere 82:199–203
Amyot M, Mierle G, Lean DRS, McQueen DJ (1994) Sunlight-induced formation of dissolved gaseous mercury in lake waters. Environ Sci Technol 28:2366–2371
Siciliano SD, O’Driscoll NJ, Tordon R, Hill J, Beauchamp S, Lean DRS (2005) Abiotic production of methylmercury by solar radiation. Environ Sci Technol 39:1071–1077
Jeremiason JD, Portner JC, Aiken GR, Hiranaka AJ, Dvorak MT, Tran KT, Latch DE (2015) Photoreduction of Hg(II) and photodemethylation of methylmercury: the key role of thiol sites on dissolved organic matter. Environ Sci Process Impacts 17:1892–1903
Lehnherr I, St. Louis VL (2009) Importance of ultraviolet radiation in the photodemethylation of methylmercury in freshwater ecosystems. Environ Sci Technol 43:5692–5698
Chen J, Pehkonen SO, Lin CJ (2003) Degradation of monomethylmercury chloride by hydroxyl radicals in simulated natural waters. Water Res 37:2496–2504
Derendorp L, Quist JB, Holzinger R, Rockmann T (2011) Emissions of H2 and CO from leaf litter of Sequoiadendron giganteum, and their dependence on UV radiation and temperature. Atmos Environ 45:7520–7524
Lee H, Rahn T, Throop HL (2012) A novel source of atmospheric H2: abiotic degradation of organic material. Biogeosciences 9:4411–4419
Vahatalo AV, Salonen K, Munster U, Jarvinen M, Wetzel RG (2003) Photochemical transformation of allochthonous organic matter provides bioavailable nutrients in a humic lake. Arch Hydrobiol 156:287–314
Francko DA, Heath RT (1983) Abiotic uptake and photodependent release of phosphate from high-molecular-weight humic-iron complexes in bog lakes. In: Christman RF, Gjessing ET (eds) Aquatic and terrestrial humic materials. Ann Arbor Science, Ann Arbor
Francko DA, Heath RT (1982) UV-sensitive complex phosphorus: association with dissolved humic material and iron in a bog lake. Limnol Oceanogr 27:564–569
Serpone N, Borgarello E, Gratzel M (1984) Visible light induced generation of hydrogen from H2S in mixed semiconductor dispersions; improved efficiency through inter-particle electron transfer. J Chem Soc, Chem Commun 6:243–344
Chaudhari NS, Warule SS, Muduli S, Kale BB, Jouen S, Lefez B, Hannoyer B, Ogale SB (2011) Maghemite (hematite) core (shell) nanorods via thermolysis of a molecular solid of Fe-complex. Dalton T 40:8003–8011
Frank SN, Bard AJ (1977) Heterogeneous photocatalytic oxidation of cyanide and sulfite in aqueous solutions at semiconductor powders. J Phys Chem 81:1484–1488
Faust BC, Hoffmann MR, Bahnemann DW (1989) Photocatalytic oxidation of sulfur dioxide in aqueous suspensions of α-Fe2O3. J Phys Chem 93:6371–6381
Baltrusaitis J, Jayaweera PM, Grassian VH (2011) Sulfur dioxide adsorption on TiO2 nanoparticles: influence of particle size, coadsorbates, sample pretreatment, and light on surface speciation and surface coverage. J Phys Chem C 115:492–500
Nanayakkara CE, Pettibone J, Grassian VH (2012) Sulfur dioxide adsorption and photooxidation on isotopically labelled titanium dioxide nanoparticle surfaces: roles of surface hydroxyl groups and adsorbed water in the formation and stability of adsorbed sulfite and sulfate. Phys Chem Chem Phys 14:6957–6966
Grosjean D (1984) Photooxidation of methyl sulfide, ethyl sulfide, and methanethiol. Environ Sci Technol 18:460–468
Zepp RG, Skurlatov YI, Pierce JT (1987) Algal-induced decay and formation of hydrogen peroxide in water: its possible role in oxidation of anilines by algae. In: Zika RG, Wopper WJ (eds) Photochemistry of environmental aquatic systems. American Chemical Society, Washington DC
Faust BC, Zepp RG (1993) Photochemistry of aqueous iron(III) polycarboxylate complexes—roles in the chemistry of atmospheric and surface waters. Environ Sci Technol 27:2517–2522
Premkumar J, Ramaraj R (1999) Photoreduction of dioxygen to hydrogen peroxide at porphyrins and phthalocyanines adsorbed Nafion membrane. J Mol Catal A 142:153–162
Kiwi J, Gratzel M (1987) Light-induced hydrogen formation and photo-uptake of oxygen in colloidal suspensions of α-Fe2O3. J Chem Soc, Faraday Trans 83:1101–1108
Kiwi J, Gratzel M (1987) Characterization of tunstosilicate/titania suspensions and their activity in the evolution of hydrogen from water. J Phys Chem 91:6673–6677
Kuznichi SM, Eyring EM (1978) “Water splitting” by titanium exchanged zeolite A. J Am Chem Soc 100:6790–6791
Abe R (2010) Recent progress on photocatalytic and photoelectrochemical water splitting under visible light irradiation. J Photochem Photobiol, C 11:179–209
Yeh TF, Syu JM, Cheng C, Chang TH, Teng HS (2010) Graphite oxide as a photocatalyst for hydrogen production from water. Adv Funct Mater 20:2255–2262
Ismail AA, Bahnemann DW (2014) Photochemical splitting of water for hydrogen production by photocatalysis: a review. Sol Energy Mater Sol Cells 128:85–101
Buxton GV, Wilford SP, Williams RJ (1962) The photo-oxidation of water by ferric ion at 25°. J Chem Soc 1962:4957–4962
Ohmori T, Takahashi H, Mametsuka H, Suzuki E (2000) Photocatalytic oxygen evolution on alpha-Fe2O3 films using Fe3+ ion as a sacrificial oxidizing agent. Phys Chem Chem Phys 2:3519–3522
Silva CG, Boulzi Y, Fornes V, Garcia H (2009) Layered double hydroxides as highly efficient photocatalysts for visible light oxygen generation from water. J Am Chem Soc 131:13833–13839
Jiao F, Frei H (2010) Nanostructured cobalt and manganese oxide clusters as efficient water oxidation catalysts. Energy Environ Sci 3:1018–1027
Robinson DM, Go YB, Mui M, Gardner G, Zhang ZJ, Mastrogiovanni D, Garfunkel E, Li J, Greenblatt M, Dismukes GC (2013) Photochemical water oxidation by crystalline polymorphs of manganese oxides: structural requirements for catalysis. J Am Chem Soc 135:3494–3501
Xu SM, Pan T, Dou YB, Yan H, Zhang ST, Ning FY, Shi WY, Wei M (2015) Theoretical and experimental study on M(II)M(III)-layered double hydroxides as efficient photocatalysts toward oxygen evolution from water. J Phys Chem 119:18823–18834
Ashokkumar M, Marignier JL (1999) Hydrogen and oxygen evolution from water using Ag and AgCl colloids. Int J Hydrog Energy 24:17–20
Kudo A, Omori K, Kato H (1999) A novel aqueous process for preparation of crystal form-controlled and highly crystalline BiVO4 powder from layered vanadates at room temperature and its photocatalytic and photophysical properties. J Am Chem Soc 121:11459–11467
Jacobs PA, Uytterhoeven JB, Beyer HK (1977) Cleavage of water over zeolites. J Chem Soc Chem Comm 4:128–129
Reddy VR, Hwang DW, Lee JS (2003) Photocatalytic water splitting over ZrO2 prepared by precipitation method. Korean J Chem Eng 20:1026–1029
Hara M, Kondo T, Komoda M, Ikeda S, Shinohara K, Tanaka A, Kondo JN, Domen K (1998) Cu2O as a photocatalyst for overall water splitting under visible light irradiation. Chem Commun 1998:357–358
Bhandari N, Reeder RJ, Strongin DR (2011) Photoinduced oxidation of arsenite to arsenate on ferrihydrite. Environ Sci Technol 46:8044–8051
Ding W, Wang YJ, Yu YT, Zhang XZ, Li JJ, Wu F (2015) Photooxidation of arsenic(III) to arsenic(V) on the surface of kaolinite clay. J Environ Sci-China 36:29–37
Zoppi M, Pratesi G (2012) The dual behavior of the β-As4S4 altered by light. Am Mineral 97:890–896
Berkes JS, Ing SW Jr, Hillegas WJ (1971) Photodecomposition of amorphous As2Se3 and As2S3. J Appl Phys 42:4908–4916
Atkinson R (2000) Atmospheric chemistry of VOCs and NOx. Atmos Environ 34:2063–2101
Oum KW, Lakin MJ, DeHaan DO, Brauers T, Finlayson-Pitts BJ (1998) Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science 279:74–77
George IJ, Anastasio C (2007) Release of gaseous bromine from the photolysis of nitrate and hydrogen peroxide in simulated sea-salt solutions. Atmos Environ 41:543–553
Acknowledgements
It is a pleasure to acknowledge discussions with colleagues and guidance from the editor and three reviewers which helped me considerably improve this paper.
Competing interests
I have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Doane, T.A. A survey of photogeochemistry. Geochem Trans 18, 1 (2017). https://doi.org/10.1186/s12932-017-0039-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12932-017-0039-y