
Abstract
Extracellular traps (ETs) are a specialized form of innate immune defense in which leukocytes release ETs composed of chromatin and active proteins to eliminate pathogenic microorganisms. In addition to the anti-infection effect of ETs, researchers have also discovered their involvement in the pathogenesis of inflammatory disease, tumors, autoimmune disease, and allergic disease. Asthma is a chronic airway inflammatory disease involving multiple immune cells. The increased level of ETs in asthma patients suggests that ETs play an important role in the pathogenesis of asthma. Here we review the research work on the formation mechanism, roles, and therapeutic strategies of ETs released by neutrophils, eosinophils, and macrophages in asthma.
Similar content being viewed by others
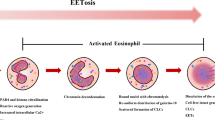

Background
Asthma is a common respiratory disease characterized by airway inflammation, mucus overproduction, and airway remodeling [1]. A global survey revealed that there are 358 million asthma patients worldwide [2], with a total of 45.7 million asthma patients in Chinese aged 20 and above [3]. Recent studies have shown an increased production of extracellular traps (ETs) in asthma patients [4], suggesting a potential role of ETs in asthma. ETs are a complex net-like structure composed of DNA and various active proteins. ETs can be produced by activated neutrophils, eosinophils, basophils, macrophages, mast cells, dentritic cells, and monocytes. The release of ETs is usually accompanied by the lysis of the cell, which is known as extracellular traps cell death (ETosis) [5, 6]. ETs can serve as an immune defense function by binding and killing pathogens such as bacteria, fungi, viruses, and parasites [7], but also participate in disease progression. This review mainly focuses on the role of the ETs derived from neutrophils, eosinophils, and macrophages in asthma.
Neutrophil extracellular traps (NETs)
In 2004, Brinkmann and colleagues first discovered that neutrophils can release a net-like structure composed of chromatin and granule proteins in response to LPS or IL-8 stimulation. They named the net-like structure as neutrophil extracellular traps (NETs) [8]. The formation process of NETs is accompanied by breakdown of nulear and plasma membranes, and this novel form of rapid cell death was later named as neutrophil extracellular trap cell death (NETosis) [9].
Initially recognized as a tool used by neutrophils to kill pathogenic microorganisms [8], NETs possess an intact net-like structure to faciliate pathogen clearance [10, 11]. NETs can also enhance the phagocytic activity and antimicrobial function of macrophages, ultimately boosting their ability in host immune responses [12]. However, excessive NETs can result in a shift from their protective role to a harmful effect [13]. The components of NETs can act as autoantigens and contribute to autoimmune disease by activating immune cells such as B lymphocytes and T lymphocytes and disrupting immune tolerance [14]. NETs also contribute to tissue damage and inflammatory response [15, 16]. It has been demonstrated that NETs are involved in the pathogenesis of respiratory diseases including asthma, chronic obstructive pulmonary disease, acute lung injury, pneumonia, and lung tumors [17].
Composition and structure of NETs
Neutrophils, as the most abundant type of leukocytes and the primary effector cell of the host’s first line of immune defense, perform functions such as phagocytosis, degranulation, and releasing NETs [18].
NETs possess a complex three-dimensional spatial structure. Using high-resolution scanning electron microscopy, NETs are composed of smooth fibers with a diameter of 15- to 17-nm and globular domains measuring 25- to 50-nm in diameter [8, 19]. The principal active proteins in NETs are neutrophil elastase (NE), cathepsin G, myeloperoxidase (MPO), citrullinated histone 3 (CitH3), calprotectin, α-defensins, and antimicrobial peptides [8, 20]. Histone is the most abundant protein, accounting for approximately 70% of all NETs-related proteins, followed by NE [19]. Given the composition of NETs, cell-free DNA(dsDNA), MPO-DNA, and CitH3 can serve as biological markers of NETs [21, 22].
Formation and release of NETs
Various pathogens and inflammatory factors can induce neutrophils to release NETs, including bacteria, fungi, viruses, phorbol-12-myristate-13-acetate (PMA), interleukin-8 (IL-8), tumor necrosis factor-α (TNF-α), lipopolysaccharide (LPS), and granulocyte-macrophage colony-stimulating factor (GM-CSF) [22]. NETs are formed in two pathways, including suicidal NETosis and vital NETosis [23] (Fig. 1). Initially, the formation of NETs was thought as a form of cell death [20], and was termed as suicidal NETosis. Suicidal NETosis is the predominant way of NETs formation and is mainly regulated by nicotinamide adenine dinucleotide phosphate (NADPH) and Raf-MEK-ERK pathways [24]. The process of suicidal NETosis involves several complicated steps, summarized as chromatin decondensation, chromatin swelling into the cytoplasm, disintegration of nuclear and plasma membranes, mixing of cytoplasmic and nuclear contents, and release of the nuclear-cytoplasmic mixture [9, 25]. Different from other forms of cell death including cell necrosis or apoptosis, the prominent morphological features of NETs are the dissolution of neutrophil nuclear and cytoplasmic granular membranes, the fusion of chromatin and cytoplasmic contents, and the disappearance of cytoplasmic organelles [9]. Nevertheless, there is another pathway for NETs formation with a non-lethal outcome, known as vital NETosis. Vital NETosis is characterized by the release of chromatin and proteases extracellularly via vesicle budding while the plasma membranes remain intact [26].

Schematic diagram of NETs formation. There are two pathways of NETs formation. Suicidal NETosis includes chromatin decondensation, the disintegration of nuclear membranes, and the release of the nuclear-cytoplasmic mixture. Vital NETosis is characterized by releasing eDNA via vesicle budding. PMA, phorbol-12-myristate-13-acetate; IL-8, interleukin-8; LPS, lipopolysaccharide; NE, neutrophil elastase; Cath G, cathepsin G; MPO, myeloperoxidase; CitH3, citrullinated histone 3
Suicidal NETosis is NADPH oxidase-dependent whereas vital NETosis is NADPH oxidase-independent. NADPH oxidase activation contributes to the production of reactive oxygen species (ROS), leading to chromatin decondensation and nuclear membrane rupture, thus initiating the release of NETs [27]. Diphenylene iodonium (DPI), an inhibitor of NADPH oxidase, suppresses both ROS and NETs formation in neutrophils [9]. PMA, as the most potent inducer of NETs, enhances NETs release by stimulating ROS production [28]. However, NADPH oxidase is not required for vital NETosis. During the infection of Staphylococcus aureus, neutrophils release chromatin and granule proteins into extracellular space via vesicle budding in a NADPH oxidase-independent manner [26].
The common enzymes NE and MPO in NETs contribute to NETosis. NE is translocated from the cytoplasm to the nucleus and binds to chromatin, resulting in chromatin decondensation through histone degradation, thereby facilitating the release of nuclear DNA into the extracellular space [29, 30]. MPO converts hypochlorous acid into hydrogen peroxide which then activates NE. Activated NE can degrade the cytoskeleton and nuclear membrane, leading to the mixing of nuclear and cytoplasmic contents [25, 31].
Peptidyl arginine deiminase 4 (PAD4) is an enzyme responsible for catalyzing the citrullination of histones, a process that has been shown in multiple studies to promote the formation and release of NETs [32, 33]. Histone citrullination affects DNA binding to histones, leading to chromatin decondensation and subsequently resulting in NETs release [34]. Nitric oxide has been implicated in the formation of NETs, potentially through the upregulation of active nitrogen production under oxidative stress conditions, which in turn leads to histone citrullination [35]. Histones, as the most abundant protein in NETs, can activate neutrophils and further promote NETs release in a feedback manner [36]. Another study has identified histamine as an inducer of NETs release in bovine neutrophils through the NADPH oxidase, ERK, and p38 pathways [37]. Autophagy may also play a role in NETs formation and release [27, 38]. The formation of NETs was significantly increased after pretreatment with autophagy inducer trehalose followed by PMA intervention, while the autophagy inhibitor hydroxychloroquine sulfate inhibited NETs formation in cultured human neutrophils. Similar results were observed in OVA-induced mouse model of allergic airway inflammation [39].
Interestingly, gender and age have been associated with differences in NETs formation. Females show a stronger ability for NETs formation compared to males, while elderly individuals exhibit diminished ability of NETs formation in response to inflammatory stimuli compared to younger individuals [40]. Despite extensive research on NETs, the regulatory mechanism of NETs formation remains incompletely understood, including the stimulating factors and underlying signaling pathways.
NETs and asthma
Neutrophils can infiltrate the airway, release inflammatory cytokines, and modulate the function of other cells, thereby playing a crucial role in asthma [41]. Based on the infiltration of inflammatory cells in induced sputum, asthma can be classified into four different inflammatory phenotypes: eosinophilic asthma, neutrophilic asthma, mixed granulocytic asthma, and paucigranulocytic asthma [42]. Neutrophilic asthma accounts for approximately 20–30% of the asthma patients population and is often associated with corticosteroid-resistant severe asthma [43, 44]. NETs may play a crucial role in asthma especially in neutrophilic asthma or severe asthma.
Increased NETs in human asthma
Several studies have shown the level of NETs is significantly increased in clinical specimens from asthma patients, and is positively correlated with the severity of asthma. Dworski et al. performed immunofluorescence staining on bronchial biopsy specimens from patients with neutrophilic asthma and observed the co-localization of dsDNA with NE [45], suggesting the presence of NETs in asthmatic airways. In a study comparing induced sputum samples from healthy individuals and asthma patients, significantly higher levels of extracellular DNA (eDNA), antimicrobial peptide LL-37, α-defensin, and NE were found in the asthma group, especially in those with neutrophilic asthma [46]. Plasma levels of CitH3 and dsDNA were also found to be elevated in asthma patients compared to healthy controls, and a negative correlation was observed between the concentration of CitH3 and forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) ratio [22]. Additionally, asthma patients with severe respiratory symptoms and impaired lung function tend to have higher plasma NETs levels [47]. Collectively, these findings indicate that increased release of NETs may be involved in the pathogenesis of asthma.
The levels of NETs were also elevated in corticosteroid-resistant severe asthma. It was reported that patients with severe asthma have markedly higher circulating NETs levels compared to mild-to-moderate asthma patients and healthy controls. Besides, the increased NETs levels were negatively correlated with the FEV1/FVC ratio in patients with severe asthma [48, 49]. Inhaled corticosteroids (ICS) are the main therapeutic drugs for asthma [50], and patients who regularly use ICS show significantly lower average plasma NETs levels compared to those who do not use or only occasionally use ICS [47]. Furthermore, circulating levels of NETs were significantly higher in patients with corticosteroid-resistant asthma compared to patients with corticosteroid-sensitive asthma [51]. These findings suggest that the abundance of NETs is associated with corticosteroid responsiveness, and could serve as an indicator to predict the effectiveness of ICS in asthma patients.
NETs in mouse models of allergic airway inflammation
NETs release is increased in mouse models of allergic airway inflammation. In a mouse model of neutrophilic airway inflammation, the levels of NETs in bronchoalveolar lavage fluid (BALF) were significantly increased and were associated with the severtity of airway inflammation [52]. OVA/LPS-induced neutrophilic airway inflammation mouse model showed a significant release of NETs in lung tissue. Interestingly, clearing NETs or inhibiting the release of NETs can alleviate airway inflammation and hyperresponsiveness [53]. Released NETs contribute to airway inflammation and mucus secretion by enhancing the ability of antigen presenting of dendritic cells and promoting Th2 immune responses in house dust mite (HDM)/LPS-induced mouse model of neutrophilic airway inflammation [54]. In the Sendai virus-induced mouse model, airway NETs release is associated with airway inflammation and hyperresponsiveness [55]. Taken together, animal studies have provided evidence supporting the potential role of NETs in the pathogenesis of asthma.
NETs in the pathogenesis of asthma
NETs can damage airway epithelialium. It was shown that NETs leads to the disruption of airway barrier integrity and exacerbation of symptoms in asthma [49]. This occurs through the inducing apoptosis in airway epithelial cells, suppressing tight junction-associated genes including claudins 4, 8, and 11, and promoting degradation of E-cadherin [49, 56, 57]. It was also reported that NETs induced by LPS can damage the tight junction protein occludin in lung epithelium, exacerbating the severity of neutrophilic airway inflammation [58]. The impaired ability of alveolar macrophages to clear NETs leads to the prolonged presence of NETs, contributing to chronic airway inflammation in neutrophilic asthma [59]. In severe asthma, excessive NETs can activate inflammasomes and enhance IL-1β secretion by monocyte-macrophages [60, 61]. NETs also can induce the release of thymic stromal lymphopoietin(TSLP), IL-33, and IL-25 by epithelial cells, which further activates the downstream immune responses [62]. Furthermore, NETs can stimulate airway epithelium to secrete cytokines such as IL-1, IL-6, and IL-8, promoting neutrophil infiltration in airway [63]. NETs also induce the secretion of CXCL1, CXCL2, and CXCL8 chemokines by the Toll-like receptor 4 (TLR4) / nuclear factor kappa-B (NF-κB) pathway in airway epithelial cells, leading to enhanced neutrophil aggregation and release of inflammatory cytokines [64]. NETs can promote the secretion of TNF-α and IL-6 by recruiting and activating CD4+ and CD8+ T cells, which in turn aggravates airway inflammation [55]. In addition, the proteases from NETs could activate IL-1 and IL-36 in neutrophilic asthma [62]. These findings indicate that NETs play an important role in the pathogenesis of asthmatic airway inflammation (Fig. 2).

The role of NETs in asthmatic airway inflammation. NETs are involved in asthmatic airway inflammation by mediating direct airway epithelial barrier injury, promoting the secretion of cytokines from epithelial cells, enhancing the recruitment of T cells and the secretion of TNF-α and IL-6 by T cells. TNF-α, tumor necrosis factor-α; IL-1/6/8/25/33, interleukin-1/6/8/25/33; TSLP, thymic stromal lymphopoietin; CXCL1/2/8, CXC motif chemokine ligand 1/2/8
NETs are associated with acute exacerbation of asthma and mucus hypersecretion. During asthma exacerbation induced by rhinovirus, IL-33 levels in nasal secretions of asthma patients were found to be associated with the levels of NE and dsDNA. Blocking IL-33 can alleviate rhinovirus-induced neutrophilia and suppress the formation of NETs in a mouse model [65]. This suggests that IL-33 is not only induced by NETs, but also promotes the NETs formation. Excessive mucus secretion in the airways is a hallmark of asthma. NETs can induce airway mucus hypersecretion by promoting MUC5AC gene expression [66]. Additionally, NETs can activate the TLR4/NF-κB signaling pathway, leading to airway mucus secretion [67].
Both asthma patients and mouse models showed increased formation of NETs, which were associated with airway inflammation, hyperresponsiveness, mucus secretion, and lung function. It is clear that NETs play a critical role in the pathogenesis of asthma, but the underlying molecular mechanisms remain to be further studied.
NETs as a therapeutic target for asthma
The significant correlation between elevated NETs and the severity of asthma suggests inhibiting the formation of NETs or promoting their degradation may represent promising therapeutic approaches for asthma.
Intraperitoneal injection of Nec-1, an inhibitor of NETs formation, decreased MPO activity and inflammatory cytokine levels, and alleviated airway inflammation in mouse model of allergic airway inflammation [52]. BB-Cl-amidine, an inhibitor of PAD4, has been reported to effectively alleviates airway inflammatory cell infiltration in mice [68]. In Sendai virus-induced mouse model, the release of NETs was increased during the early stage of viral infection. Intraperitoneal injection of Cl-amidine effectively suppressed the recruitment of inflammatory cells and the secretion of inflammatory cytokines [55].
The degradation of NETs also represents a potential therapy for asthma. In a rhinovirus-induced mouse model with increased release of NETs and airway inflammation, treatment with deoxyribonuclease (DNase) to degrade NETs alleviated airway inflammation and mucus secretion [69]. In toluene diisocyanate-induced mouse model of neutrophilic airway inflammation, administration of DNase I also ameliorated airway inflammation by degrading NETs [70].
In human, as a mucolytic agent that can reduce the viscosity of mucus by degarding DNA in sputum, recombinant human deoxyribonuclease (rhDNase) is widely used in cystic fibrosis therapy [71, 72]. Several clinical trials have evaluated the therapeutic effect of rhDNase on asthma. It is reported that treatment with rhDNase can rapidly alleviate airway mucus obstruction in asthma [73]. Additionally, rhDNase can improve respiratory symptoms and lung ventilation in severely ill, non-intubated adult asthmatics refractory to bronchodilators [74, 75]. However, nebulized rhDNase treatment did not provide significant benefits for children with acute asthma [76] or asthmatic children with persistent small airway obstruction [77]. One possibility is that rhDNase can alleviate airway obstruction caused by mucus plug, but it can not improve airway obstruction caused by smooth muscle contraction. Besides the beneficial effect on airwy mucus obstruction, rhDNase may also ameliorate airway inflammation by degrading NETs as suggested by the findings from mouse experiments. The therapeutic effects of azithromycin in severe asthma have been reported [78, 79]. Several studies have concluded that the administration of macrolide antibiotics, particularly azithromycin, can ameliorate asthma symptoms by inhibiting NETs formation [80, 81].
Eosinophil extracellular traps (EETs)
EETs are a part of the innate immune response and can be present in various infectious, allergic, and autoimmune diseases [45, 82, 83]. Similar to NETs, EETs can resist pathogen invasion by capturing and eliminating pathogens, particularly when the epithelial barrier is impaired [84, 85].
The composition of EETs
Simon and colleagues observed that eosinophils release mitochondrial DNA (mtDNA) into the extracellular space, and first proposed the concept of EETs in 2008 [84]. Multiple factors including IL-5, LPS, IgG, IgA, platelet-activating factors, calcium ionophores, PMA, and sorbitol acetate are involved in cell lysis and the release of DNA during the formation of EETs [86,87,88].
EETs are composed of 25–35 nm chromatin fibers which are thicker than the fibers in NETs (15- to 17-nm). In contast to graule lysis and release of protease in neutrophil, intact eosinophil granules composing of eosinophilic cationic protein, eosinophil peroxidase and major basic protein are released [45, 84, 86]. Regarding the source of eDNA in EETs, multiple studies showed that the eDNA in EETs mainly originates from nuclear DNA [86, 89,90,91,92,93], and one study revealed that mtDNA constitutes 8.72 ± 1.99% of the eDNA of EETs [89]. It is reported fast-forming EETs consist of mitochondrial DNA and cationic proteins derived from cytoplasmic secondary granules, whereas slow-forming EETs predominantly consist of nuclear DNA without cationic proteins [94]. Compared to NETs, EETs have less protease content and are less susceptible to degradation. Therefore, EETs exhibit more stable and concentrated chromatin structures when compared with NETs [90, 95, 96]. Due to their stability and resistance to degradation, EETs can function in the body for a longer time than NETs [97].
Factors involved in EETs formation and release
The process of EETs formation is termed as EETosis, a form of eosinophil death [98]. Mutiple studies demonstrated that nuclear and plasma membrane rupture and disintegration occur during EETosis, ultimately leading to cell death [90, 92, 99, 100]. However, several studies showed that eosinophils remain alive after the release of EETs without significant cell necrosis or apoptosis [84, 101]. One study demonstrated that the fate of eosinophil after the release of EETs may depend on the stimulation factor [102]. It has been reported that IL-5, LPS, ROS, autophagy, bacteria, and lysophosphatidylserine (LysoPS) can trigger the release of EETs [84, 92, 103]. Toll-like receptors and adhesion receptors are involved in the release of EETs [104]. TSLP can induce eosinophils to release EETs and contributes to the pathogenesis of asthma [89, 105, 106].
Eosinophils release EETs through a NADPH oxidase-dependent pathway, which is associated with ROS production [90]. In mouse model of allergic airway inflammation, inhibition of NADPH oxidase by DPI reduced ROS and EETs formation [107]. These observations suggest that NADPH oxidase is essential for EETs formation, but alternative NADPH oxidase-independent mechanisms exist. For instance, LysoPS-induced EETs formation is PAD4-dependent, rather than NADPH oxidase-dependent [92]. Inhibition of PAD4 in human eosinophils led to a decrease in histone citrullination and EETs formation [89]. This indicates that PAD4 also plays a crucial role in regulating EETs formation.
The formation of EETs is generally believed to be similar to that of NETs. However, autophagy promotes human neutrophils to release NETs [27, 49, 108] but reduces the release of EETs by eosinophils. Adhesion-induced eosinophil cytolysis mediated by the activation of receptor-interacting protein kinase 3 - mixed lineage kinase-like signaling pathway could be inhibited by rapamycin, an inducer of autophagy [109]. Consistent with this, autophagy may confer protection against eosinophil cytolysis and the release of EETs [110]. However, inhibiting autophagy with 3-methyladenine (3-MA) can reduce the release of EETs in a mouse model of allergic airway inflammation [103]. However, it is reported that 3-MA inhibits the release of EETs in eosinophils by suppressing ROS production, rather than blocking autophagy. Besides, the release of EETs of eosinophils from autophagy-related gene 5 knockout mice remains unaffected [111]. Therefore, the role of autophagy in EETs requires further study.
EETs in asthma
Eosinophils are the core inflammatory cells in asthma [112, 113]. Elevated eosinophil count in the blood or airways is not only associated with the frequency of asthma attacks but also indicates the severity of asthma [114]. EETs are implicated in the pathogenesis of asthma. Analysis of airway biopsies and BALF samples from asthma patients revealed the presence of DNA colocalized with major basic protein, which is associated with eosinophilic infiltration [45]. In BALF samples from OVA-induced mouse model, a significant increase in eosinophil counts and eDNA content was observed. Immunofluorescence staining further revealed co-localization of eDNA with eosinophil peroxidase [89, 115]. The existence of EETs in both asthmatic patients and mouse model suggests a role of EETs in asthma pathogenesis.
EETs contribute to the airway inflammation of asthma (Fig. 3). Eosinophil cationic protein in EETs can directly damage the airway epithelium, thus worsening airway inflammation [88]. EETs had the potential to stimulate the secretion of neurotransmitters and neuropeptides from pulmonary neuroendocrine cells, resulting in the recruitment of inflammatory cells and the release of inflammatory cytokines in the airway [89]. Several studies demonstrated that elevated levels of EETs could interact with type 2 immune cells to enhance Th2 immune responses in severe eosinophilic asthma [88, 116, 117]. In a mouse model of allergic airway inflammation, exogenous injection of EETs induced the expression of airway epithelial-derived cytokines including IL-1α, IL-1β, IL-33, and TSLP in BALF, and increased the counts of type 2 innate lymphoid cells (ILC2) in lung tissue [118]. On the other hand, EETs trigger the airway epithelium to release IL-33 and TSLP which in turn induce ILC2 activation as well as eosinophil degranulation, thus aggravating airway inflammation [119].

The role of EETs in asthmatic airway inflammation. EETs promotes airway inflammation in asthma through several mechanisms, including mediating damage to the airway epithelial barrier, activating the secretion of neurotransmitters and neuropeptides in neuroendocrine cells, stimulating the secretion of airway epithelial-derived cytokines (TSLP, IL-33, IL-1α, IL-1β) to induce eosinophil degranulation and activate type 2 immune cells. ILC2, type 2 innate lymphoid cells; IL-1α/1β, interleukin-1α/1β; IL-13/4/5/33, interleukin-13/4/5/33; TSLP, thymic stromal lymphopoietin
EETs play a role in the acute exacerbation of asthma. Respiratory syncytial virus is a common trigger of asthma exacerbation and it can stimulate the release of EETs [120]. Moreover, elevated release of EETs could result in airway obstruction and impairment of lung function by increasing mucus viscosity in asthma [107]. Although the precise mechanism underlying EETs’ contribution to asthma remains elusive, the role of EETs in asthma should not be underestimated.
Significance of EETs as a target for asthma therapy
EETs as a potential therapeutic target for asthma can be explored from two perspectives: inhibiting EETs formation and eliminating components of EETs. So far, there has been limited research in this field.
DNase has been shown to cleave eDNA and degrade EETs [115, 121]. Treatment with rhDNase resulted in a significant decrease in EETs formation in BALF and lung tissue and improvement in airway resistance and lung function in OVA-induced mouse model [122]. Surfactant protein-D (SP-D), a product of airway epithelial cells, can bind directly to the membrane of eosinophils to inhibit EETs formation, thus reducing the exacerbation of asthma [123]. TSLP secreted by epithelial cells can regulate immune cell activation and the release of type 2 cytokines, contributing to airway inflammation in asthma [124]. Anti-TSLP antibodies effectively inhibit the release of EETs, thereby alleviating airway hyperresponsiveness [118].
Macrophage extracellular traps (METs)
Macrophages have diverse functions including support for development, maintenance of homeostasis, immune surveillance, and regulation of tissue repair [125]. Macrophages are also capable of releasing a net-like structure known as METs to capture and eliminate microorganisms [126].
Definition of METs
In 2010, Chow and colleagues first reported that RAW264.7 cells can release DNA to form METs when stimulated by PMA [127]. Similar to NETs and EETs, METs are composed of eDNA and active proteins including histones, elastase, and MPO [126, 128,129,130]. However, METs released by THP-1 cells lack protease when stimulated by TNF-α, hypochlorous acid, and nigerin [131]. As the central scaffold of METs, the eDNA of METs mainly originates from nuclear DNA [129, 132,133,134,135]. However, several studies suggest that the origin of eDNA in METs is from mitochondria [131, 136].
Formation and release of METs
The formation of METs is generally accompanied by macrophages death, a process known as METosis [127, 134, 137, 138]. Pathogenic microorganisms, hypochlorous acid, PMA, IL-8, and TNF-α can induce macrophages to release METs [134, 139]. The studies on the mechanisms underlying METs formation are currently limited. Similar to NETs, METs formation can be classified into NADPH oxidase-dependent and NADPH oxidase-independent pathway. Inhibitors of NADPH oxidase, such as DPI or apocynin, can decrease METs formation by inhibiting ROS production [132, 140, 141]. Exposure of alveolar macrophages to cigarette smoke extracts could increase the production of ROS and METs, suggesting a positive role for ROS in METs formation [142]. Human alveolar macrophages producing METs showed higher ROS fluorescence intensity compared to non-producing macrophages, and NADPH oxidase inhibitors significantly inhibit the release of METs [141]. However, the formation of METs induced by some pathogens is independent of NADPH oxidase [129, 133, 135, 136].
PAD enzymes are responsible for catalyzing the citrullination of histones, a process critical for the formation of ETs [143]. The release of METs was associated with the citrullination and partial cleavage of histone H3 [144]. The citrullination of histones in METs may involve the activation of either PAD4 or PAD2. The release of METs was accompanied by the upregulation of PAD4 gene, and knockdown of PAD4 with small interfering RNA could inhibit METs release in THP-1 cells [145]. Citrullination of histone H4 is dependent on PAD2 in METs induced by TNF-α [137].
The difference in METs release is associated with the type of stimulus and the polarization state of the macrophages [146, 147]. For instance, M1 macrophages induced by interferon-γ and LPS released METs, while M2 macrophages induced by IL-4 did not release METs [145]. Treating bovine macrophages with 100µM PMA could induce METs formation [132], whereas there was no MET release in murine J774A.1 macrophages stimulated by 100nM PMA [136]. Regarding pathogenic microbial stimulation, bacteria, parasites, fungi, and acid-fast bacilli all can induce METs release [148]. In the formation of METs induced by pathogens, the proportion of macrophages that release METs increases with the diversity of infectious pathogens [149].
METs in asthma
Macrophages, as the most abundant immune cells in the airway, play a critical role in chronic airway inflammation and remodeling in asthma [150]. However, there is limited research on METs in asthma. Using LPS/ IFNγ to stimulate the macrophages isolated from the peripheral blood of asthma patients, the level of M1-like macrophage ETs (M1ETs) in the severe asthma is significantly higher than those in the non-severe asthma group, and the level of M1ETs is negatively correlated with FEV1% pred. M1ETs was shown to activate airway epithelial cells, neutrophils, ILC1, and ILC3 to promote the migration of inflammatory cells and the release of inflammatory cytokines, thereby enhancing airway inflammation [151]. This suggests that METs are involved in the airway inflammation of severe asthma.
limited research work on the role of METs in asthma makes it hard to evaluate their potential as a novel target for asthma therapy. DNase I can effectively disrupt the main structure of METs by digesting the eDNA strands [142]. However, DNase is unable to eliminate histones and pro-inflammatory proteins in METs [146]. PAD mediates histone citrullination during the formation of METs, indicating that PAD inhibitors may have therapeutic effect for asthma [152].
Conclusion
Extracellular traps play a beneficial role in innate immune defense against pathogens, but their enzymes and active proteins can also contribute to tissue damage and inflammatory response, making them a double-edged sword. The elevated production of NETs, EETs, and METs in asthma patients suggests their potential role in regulating pathological processes including airway inflammation, airway hyperreactivity, and mucus secretion.
Most of the studies focused on the basic structure and classical formation pathways of ETs. The specific mechanisms of ETs in asthma remain largely unexplored, posing significant challenges for the development of drugs targeting ETs in asthma. Currently, there are some unresolved questions about NETs, EETs, and METs that require further research. For example, what is the threshold level of ETs release when it becomes pathogenic? What is the roles of active proteins and granzymes from ETs in asthma? What are the mechanisms underlying the increased formation of NETs, EETs, and METs in asthma patients? Deeper insights into ETs and their formation mechanisms are necessary to address these questions. As the studies of ETs in asthma continue to grow, NETs, EETs, and METs may emerge as effective therapeutic targets for asthma in the future.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ETs:
-
Extracellular traps
- NETs:
-
Neutrophil extracellular traps
- NE:
-
Neutrophil elastase
- MPO:
-
Myeloperoxidase
- CitH3:
-
Citrullinated histone 3
- dsDNA:
-
Double-stranded DNA
- PMA:
-
Phorbol-12-myristate-13-acetate
- IL-8:
-
Interleukin-8
- TNF-α:
-
Tumor necrosis factorα
- LPS:
-
Lipopolysaccharide
- GM-CSF:
-
Granulocyte/macrophage colony-stimulating factor
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- ROS:
-
Reactive oxygen species
- PAD4:
-
Peptidyl arginine deiminase 4
- EA:
-
Eosinophilic asthma
- NA:
-
Neutrophilic asthma
- eDNA:
-
Extracellular DNA
- ICS:
-
Inhaled corticosteroids
- BALF:
-
Bronchoalveolar lavage fluid
- HDM:
-
House dust mite
- TLR4:
-
Toll-like receptor 4
- NF-κB:
-
Nuclear factor kappa-B
- TSLP:
-
Thymic stromal lymphopoietin
- DNase:
-
Deoxyribonuclease
- rhDNase:
-
Recombinant human deoxyribonuclease
- EETs:
-
Eosinophil extracellular traps
- mtDNA:
-
Mitochondrial DNA
- LysoPS:
-
Lysophosphatidylserine
- 3-MA:
-
3-Methyladenine
- ILC2:
-
Type 2 innate lymphoid cells
- RSV:
-
Respiratory syncytial virus
- SP-D:
-
Surfactant protein-D
- METs:
-
Macrophage extracellular traps
- M1ETs:
-
M1-like macrophage extracellular traps
References
Ray A, Camiolo M, Fitzpatrick A, Gauthier M, Wenzel SE. Are we meeting the Promise of Endotypes and Precision Medicine in Asthma? Physiol Rev. 2020;100:983–1017.
Global regional. National deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the global burden of Disease Study 2015. Lancet Respir Med. 2017;5:691–706.
Huang K, Yang T, Xu J, Yang L, Zhao J, Zhang X, Bai C, Kang J, Ran P, Shen H, et al. Prevalence, risk factors, and management of asthma in China: a national cross-sectional study. Lancet. 2019;394:407–18.
Kuczia P, Zuk J, Iwaniec T, Soja J, Dropinski J, Malesa-Wlodzik M, Zareba L, Bazan JG, Undas A, Bazan-Socha S. Citrullinated histone H3, a marker of extracellular trap formation, is increased in blood of stable asthma patients. Clin Transl Allergy. 2020;10:31.
Pertiwi KR, de Boer OJ, Mackaaij C, Pabittei DR, de Winter RJ, Li X, van der Wal AC. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J Pathol. 2019;247:505–12.
Nija RJ, Sanju S, Sidharthan N, Mony U. Extracellular trap by blood cells: clinical implications. Tissue Eng Regenerative Med. 2020;17:141–53.
Cheng OZ, Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Front Immunol. 2013;4:1.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41.
Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, Bilyy R, Krenn V, Renné C, Renné T, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358:1202–6.
Halverson TW, Wilton M, Poon KK, Petri B, Lewenza S. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 2015;11:e1004593.
Monteith AJ, Miller JM, Maxwell CN, Chazin WJ, Skaar EP. Neutrophil extracellular traps enhance macrophage killing of bacterial pathogens. Sci Adv. 2021;7:eabj2101.
Brinkmann V. Neutrophil Extracellular traps in the second decade. J Innate Immun. 2018;10:414–21.
Fousert E, Toes R, Desai J. Neutrophil Extracellular traps (NETs) take the Central Stage in driving autoimmune responses. Cells 2020, 9.
Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7:e32366.
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–52.
Twaddell SH, Baines KJ, Grainge C, Gibson PG. The emerging role of Neutrophil Extracellular traps in Respiratory Disease. Chest. 2019;156:774–82.
Abu Abed U, Brinkmann V. Immunofluorescence labelling of Human and Murine Neutrophil Extracellular traps in paraffin-embedded tissue. J Vis Exp 2019.
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639.
Yang LY, Shen XT, Sun HT, Zhu WW, Zhang JB, Lu L. Neutrophil extracellular traps in hepatocellular carcinoma are enriched in oxidized mitochondrial DNA which is highly pro-inflammatory and pro-metastatic. J Cancer. 2022;13:1261–71.
Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5.
Varricchi G, Modestino L, Poto R, Cristinziano L, Gentile L, Postiglione L, Spadaro G, Galdiero MR. Neutrophil extracellular traps and neutrophil-derived mediators as possible biomarkers in bronchial asthma. Clin Exp Med. 2022;22:285–300.
Chen F, Yu M, Zhong Y, Wang L, Huang H. Characteristics and role of Neutrophil Extracellular traps in Asthma. Inflammation. 2022;45:6–13.
Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75–7.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91.
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–25.
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304.
Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol. 2012;92:841–9.
Gavillet M, Martinod K, Renella R, Wagner DD, Williams DA. A key role for Rac and Pak signaling in neutrophil extracellular traps (NETs) formation defines a new potential therapeutic target. Am J Hematol. 2018;93:269–76.
Li M, Lin C, Leso A, Nefedova Y. Quantification of Citrullinated histone H3 bound DNA for detection of Neutrophil Extracellular traps. Cancers (Basel) 2020, 12.
Bjornsdottir H, Welin A, Michaelsson E, Osla V, Berg S, Christenson K, Sundqvist M, Dahlgren C, Karlsson A, Bylund J. Neutrophil NET formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radic Biol Med. 2015;89:1024–35.
Liu X, Arfman T, Wichapong K, Reutelingsperger CPM, Voorberg J, Nicolaes GAF. PAD4 takes charge during neutrophil activation: impact of PAD4 mediated NET formation on immune-mediated disease. J Thromb Haemost. 2021;19:1607–17.
Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, Bicker KL, Bingham RP, Campbell M, Chen YH, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11:189–91.
Ansari J, Vital SA, Yadav S, Gavins FNE. Regulating Neutrophil PAD4/NOX-Dependent Cerebrovasular Thromboinflammation. Int J Biol Sci. 2023;19:852–64.
Li WX, Wang F, Zhu YQ, Zhang LM, Zhang ZH, Wang XM. Inhibitors of nitric oxide synthase can reduce extracellular traps from neutrophils in asthmatic children in vitro. Pediatr Pulmonol. 2020;55:68–75.
Shrestha B, Ito T, Kakuuchi M, Totoki T, Nagasato T, Yamamoto M, Maruyama I. Recombinant thrombomodulin suppresses histone-Induced Neutrophil Extracellular trap formation. Front Immunol. 2019;10:2535.
Zhou E, Wu Z, Zhu X, Li P, Wang J, Yang Z. Histamine triggers the formation of neutrophil extracellular traps via NADPH oxidase, ERK and p38 pathways. Vet Immunol Immunopathol. 2021;235:110234.
Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C. Autophagy and reactive oxygen species are involved in Neutrophil Extracellular traps Release Induced by C. Albicans Morphotypes. Front Microbiol. 2016;7:879.
Guo Y, Gao F, Wang X, Pan Z, Wang Q, Xu S, Pan S, Li L, Zhao D, Qian J. Spontaneous formation of neutrophil extracellular traps is associated with autophagy. Sci Rep. 2021;11:24005.
Gupta S, Nakabo S, Blanco LP, O’Neil LJ, Wigerblad G, Goel RR, Mistry P, Jiang K, Carmona-Rivera C, Chan DW, et al. Sex differences in neutrophil biology modulate response to type I interferons and immunometabolism. Proc Natl Acad Sci U S A. 2020;117:16481–91.
Hosoki K, Ying S, Corrigan C, Qi H, Kurosky A, Jennings K, Sun Q, Boldogh I, Sur S. Analysis of a panel of 48 cytokines in BAL fluids specifically identifies IL-8 levels as the only cytokine that distinguishes controlled asthma from uncontrolled asthma, and correlates inversely with FEV1. PLoS ONE. 2015;10:e0126035.
Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology. 2006;11:54–61.
Ray A, Kolls JK. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol. 2017;38:942–54.
Chung KF. Neutrophilic asthma: a distinct target for treatment? Lancet Respir Med. 2016;4:765–7.
Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol. 2011;127:1260–6.
Wright TK, Gibson PG, Simpson JL, McDonald VM, Wood LG, Baines KJ. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology. 2016;21:467–75.
Gál Z, Gézsi A, Pállinger É, Visnovitz T, Nagy A, Kiss A, Sultész M, Csoma Z, Tamási L, Gálffy G, Szalai C. Plasma neutrophil extracellular trap level is modified by disease severity and inhaled corticosteroids in chronic inflammatory lung diseases. Sci Rep. 2020;10:4320.
Granger V, Taille C, Roach D, Letuve S, Dupin C, Hamidi F, Noel B, Neukirch C, Aubier M, Pretolani M, et al. Circulating neutrophil and eosinophil extracellular traps are markers of severe asthma. Allergy. 2020;75:699–702.
Pham DL, Ban GY, Kim SH, Shin YS, Ye YM, Chwae YJ, Park HS. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin Exp Allergy. 2017;47:57–70.
Reddel HK, Bacharier LB, Bateman ED, Brightling CE, Brusselle GG, Buhl R, Cruz AA, Duijts L, Drazen JM, FitzGerald JM, et al. Global Initiative for Asthma Strategy 2021: executive Summary and Rationale for Key Changes. Am J Respir Crit Care Med. 2022;205:17–35.
Tsai CH, Lai AC, Lin YC, Chi PY, Chen YC, Yang YH, Chen CH, Shen SY, Hwang TL, Su MW, et al. Neutrophil extracellular trap production and CCL4L2 expression influence corticosteroid response in asthma. Sci Transl Med. 2023;15:eadf3843.
Han XA, Jie HY, Wang JH, Zhang XM, Wang J, Yu CX, Zhang JL, He J, Chen JQ, Lai KF, Sun EW. Necrostatin-1 ameliorates neutrophilic inflammation in Asthma by suppressing MLKL Phosphorylation to Inhibiting NETs Release. Front Immunol. 2020;11:666.
Xia M, Xu F, Ni H, Wang Q, Zhang R, Lou Y, Zhou J. Neutrophil activation and NETosis are the predominant drivers of airway inflammation in an OVA/CFA/LPS induced murine model. Respir Res. 2022;23:289.
Radermecker C, Sabatel C, Vanwinge C, Ruscitti C, Marechal P, Perin F, Schyns J, Rocks N, Toussaint M, Cataldo D, et al. Locally instructed CXCR4(hi) neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nat Immunol. 2019;20:1444–55.
Akk A, Springer LE, Pham CT. Neutrophil Extracellular traps enhance early inflammatory response in Sendai Virus-Induced Asthma phenotype. Front Immunol. 2016;7:325.
Hudock KM, Collins MS, Imbrogno MA, Kramer EL, Brewington JJ, Ziady A, Zhang N, Snowball J, Xu Y, Carey BC, et al. Alpha-1 antitrypsin limits neutrophil extracellular trap disruption of airway epithelial barrier function. Front Immunol. 2022;13:1023553.
Li Y, Yang Y, Gan T, Zhou J, Hu F, Hao N, Yuan B, Chen Y, Zhang M. Extracellular RNAs from lung cancer cells activate epithelial cells and induce neutrophil extracellular traps. Int J Oncol. 2019;55:69–80.
Ma Q, Qian Y, Jiang J, Wu J, Song M, Li X, Chen Z, Wang Z, Zhu R, Sun Z, et al. IL-33/ST2 axis deficiency exacerbates neutrophil-dominant allergic airway inflammation. Clin Transl Immunol. 2021;10:e1300.
Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol. 2013;191:2647–56.
Lachowicz-Scroggins ME, Dunican EM, Charbit AR, Raymond W, Looney MR, Peters MC, Gordon ED, Woodruff PG, Lefrancais E, Phillips BR, et al. Extracellular DNA, Neutrophil Extracellular traps, and Inflammasome activation in severe asthma. Am J Respir Crit Care Med. 2019;199:1076–85.
Chen X, Li Y, Qin L, He R, Hu C. Neutrophil Extracellular Trapping Network promotes the Pathogenesis of Neutrophil-associated asthma through macrophages. Immunol Invest. 2021;50:544–61.
Stojkov D, Yousefi S, Simon HU. NETs: important players in asthma? J Allergy Clin Immunol. 2024;153:100–2.
Hudock KM, Collins MS, Imbrogno M, Snowball J, Kramer EL, Brewington JJ, Gollomp K, McCarthy C, Ostmann AJ, Kopras EJ, et al. Neutrophil extracellular traps activate IL-8 and IL-1 expression in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. 2020;319:L137–47.
Wan R, Jiang J, Hu C, Chen X, Chen C, Zhao B, Hu X, Zheng Z, Li Y. Neutrophil extracellular traps amplify neutrophil recruitment and inflammation in neutrophilic asthma by stimulating the airway epithelial cells to activate the TLR4/ NF-κB pathway and secrete chemokines. Aging. 2020;12:16820–36.
Curren B, Ahmed T, Howard DR, Ashik Ullah M, Sebina I, Rashid RB, Al Amin Sikder M, Namubiru P, Bissell A, Ngo S, et al. IL-33-induced neutrophilic inflammation and NETosis underlie rhinovirus-triggered exacerbations of asthma. Mucosal Immunol. 2023;16:671–84.
Chen Y, Garvin LM, Nickola TJ, Watson AM, Colberg-Poley AM, Rose MC. IL-1beta induction of MUC5AC gene expression is mediated by CREB and NF-kappaB and repressed by dexamethasone. Am J Physiol Lung Cell Mol Physiol. 2014;306:L797–807.
Chen L, Ran D, Xie W, Xu Q, Zhou X. Cold-inducible RNA-binding protein mediates cold air inducible airway mucin production through TLR4/NF-kappaB signaling pathway. Int Immunopharmacol. 2016;39:48–56.
Zhang H, Liu J, Zhou Y, Qu M, Wang Y, Guo K, Shen R, Sun Z, Cata JP, Yang S, et al. Neutrophil extracellular traps mediate m(6)a modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Biol Sci. 2022;18:3337–57.
Toussaint M, Jackson DJ, Swieboda D, Guedan A, Tsourouktsoglou TD, Ching YM, Radermecker C, Makrinioti H, Aniscenko J, Bartlett NW, et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med. 2017;23:681–91.
Peng X, Li Y, Zhao W, Yang S, Huang J, Chen Y, Wang Y, Gong Z, Chen X, Yu C, et al. Blockade of neutrophil extracellular traps ameliorates toluene diisocyanate-induced steroid-resistant asthma. Int Immunopharmacol. 2023;117:109719.
VanDevanter DR, Craib ML, Pasta DJ, Millar SJ, Morgan WJ, Konstan MW. Cystic fibrosis clinical characteristics associated with dornase alfa treatment regimen change. Pediatr Pulmonol. 2018;53:43–9.
Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci U S A. 1990;87:9188–92.
Greally P. Human recombinant DNase for mucus plugging in status asthmaticus. Lancet. 1995;346:1423–4.
Silverman RA, Foley F, Dalipi R, Kline M, Lesser M. The use of rhDNAse in severely ill, non-intubated adult asthmatics refractory to bronchodilators: a pilot study. Respir Med. 2012;106:1096–102.
Patel A, Harrison E, Durward A, Murdoch IA. Intratracheal recombinant human deoxyribonuclease in acute life-threatening asthma refractory to conventional treatment. Br J Anaesth. 2000;84:505–7.
Boogaard R, Smit F, Schornagel R, Vaessen-Verberne AA, Kouwenberg JM, Hekkelaan M, Hendriks T, Feith SW, Hop WC, de Jongste JC, Merkus PJ. Recombinant human deoxyribonuclease for the treatment of acute asthma in children. Thorax. 2008;63:141–6.
Bakker EM, van der Wiel-Kooij EC, Müllinger B, Kroneberg P, Hop WC, Tiddens HA. Small-airways deposition of dornase alfa in children with asthma and persistent airway obstruction. J Allergy Clin Immunol. 2013;132:482–e485410.
Gibson PG, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Jenkins C, Peters MJ, Marks GB, Baraket M, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:659–68.
Niessen NM, Gibson PG, Baines KJ, Barker D, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Jenkins C, et al. Sputum TNF markers are increased in neutrophilic and severe asthma and are reduced by azithromycin treatment. Allergy. 2021;76:2090–101.
Keir HR, Shoemark A, Dicker AJ, Perea L, Pollock J, Giam YH, Suarez-Cuartin G, Crichton ML, Lonergan M, Oriano M, et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med. 2021;9:873–84.
Bystrzycka W, Manda-Handzlik A, Sieczkowska S, Moskalik A, Demkow U, Ciepiela O. Azithromycin and Chloramphenicol Diminish Neutrophil Extracellular traps (NETs) release. Int J Mol Sci 2017, 18.
Simon D, Hoesli S, Roth N, Staedler S, Yousefi S, Simon HU. Eosinophil extracellular DNA traps in skin diseases. J Allergy Clin Immunol. 2011;127:194–9.
Hashimoto T, Ueki S, Kamide Y, Miyabe Y, Fukuchi M, Yokoyama Y, Furukawa T, Azuma N, Oka N, Takeuchi H, et al. Increased circulating cell-free DNA in Eosinophilic Granulomatosis with Polyangiitis: implications for Eosinophil Extracellular traps and Immunothrombosis. Front Immunol. 2021;12:801897.
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53.
Yousefi S, Simon D, Stojkov D, Karsonova A, Karaulov A, Simon HU. In vivo evidence for extracellular DNA trap formation. Cell Death Dis. 2020;11:300.
Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF. Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood. 2013;121:2074–83.
Sim MS, Kim HJ, Bae I, Kim C, Chang HS, Choi Y, Lee DH, Park HS, Chung IY. Calcium ionophore-activated platelets induce eosinophil extracellular trap formation. Allergol Int. 2023;72:466–76.
Choi Y, Le Pham D, Lee DH, Lee SH, Kim SH, Park HS. Biological function of eosinophil extracellular traps in patients with severe eosinophilic asthma. Exp Mol Med. 2018;50:1–8.
Lu Y, Huang Y, Li J, Huang J, Zhang L, Feng J, Li J, Xia Q, Zhao Q, Huang L, et al. Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat Cell Biol. 2021;23:1060–72.
Ueki S, Konno Y, Takeda M, Moritoki Y, Hirokawa M, Matsuwaki Y, Honda K, Ohta N, Yamamoto S, Takagi Y, et al. Eosinophil extracellular trap cell death-derived DNA traps: their presence in secretions and functional attributes. J Allergy Clin Immunol. 2016;137:258–67.
Muniz VS, Silva JC, Braga YAV, Melo RCN, Ueki S, Takeda M, Hebisawa A, Asano K, Figueiredo RT, Neves JS. Eosinophils release extracellular DNA traps in response to Aspergillus Fumigatus. J Allergy Clin Immunol. 2018;141:571–e585577.
Kim HJ, Sim MS, Lee DH, Kim C, Choi Y, Park HS, Chung IY. Lysophosphatidylserine induces eosinophil extracellular trap formation and degranulation: implications in severe asthma. Allergy. 2020;75:3159–70.
Barroso MV, Gropillo I, Detoni MAA, Thompson-Souza GA, Muniz VS, Vasconcelos CRI, Figueiredo RT, Melo RCN, Neves JS. Structural and signaling events driving aspergillus fumigatus-Induced Human Eosinophil Extracellular Trap Release. Front Microbiol. 2021;12:633696.
Germic N, Fettrelet T, Stojkov D, Hosseini A, Horn MP, Karaulov A, Simon D, Yousefi S, Simon HU. The release kinetics of Eosinophil peroxidase and mitochondrial DNA is different in Association with Eosinophil Extracellular trap formation. Cells 2021, 10.
Ueki S, Tokunaga T, Fujieda S, Honda K, Hirokawa M, Spencer LA, Weller PF. Eosinophil ETosis and DNA traps: a New look at eosinophilic inflammation. Curr Allergy Asthma Rep. 2016;16:54.
Ueki S, Hebisawa A, Kitani M, Asano K, Neves JS. Allergic bronchopulmonary Aspergillosis-A luminal hypereosinophilic Disease with Extracellular Trap Cell Death. Front Immunol. 2018;9:2346.
Thompson-Souza GA, Vasconcelos CRI, Neves JS. Eosinophils: focus on DNA extracellular traps. Life Sci. 2022;311:121191.
Appelgren D, O’Sullivan KM. Editorial: the role of leukocyte extracellular traps in inflammation and autoimmunity. Front Immunol. 2022;13:1075026.
Hwang CS, Park SC, Cho H-J, Park D-J, Yoon J-H, Kim C-H. Eosinophil extracellular trap formation is closely associated with disease severity in chronic rhinosinusitis regardless of nasal polyp status. Sci Rep 2019, 9.
Neves VH, Palazzi C, Bonjour K, Ueki S, Weller PF, Melo RCN. In vivo ETosis of human eosinophils: the ultrastructural signature captured by TEM in Eosinophilic diseases. Front Immunol. 2022;13:938691.
Ueki S, Tokunaga T, Melo RCN, Saito H, Honda K, Fukuchi M, Konno Y, Takeda M, Yamamoto Y, Hirokawa M, et al. Charcot-Leyden crystal formation is closely associated with eosinophil extracellular trap cell death. Blood. 2018;132:2183–7.
Rosenberg HF, Foster PS. Reply to eosinophil cytolysis and release of cell-free granules. Nat Rev Immunol. 2013;13:902.
Silveira JS, Antunes GL, Kaiber DB, da Costa MS, Ferreira FS, Marques EP, Schmitz F, Gassen RB, Breda RV, Wyse ATS, et al. Autophagy induces eosinophil extracellular traps formation and allergic airway inflammation in a murine asthma model. J Cell Physiol. 2020;235:267–80.
Yousefi S, Simon D, Simon HU. Eosinophil extracellular DNA traps: molecular mechanisms and potential roles in disease. Curr Opin Immunol. 2012;24:736–9.
Dash B, Sun X. Eosinophils set DNA traps in allergic asthma. Nat Cell Biol. 2021;23:1057–9.
Morshed M, Yousefi S, Stöckle C, Simon HU, Simon D. Thymic stromal lymphopoietin stimulates the formation of eosinophil extracellular traps. Allergy. 2012;67:1127–37.
Silveira JS, Antunes GL, Kaiber DB, da Costa MS, Marques EP, Ferreira FS, Gassen RB, Breda RV, Wyse ATS, Pitrez P, da Cunha AA. Reactive oxygen species are involved in eosinophil extracellular traps release and in airway inflammation in asthma. J Cell Physiol. 2019;234:23633–46.
Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, Park SH, Ahn WG, Kim S, Lee MG, et al. Autophagy primes neutrophils for Neutrophil Extracellular trap formation during Sepsis. Am J Respir Crit Care Med. 2017;196:577–89.
Radonjic-Hoesli S, Wang X, de Graauw E, Stoeckle C, Styp-Rekowska B, Hlushchuk R, Simon D, Spaeth PJ, Yousefi S, Simon HU. Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counterregulated by autophagy. J Allergy Clin Immunol. 2017;140:1632–42.
Esnault S, Fichtinger PS, Barretto KT, Fogerty FJ, Bernau K, Mosher DF, Mathur SK, Sandbo N, Jarjour NN. Autophagy protects against Eosinophil Cytolysis and Release of DNA. Cells 2022, 11.
Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU. Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology. 2017;152:517–25.
Frigas E, Gleich GJ. The eosinophil and the pathophysiology of asthma. J Allergy Clin Immunol. 1986;77:527–37.
Korevaar DA, Westerhof GA, Wang J, Cohen JF, Spijker R, Sterk PJ, Bel EH, Bossuyt PM. Diagnostic accuracy of minimally invasive markers for detection of airway eosinophilia in asthma: a systematic review and meta-analysis. Lancet Respir Med. 2015;3:290–300.
Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, Gerard C. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–9.
Cunha AA, Porto BN, Nuñez NK, Souza RG, Vargas MH, Silveira JS, Souza TT, Jaeger N, Pitrez PM. Extracellular DNA traps in bronchoalveolar fluid from a murine eosinophilic pulmonary response. Allergy. 2014;69:1696–700.
Choi Y, Sim S, Park HS. Distinct functions of eosinophils in severe asthma with type 2 phenotype: clinical implications. Korean J Intern Med. 2020;35:823–33.
Mukherjee M, Lacy P, Ueki S. Eosinophil Extracellular traps and Inflammatory pathologies-untangling the web! Front Immunol. 2018;9:2763.
Choi Y, Kim YM, Lee HR, Mun J, Sim S, Lee DH, Pham DL, Kim SH, Shin YS, Lee SW, Park HS. Eosinophil extracellular traps activate type 2 innate lymphoid cells through stimulating airway epithelium in severe asthma. Allergy. 2020;75:95–103.
Whetstone CE, Ranjbar M, Omer H, Cusack RP, Gauvreau GM. The role of Airway Epithelial Cell alarmins in Asthma. Cells 2022, 11.
Silveira JS, Antunes GL, Gassen RB, Breda RV, Stein RT, Pitrez PM, da Cunha AA. Respiratory syncytial virus increases eosinophil extracellular traps in a murine model of asthma. Asia Pac Allergy. 2019;9:e32.
Shah PL, Scott SF, Knight RA, Marriott C, Ranasinha C, Hodson ME. In vivo effects of recombinant human DNase I on sputum in patients with cystic fibrosis. Thorax. 1996;51:119–25.
da Cunha AA, Nunez NK, de Souza RG, Moraes Vargas MH, Silveira JS, Antunes GL, Durante Lda S, Porto BN, Marczak ES, Jones MH, Pitrez PM. Recombinant human deoxyribonuclease therapy improves airway resistance and reduces DNA extracellular traps in a murine acute asthma model. Exp Lung Res. 2016;42:66–74.
Yousefi S, Sharma SK, Stojkov D, Germic N, Aeschlimann S, Ge MQ, Flayer CH, Larson ED, Redai IG, Zhang S, et al. Oxidative damage of SP-D abolishes control of eosinophil extracellular DNA trap formation. J Leukoc Biol. 2018;104:205–14.
Murrison LB, Ren X, Preusse K, He H, Kroner J, Chen X, Jenkins S, Johansson E, Biagini JM, Weirauch MT, et al. TSLP disease-associated genetic variants combined with airway TSLP expression influence asthma risk. J Allergy Clin Immunol. 2022;149:79–88.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–55.
Okubo K, Kurosawa M, Kamiya M, Urano Y, Suzuki A, Yamamoto K, Hase K, Homma K, Sasaki J, Miyauchi H, et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med. 2018;24:232–8.
Chow OA, von Köckritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, Gallo RL, Monestier M, Wang Y, Glass CK, Nizet V. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe. 2010;8:445–54.
El Shikh MEM, El Sayed R, Nerviani A, Goldmann K, John CR, Hands R, Fossati-Jimack L, Lewis MJ, Pitzalis C. Extracellular traps and PAD4 released by macrophages induce citrullination and auto-antibody production in autoimmune arthritis. J Autoimmun. 2019;105:102297.
Wong KW, Jacobs WR Jr. Mycobacterium tuberculosis exploits human interferon γ to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis. 2013;208:109–19.
Wei Z, Wang Y, Zhang X, Wang X, Gong P, Li J, Taubert A, Hermosilla C, Zhang X, Yang Z. Bovine macrophage-derived extracellular traps act as early effectors against the abortive parasite Neospora Caninum. Vet Parasitol. 2018;258:1–7.
Jensen M, Thorsen NW, Hallberg LAE, Hägglund P, Hawkins CL. New insight into the composition of extracellular traps released by macrophages exposed to different types of inducers. Free Radic Biol Med. 2023;202:97–109.
Aulik NA, Hellenbrand KM, Czuprynski CJ. Mannheimia haemolytica and its leukotoxin cause macrophage extracellular trap formation by bovine macrophages. Infect Immun. 2012;80:1923–33.
Kalsum S, Braian C, Koeken V, Raffetseder J, Lindroth M, van Crevel R, Lerm M. The Cording phenotype of Mycobacterium tuberculosis induces the formation of Extracellular traps in Human macrophages. Front Cell Infect Microbiol. 2017;7:278.
Rayner BS, Zhang Y, Brown BE, Reyes L, Cogger VC, Hawkins CL. Role of hypochlorous acid (HOCl) and other inflammatory mediators in the induction of macrophage extracellular trap formation. Free Radic Biol Med. 2018;129:25–34.
Je S, Quan H, Yoon Y, Na Y, Kim BJ, Seok SH. Mycobacterium massiliense induces macrophage extracellular traps with facilitating bacterial growth. PLoS ONE. 2016;11:e0155685.
Liu P, Wu X, Liao C, Liu X, Du J, Shi H, Wang X, Bai X, Peng P, Yu L, et al. Escherichia coli and Candida albicans induced macrophage extracellular trap-like structures with limited microbicidal activity. PLoS ONE. 2014;9:e90042.
Mohanan S, Horibata S, McElwee JL, Dannenberg AJ, Coonrod SA. Identification of macrophage extracellular trap-like structures in mammary gland adipose tissue: a preliminary study. Front Immunol. 2013;4:67.
Mónaco A, Canales-Huerta N, Jara-Wilde J, Härtel S, Chabalgoity JA, Moreno M, Scavone P. Salmonella Typhimurium triggers Extracellular traps Release in Murine macrophages. Front Cell Infect Microbiol. 2021;11:639768.
Zhang Y, Rayner BS, Jensen M, Hawkins CL. In Vitro Stimulation and visualization of Extracellular Trap Release in Differentiated Human Monocyte-derived macrophages. J Vis Exp 2019.
Perez D, Munoz MC, Molina JM, Munoz-Caro T, Silva LM, Taubert A, Hermosilla C, Ruiz A. Eimeria Ninakohlyakimovae induces NADPH oxidase-dependent monocyte extracellular trap formation and upregulates IL-12 and TNF-alpha, IL-6 and CCL2 gene transcription. Vet Parasitol. 2016;227:143–50.
King PT, Sharma R, O’Sullivan K, Selemidis S, Lim S, Radhakrishna N, Lo C, Prasad J, Callaghan J, McLaughlin P, et al. Nontypeable Haemophilus influenzae induces sustained lung oxidative stress and protease expression. PLoS ONE. 2015;10:e0120371.
King PT, Sharma R, O’Sullivan KM, Callaghan J, Dousha L, Thomas B, Ruwanpura S, Lim S, Farmer MW, Jennings BR, et al. Deoxyribonuclease 1 reduces pathogenic effects of cigarette smoke exposure in the lung. Sci Rep. 2017;7:12128.
Yang ML, Sodré FMC, Mamula MJ, Overbergh L. Citrullination and PAD Enzyme Biology in Type 1 diabetes - regulators of inflammation, autoimmunity, and Pathology. Front Immunol. 2021;12:678953.
Kummarapurugu AB, Zheng S, Ma J, Ghosh S, Hawkridge A, Voynow JA. Neutrophil elastase triggers the release of Macrophage Extracellular traps: relevance to cystic fibrosis. Am J Respir Cell Mol Biol. 2022;66:76–85.
Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, Atsumi T, Ishizu A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun. 2016;67:19–28.
Rasmussen KH, Hawkins CL. Role of macrophage extracellular traps in innate immunity and inflammatory disease. Biochem Soc Trans. 2022;50:21–32.
Bonne-Annee S, Kerepesi LA, Hess JA, Wesolowski J, Paumet F, Lok JB, Nolan TJ, Abraham D. Extracellular traps are associated with human and mouse neutrophil and macrophage mediated killing of larval Strongyloides stercoralis. Microbes Infect. 2014;16:502–11.
Doster RS, Rogers LM, Gaddy JA, Aronoff DM. Macrophage extracellular traps: a scoping review. J Innate Immun. 2018;10:3–13.
Loureiro A, Pais C, Sampaio P. Relevance of Macrophage Extracellular traps in C. Albicans Killing. Front Immunol. 2019;10:2767.
Britt RD Jr., Ruwanpathirana A, Ford ML, Lewis BW. Macrophages Orchestrate Airway Inflammation, remodeling, and Resolution in Asthma. Int J Mol Sci 2023, 24.
Quoc QL, Cao TBT, Moon JY, Jang JH, Shin YS, Choi Y, Ryu MS, Park HS. Contribution of monocyte and macrophage extracellular traps to neutrophilic airway inflammation in severe asthma. Allergol Int. 2024;73:81–93.
Zhang X, Zhang L, Tan YM, Liu YP, Li JJ, Deng QM, Yan SB, Zhang W, Han L, Zhong M. Hepcidin gene silencing ameliorated inflammation and insulin resistance in adipose tissue of db/db mice via inhibiting METs formation. Mol Immunol. 2021;133:110–21.
Acknowledgements
We thank FigDraw for assistance with the cartographic work.
Funding
The work was supported by the National Natural Science Foundation of China (82170036).
Author information
Authors and Affiliations
Contributions
W.G. and G.Z designed the research. G.C and L.Z collected the data. H.J. and Z.L. prepared the materials. G.C., C.H., W.G., and G.Z wrote the manuscript. All authors read and reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gu, W., Huang, C., Chen, G. et al. The role of extracellular traps released by neutrophils, eosinophils, and macrophages in asthma. Respir Res 25, 290 (2024). https://doi.org/10.1186/s12931-024-02923-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-024-02923-x