
Abstract
Background
Interstitial lung diseases (ILD) comprise a heterogeneous group of mainly chronic lung diseases with different disease trajectories. Progression (PF-ILD) occurs in up to 50% of patients and is associated with increased mortality.
Methods
The EXCITING-ILD (Exploring Clinical and Epidemiological Characteristics of Interstitial Lung Diseases) registry was analysed for disease trajectories in different ILD. The course of disease was classified as significant (absolute forced vital capacity FVC decline > 10%) or moderate progression (FVC decline 5–10%), stable disease (FVC decline or increase < 5%) or improvement (FVC increase ≥ 5%) during time in registry. A second definition for PF-ILD included absolute decline in FVC % predicted ≥ 10% within 24 months or ≥ 1 respiratory-related hospitalisation. Risk factors for progression were determined by Cox proportional-hazard models and by logistic regression with forward selection. Kaplan-Meier curves were utilised to estimate survival time and time to progression.
Results
Within the EXCITING-ILD registry 28.5% of the patients died (n = 171), mainly due to ILD (n = 71, 41.5%). Median survival time from date of diagnosis on was 15.5 years (range 0.1 to 34.4 years). From 601 included patients, progression was detected in 50.6% of the patients (n = 304) with shortest median time to progression in idiopathic NSIP (iNSIP; median 14.6 months) and idiopathic pulmonary fibrosis (IPF; median 18.9 months). Reasons for the determination as PF-ILD were mainly deterioration in lung function (PFT; 57.8%) and respiratory hospitalisations (40.6%). In multivariate analyses reduced baseline FVC together with age were significant predictors for progression (OR = 1.00, p < 0.001). Higher GAP indices were a significant risk factor for a shorter survival time (GAP stage III vs. I HR = 9.06, p < 0.001). A significant shorter survival time was found in IPF compared to sarcoidosis (HR = 0.04, p < 0.001), CTD-ILD (HR = 0.33, p < 0.001), and HP (HR = 0.30, p < 0.001). Patients with at least one reported ILD exacerbation as a reason for hospitalisation had a median survival time of 7.3 years (range 0.1 to 34.4 years) compared to 19.6 years (range 0.3 to 19.6 years) in patients without exacerbations (HR = 0.39, p < 0.001).
Conclusion
Disease progression is common in all ILD and associated with increased mortality. Most important risk factors for progression are impaired baseline forced vital capacity and higher age, as well as acute exacerbations and respiratory hospitalisations for mortality. Early detection of progression remains challenging, further clinical criteria in addition to PFT might be helpful.
Similar content being viewed by others


Background
Interstitial lung diseases (ILD) comprise more than 200 mainly chronic diseases affecting the lung parenchyma due to inflammation and/or fibrosis [1, 2]. The multidisciplinary diagnostic process considers clinical, radiological, and pathological aspects of the disease [2, 3]. A relevant number of both, inflammatory and fibrotic ILD patients shows a progressive course of disease (PF-ILD) characterised by worsening of respiratory symptoms and decline in lung functional parameters such as vital capacity (VC). Recently a definition of progressive pulmonary fibrosis (PPF) has been published considering radiological evidence of progression as well as worsening of respiratory symptoms and functional decline [4]. Progression, at least in fibrotic ILD (fILD), is associated with increased mortality [5, 6]. Close monitoring is therefore important to detect progression as early as possible [7]. This is also relevant with regard to new therapeutic options, e.g. antifibrotic therapy in PPF. In the past years, antifibrotic drugs have been investigated in PPF [8,9,10]. Within the INBUILD trial, nintedanib showed efficacy in attenuation of FVC decline [8]. The RELIEF study investigated the efficacy of pirfenidone in PPF, and although the trial was stopped early due to under-recruitment, it showed a significant slowing of disease progression [9]. Another study studied pirfenidone in unclassifiable ILD (uILD) with progressive fibrosis. Although the primary endpoint based on home spirometry was not met, secondary endpoints such as on-site FVC, diffusing capacity for carbon monoxide (DLCO) and 6-MWD (6 min walking distance) were suggestive of effect of pirfenidone treatment [10].
In order to ensure early detection of progression in different ILD subtypes with a high degree of diagnostic certainty, further characterisation and understanding of disease behaviour is essential. Registries allow important insights on such aspects. The Canadian Registry for Pulmonary Fibrosis (CARE-PF) enrolled patients with fILD of any subtype prospectively, identified associated baseline factors, clinical characteristics and outcomes [11]. Progression was common in this cohort, and similarly prevalent in idiopathic pulmonary fibrosis (IPF) and hypersensitivity pneumonitis (HP) [11]. In line with this, prevalence of PF-ILD was reported to be 27% of al non-IPF patients in the retrospective PROGRESS study [12]. An international survey estimated progression in 14–32% of patiens with ILD other than IPF [13].
Our analyses are based on the “Exploring Clinical and Epidemiological Characteristics of Interstitial Lung Diseases” (EXCITING-ILD) registry. This multicenter, noninterventional prospective and observational disease and outcomes registry was conducted by the German Center for Lung Research (DZL) in close collaboration with cross-sectional sites [14]. Aim of the current work was to assess the following three objectives: (a) assessing progression in different ILD subtypes including IPF and other form of fibrotic and non-fibrotic ILD, (b) assessing risk factors for ILD progression and (c) analysing association between ILD progression and mortality.
Methods
Study Design
Within the EXCITING-ILD registry, sociodemographic and medical data on all different ILD subtypes were collected. The study protocol has been published elsewhere [14]. To summarise, incident and prevalent ILD patients from various healthcare facilities including outpatient, inpatient, and academic sites, were included. All patients were followed prospectively for a minimum of 36 months and a maximum of five years. Data from baseline and follow-up visits were reported by the investigators and entered into the full analysis set (FAS): demographic data, information on ILD subtypes, diagnostic procedures, distinct comorbidities, ILD management, as well as outcomes, progression and associated factors. For further analyses all patients with a minimum of one documented post-baseline visit during a median follow-up of 3 years were considered [14, 15]. The study was approved by the Ethics Committee of the Medical Faculty of the University of Heidelberg, Germany (S-525/2013) as well as by all local ethics committees of the participating centers.
Statistical analysis
Means with standard deviations (SD) and percentages were used to analyse observational data descriptively. For graphical presentation lineplots were generated for changes from baseline in FVC, and alluvial plots for transitions of progression stages. Kaplan-Meier curves were utilised to estimate progression-free survival times (survival time, time to progression), for the comparison of two or more Kaplan-Meier curves a log-rank test was performed. For these analyses, individuals who do not experience the event until the registry was closed, who were lost to follow up or withdraw from the registry are censored. Then the earlier available date of last follow-up or discontinuation was used. Confidence intervals were set with a two-sided level of 95% [14]. To quantify the difference between groups, hazard ratios complemented by corresponding two-sided 95% confidence intervals were estimated based on a Cox proportional hazards model. P-values of the corresponding Wald-test were calculated. For data analysis the statistics software R (R version 4.1.2) was used.
The following parameters were applied for the definition of ILD progression during time in registry: (1) Progression was classified on the basis of absolute changes in FVC % predicted as established by Hoffmann-Vold et al. [16] into significant (FVC decline > 10%) or moderate progression (FVC decline 5%–10%), stable (FVC decline or increase < 5%) and improvement (FVC increase ≥ 5%). The reference time was chosen as first visit with a non-missing FVC value. Patients were considered as progressive, if progression had occurred in at least one follow-up visit. (2) A second definition for PF-ILD was set as absolute decline in FVC % predicted ≥ 10% wthin a period of 24 months or at least one respiratory hospitalisation.
ILD-GAP index was calculated for each patient. The ILD-GAP index is a point scoring stage model based on clinical and physiologic variables to predict mortality in patients with different forms of ILD. Higher ILD-GAP scores indicate worse prognosis [17].
To develop a model to predict progression and to identify variables with a significant influence on progression, logistic regression with forward selection was used. As possible predictors the following variables at baseline were considered: time since baseline visit, age at year of inclusion, sex, body mass index (BMI), familial ILD, ILD subtype, smoking behaviour, FVC in % predicted, DLCO-SB in % predicted, reflux, pulmonary hypertension, concomitant emphysema. The same model was used to predict FVC changes. First, univariable regression models were set up (5% significance level) and second multiple regression models were generated.
Survival time was defined as the time between date of diagnosis and date of death. Another analysis was made for survival time from inclusion to the registry. Outcome analyses were made for a selection of ILD subtypes of special interest; these included IPF, non-specific interstitial pneumonia (iNSIP), cryptogenic organizing pneumonia (COP), uILD, sarcoidosis, HP, rheumatic and connective tissue diseases with pulmonary involvement (CTD-ILD), and drug-related ILD (DI-ILD).
Results
Study population
The EXCITING-ILD registry included 601 patients of 32 centers with a mean age of 64.3 years (Table 1). 60.7% were male and mean FVC was 76.4% predicted. The ILD subtypes included were: 26.6% sarcoidosis, 25.3% IPF, 9.7% HP, 7.2% CTD- and RA-ILD, 7% iNSIP, 5.7% uILD, 4.2% COP, 2.7% DI-ILD, 2.3% fibrosis in emphysema patients without signs of other ILD (CPFE), 1.8% pneumoconiosis, 1.2% pulmonary lymphangioleiomyomatosis (LAM), 1.0% eosinophilic pneumonia, 1.0% radiotherapy associated-ILD (RTX-ILD), 0.8% other granulomatous lung disease (other GRAN-ILD), 0.8% desquamative interstitial pneumonia (DIP), 0.7% respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), 0.5% pulmonary alveolar proteinosis (PAP), 0.3% pulmonary Langerhans´ cell histiocytosis (PLCH), and 1.2% others.
Disease trajectories
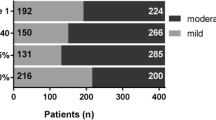
Progression based on absolute changes in FVC % predicted was detected in more than half of the patients (n = 304, 50.6%) with significant progression occurring more frequently than moderate progression, e.g. 15.5% vs. 10.3% for follow up visit 5, accordingly after 2.5 years in the registry. The rate of progressive patients increased over time while 32.4% of patients demonstrated stable FVC values or improvement - with heterogeneous individual disease trajectories (Table 2; Fig. 1).

Lineplot with individual changes from baseline in FVC [% predicted]. Progression was classified as described above and is shown in four different colours. Only individuals with non-missing values are shown (n = 558). The frequencies are shown for each follow-up visit (FU) starting with the first follow-up visit (FU 1) after 6 months, FU 2 after 12 months, FU 3 after 18 months, etc. FVC = forced vital capacity
Transition between different progression stages is shown in Fig. 2. Mainly, disease trajectory remained similar or progression occurred over time; only a minority of patients improved or had a more severe or less severe deterioration compared to the first follow-up.

Alluvial plot with transitions of progression stages. This figure shows the transition between different stages of progression. These are presented in different colours for significant progression, moderate progression, stable, improvement or missing parameters. The frequencies are shown for each follow-up visit (FU) starting with the first follow-up visit (FU 1) after 6 months, FU 2 after 12 months, FU 3 after 18 months, etc
Characterisation of progression
Median time to ILD progression based on absolute changes in FVC % predicted was 18.9 months in IPF (range 1.1 to 47.54 months) compared to 14.6 months in iNSIP (range 0.9 to 32.69 months) and 32.4 months in HP (range 4.8 to 40.28 months). Significant differences were found between sarcoidosis and IPF (HR = 0.31, p < 0.001), COP and IPF (HR = 0.5, p = 0.021), and HP and IPF (HR = 0.6, p = 0.011; Table 3; Fig. 3).

Time to ILD progression in months - Kaplan Meier curve by ILD subtypes of special interest. ILD subtypes of special interest included IPF, iNSIP, COP, uILD, sarcoidosis, HP, CTD-ILD, DI-ILD (n = 530). In addition, iNSIP, COP and uILD were grouped
Regarding the second definition of PF-ILDs, 57.8% met the PFT criterion and 40.6% were hospitalized due to a respiratory cause. In 1.6% both reasons were present. Subsequently, PF-ILD were compared with stable ILD and IPF as reference population. Compared to IPF, patients with PF-ILD were significantly less often male (p < 0.001), younger (p = 0.006), younger at onset of first symptoms (p < 0.001) less often ex-smokers (p = 0.028), and had better baseline values for TLC (p < 0.001). Baseline characteristics between PF-ILD and stable ILD showed no significant differences.
Risk factors for progression
Univariate logistic regression revealed that baseline value of FVC (OR = 1.02, p = 0.003) as well as age at inclusion (OR = 1.02, p = 0.003) were significant predictors of progression.
In multivariate analyses, significant predictors for progression were reduced baseline FVC together with age (OR = 1.00, p < 0.001). For changes in FVC, significant interaction effects were shown between time since baseline visit and BMI (p = 0.023) and time since baseline visit and the presence of the ILD subtypes IPF (p = 0.001), HP with unknown antigen (p = 0.001), iNSIP (p = 0.025) or CTD-ILD (p = 0.042). The baseline value of FVC was also significantly predictive for changes in FVC, but without a time interaction effect (p = 0.001).
Survival time
Median survival time since ILD diagnosis was 15.5 years (range 0.1 to 34.4 years), and death was observed in 171 cases (28.5%), mainly due to ILD (n = 71, 41.5%). During the time in the registry, deaths occurred early with 35.1% (n = 60) in the first 12 months and 28.1% (n = 48) in the second year (supplement table S1). Median survival time from inclusion in the registry was 58.7 months [50.1; n.e.].
Median survival time since ILD diagnosis in PF-ILD was 9.9 years (range 0.2 to 22.6). Patients with PF-ILD had a better prognosis than those with IPF (HR = 0.62, p = 0.013; Fig. 4).

Survival time - Kaplan Meier curve (years 0–10) by IPF, PF-ILD, and stable ILD. The definition for PF-ILD was set as absolute decline in FVC % predicted ≥ 10% within a period of 24 months or at least one respiratory hospitalisation
Risk factors for survival time
Higher ILD-GAP indices were a significant risk factor for shorter survival time (GAP stage II vs. I HR = 4.95, p < 0.001; GAP stage III vs. I HR = 9.06, p < 0.001; Table 3). Patients with at least one reported ILD exacerbation (AE) had a median survival time of 7.3 years (range 0.1 to 34.4 years) compared to 19.6 years (range 0.3 to 19.6 years) in patients without exacerbations (HR = 0.39, p < 0.001, Table 3, supplement figure S1). Median survival time after the diagnosis of an AE-ILD was 14.8 months (range 0.1 to 45.6 months). Patients with a long-term oxygen therapy (LTOT) showed worse prognoses with shorter survival time (HR = 0.26, p < 0.001, Table 3).
Table 3 shows median survival time (in years) and median time to progression (in months) for a selection of ILD subtypes of special interest (n = 530) as well as output of Cox regression model of survival time and time to progression analyses for ILD subtypes compared to IPF. Time to progression was defined according to the definition of progression free survival (PFS; Δ FVC ≥ 10% or Δ DLCO-SB ≥ 15% or death). In addition, output of Cox regression model of survival time was calculated for the full analysis set (FAS, n = 601) by ILD-GAP Index, acute exacerbations (AE), and long-term oxygen therapy (LTOT). AE = acute exacerbations, CI = confidence interval, COP = cryptogenic organizing pneumonia, CTD-ILD = Rheumatic and connective tissue diseases with pulmonary involvement, DI-ILD = drug-related ILD, HP = hypersensitivity pneumonitis HR = Hazard Ratio, iNSIP = non-specific interstitial pneumonia, IPF = idiopathic pulmonary fibrosis, LTOT = long-term oxygen therapy, No PFT = No pulmonary function testing, uILD = unclassifiable ILD.
Outcome analyses by ILD subtypes were performed for a selection of ILD of special interest: IPF, iNSIP, COP, uILD, sarcoidosis, HP, CTD-ILD, DI-ILD. Survival time differed between ILD subtypes (Table 3). For IPF, median survival time was 5.8 years (n = 152, range 0.6 to 14.5 years). The longest median survival time was found in CTD-ILD with 15.5 years (n = 43, range 0.6 to 15.5 years) and the shortest in uILD with 5.6 years (n = 34, range 0.2 to 5.6 years). A significant shorter survival time was found in IPF compared to sarcoidosis (HR = 0.04, p < 0.001), CTD-ILD (HR = 0.33, p < 0.001), and HP (HR = 0.30, p < 0.001, Table 3).
Discussion
In the present study we report, that within the EXCITING-ILD registry, a registry comprising all ILD entities with prevalent and incident patients, more than half of the patients demonstrated a clinically relevant progression.
Our definition of progression was based on pulmonary function parameters according to the definition used by Hoffmann-Vold et al. for systemic sclerosis-associated ILD (SSc-ILD) [16]. FVC has been used in many studies of fibrotic ILD to characterise disease progression, e.g. in the INBUILD trial [8], another trial on uILD [10], and the RELIEF trial [9]. However, especially in moderate progressions with smaller FVC declines or fluctuating values as shown for SSc-ILD [16], the addition of a further criterion might be reasonable. Furthermore, an international survey emphasised significant delays in the detection of progression in ILD resulting in 25–50% of patients witout adequate therapy [13]. Accordingly, our definition of progression also included hospitalisations known to impact mortality and other outcomes in patients with different ILD [18, 19]. An analysis for hospitalisations based on the EXCITING-ILD registry showed an association of mortality with all cause, ILD-related and respiratory-related hospitalisations for all ILD [15]. It can be assumed that the risk for non-elective hospitalisations is higher in progressive ILD and thus hospitalisation may indicate progression [15, 20].
Frequency of progression within the EXCITING-ILD registry is comparable with other registries, e.g. in the Canadian Registry for fibrosing ILD (CARE-PF), with 50% out of 2746 patients showing a progressive course of disease after two years [11]. Within the CARE-PF registry, progression was defined as FVC decline ≥ 10%, death, lung transplantation or any two of: relative FVC decline ≥ 5% and < 10%, worsening respiratory symptoms or worsening fibrosis on computed tomography of the chest, all within 24 months of diagnosis [11]. Based on the CARE-PF registry and our results also including FVC decline as main parameter for future progression, it can be stated that FVC decline is a reliable indicator of future progression in all ILD. The PROGRESS study based on a cohort of patients with progressive fibrosing ILD found criteria of progression to be fulfilled in 27% of patients over a period of seven years [12]. Progression criteria were defined as a FVC decline ≥ 10%, FVC decline between 5% and 10% associated with worsening of respiratory symptoms or increased extent of fibrosis on chest HRCT, or increased extent of fibrosis on chest HRCT with worsening of respiratory symptoms [12]. Remarkably, in the PROGRESS study, progression was detected less frequently as compared to the CARE-PF and EXCITING-ILD registries. Main reasons for these differences might be the exclusion of patients with IPF in the PROGRESS study in contrast to EXCITING-ILD and CARE-PF registry, as well as differing definitions of ILD progression without additional criteria as hospitalisations, death, or lung transplantations within the PROGRESS study.
The probability of progression increases over time [11] and a transition to more severe stages of disease progression becomes more likely. Also, within the EXCITING-ILD registry, disease trajectory remained similar or progression occurred over time. Only a minority of patients improved or had a more severe or less severe deterioration compared to the first follow-up.
Progression results in worse outcomes and higher mortality. As described by Hambly et al. for progressive fibrosing ILD, IPF showed worse outcomes compared to sarcoidosis and CTD-ILD [11]. Only unclassified ILD had shorter survival times. To complement this, Torrisi et al. reported worse prognosis for unclassifiable ILD and a progressive phenotype [21]. We here report, that mortality was caused to a large extent directly by the ILD. Again, this reflects the high burden of disease in ILD as described by a recent update of the global burden of disease study [22].
The most important predictor for progression within the EXCITING-ILD registry was a reduced baseline FVC, also in conjunction with older age. The CARE-PF registry also found highest risk for progression in patients with reduced PFT [11]. These findings are underlining the value of the ILD-GAP index, a point scoring stage model based on the ILD subtype, gender, age and pulmonary function parameters (FVC and DLCO) to predict mortality in patients with different ILD [17]. Another important risk factor for ILD progression are acute exacerbations, as also reflected in our analyses with a significantly impaired survival after AE-ILD compared to patients not experiencing AE-ILD. The mortality risk after AE is well established for IPF [23], but only sparsely reported in other ILD such as HP [24] and very recently for progressive fibrosing ILD other than IPF [25].
Here, we report a significant better prognosis in HP than in IPF. This is in contrast to the Canadian registry demonstrating similar prevalence for progression in HP and IPF [11]. This discrepancy might be explained by different inclusion criteria as the CARE-PF registry only enrolled patients with fibrosing ILD [11]. The EXCITING-ILD registry included all different ILD regardless of a fibrosing phenotype.
Further strengths of the prospective EXCITING-ILD registry include the reflection of the “real world”-situation of patients with different ILD being treated in different institutions of the health care system including general pulmonology outpatient practice to ILD expert centers. Another advantage is the broad inclusion of all ILD. The large number of patients and the availability of many different parameters allows special questions to be investigated in detail. Our findings highlight the impact of ILD progression on outcome and mortality and are therefore of high value. Our data support the value regular clinical and functional monitoring of ILD patients, especially for those with a risk for future progression [26].
However, some limitations of our approach should be mentioned. Since both incident and prevalent patients were included, no distinctions are possible in this respect. In addition, causal statements cannot be made due to the observational and nonrandomised study character. Furthermore, as baseline CTs are not available, outcomes cannot be distinguished between fibrotic and inflammatory driven ILD. Moreover, not all diagnoses, especially for sarcoidosis, were made on an interdisciplinary basis. Clinical decisions of the physicians may differ, also due to the recruitment from many different centers and therefore levels of expertise in ILD [14]. Another limitation is that comorbidities were not considered thoroughly, although they pose an important role as shown for the prediction of survival in IPF by TORVAN model considering comorbidities in addition to ILD-GAP index [27]. Because the EXCITING-ILD registry was conducted before the establishment of the ATS/ERS/JRS/ALAT Clinical Practice Guidelines on PPF [4], our definition of progression differs. In particular, radiological progression is not considered due to lack of data on CT imaging and clinical symptoms were not reported.
Conclusion
In the EXCITING-ILD registry, progression was common with more than 50%, and resulted in higher mortality. The most important risk factor was a reduced baseline forced vital capacity. Furthermore, acute exacerbations and respiratory hospitalisations were associated with a significant higher mortality. Early detection of progression remains challenging, especially in patients with only moderate FVC declines. In these cases, further clinical criteria as hospitalisations might be helpful.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AE:
-
ILD acute exacerbation of ILD
- BMI:
-
body mass index
- CARE-PF:
-
Canadian Registry for Pulmonary Fibrosis
- CI:
-
confidence interval
- CO:
-
Pcryptogenic organizing pneumonia
- CPFE:
-
Fibrosis in emphysema patients without signs of other ILD
- CTD-ILD:
-
connective tissue disease-associated ILD
- DI-ILD:
-
drug-related ILD
- DIP:
-
desquamative interstitial pneumonia
- DLCO-SB:
-
diffusing capacity for carbon monoxide (CO) – single breath
- EXCITING-ILD:
-
Exploring Clinical and Epidemiological Characteristics of Interstitial Lung Diseases
- FAS:
-
full analysis set
- FEV1:
-
forced expiratory volume in 1 s
- fILD:
-
fibrotic ILD
- FU:
-
follow up
- FVC:
-
forced vital capacity
- GRAN-ILD:
-
granulomatous ILD
- HP:
-
hypersensitivity pneumonitis
- HR:
-
Hazard ratios
- ILD:
-
interstitial lung disease
- iNSIP:
-
non-specific interstitial pneumonia
- IPF:
-
idiopathic pulmonary fibrosis
- LAM:
-
pulmonary lymphangioleiomyomatosis
- LTOT:
-
long-term oxygen therapy
- OR:
-
Odd’s ratio
- PAP:
-
pulmonary alveolar proteinosis
- PFT:
-
pulmonary function testing
- PF-ILD:
-
progressive fibrosing ILD
- PFS:
-
progression free survival
- PLCH:
-
pulmonary Langerhans´ cell histiocytosis
- PPF:
-
progressive pulmonary fibrosis
- SD:
-
standard deviation
- RB-ILD:
-
respiratory bronchiolitis-associated interstitial lung disease
- RTX-ILD:
-
radiotherapy associated ILD
- SSc:
-
systemic sclerosis
- SD:
-
standard deviations
- uILD:
-
unclassifiable ILD
- VC:
-
vital capacity
- 6-MWD:
-
6 min walking distance
References
Valeyre D, Duchemann B, Annesi-Maesano I et al. Interstitial lung diseases, in Respiratory Epidemiology, T. Welte, I. Annesi-Maesano, G. Viegi, and B. Lundbäck,Eds., vol. 65 of ERSMonograph, Chap. 6, ERS, 2014.
American Thoracic Society and European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165(2):277–304.
Travis WD, Costabel U, Hansell D. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina-Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia-Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47.
Valenzuela C, Cottin V. Epidemiology and real-life experience in progressive pulmonary fibrosis. Curr Opin Pulm Med. 2022;28(5):407–13. https://doi.org/10.1097/MCP.0000000000000908.
Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, Skowasch D, Park JS, Poonyagariyagorn HK, Wuyts W, Wells AU. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076. https://doi.org/10.1183/16000617.0076-2018.
Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung diseases. N Engl J Med. 2020;383(10):958–68. https://doi.org/10.1056/NEJMra2005230.
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme-Beaty E, Rosenstock B, Quaresma M, Haeufel T, Goeldner RG, Schlenker-Herceg R, Brown KK. INBUILD Trial investigators. Nintedanib in Progressive Fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–27. https://doi.org/10.1056/NEJMoa1908681.
Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, Bonnet R, Grohe C, Held M, Wilkens H, Hammerl P, Koschel D, Blaas S, Wirtz H, Ficker JH, Neumeister W, Schönfeld N, Claussen M, Kneidinger N, Frankenberger M, Hummler S, Kahn N, Tello S, Freise J, Welte T, Neuser P, Günther A. RELIEF investigators. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. 2021;9(5):476–86. https://doi.org/10.1016/S2213-2600(20)30554-3.
Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, Axmann J, Kirchgaessler KU, Samara K, Gilberg F, Cottin V. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020;8(2):147–57. https://doi.org/10.1016/S2213-2600(19)30341-8.
Hambly N, Farooqi MM, Dvorkin-Gheva A, Donohoe K, Garlick K, Scallan C, Chong SG, MacIsaac S, Assayag D, Johannson KA, Fell CD, Marcoux V, Manganas H, Morisset J, Comes A, Fisher JH, Shapera S, Gershon AS, To T, Wong AW, Sadatsafavi M, Wilcox PG, Halayko AJ, Khalil N, Cox G, Richeldi L, Ryerson CJ, Kolb M. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J. 2022;60(4):2102571. https://doi.org/10.1183/13993003.02571-2021.
Nasser M, Larrieu S, Si-Mohamed S, Ahmad K, Boussel L, Brevet M, Chalabreysse L, Fabre C, Marque S, Revel D, Thivolet-Bejui F, Traclet J, Zeghmar S, Maucort-Boulch D, Cottin V. Progressive fibrosing interstitial lung disease: a clinical cohort (the PROGRESS study). Eur Respir J. 2021;57(2):2002718. https://doi.org/10.1183/13993003.02718-2020.
Wijsenbeek M, Kreuter M, Olson A, Fischer A, Bendstrup E, Wells CD, Denton CP, Mounir B, Zouad-Lejour L, Quaresma M, Cottin V. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. 2019;35(11):2015–24. https://doi.org/10.1080/03007995.2019.1647040.
Kreuter M, Herth FJ, Wacker M, Leidl R, Hellmann A, Pfeifer M, Behr J, Witt S, Kauschka D, Mall M, Günther A, Markart P. Exploring clinical and epidemiological characteristics of interstitial Lung diseases: Rationale, Aims, and design of a nationwide prospective Registry–The EXCITING-ILD Registry. Biomed Res Int. 2015;2015:123876. https://doi.org/10.1155/2015/123876.
Buschulte K, Kabitz HJ, Hagmeyer L, Hammerl P, Esselmann A, Wiederhold C, Skowasch D, Stolpe C, Joest M, Veitshans S, Höffgen M, Maqhuzu P, Schwarzkopf L, Hellmann A, Pfeifer M, Behr J, Karpavicius R, Günther A, Polke M, Höger P, Somogyi V, Lederer C, Markart P, Kreuter M. Hospitalisation patterns in interstitial lung diseases: data from the EXCITING-ILD registry. Respir Res. 2024 Jan 4;25(1):5. https://doi.org/10.1186/s12931-023-02588-y. PMID: 38178212; PMCID: PMC10765927.
Hoffmann-Vold AM, Allanore Y, Alves M, Brunborg C, Airó P, Ananieva LP, Czirják L, Guiducci S, Hachulla E, Li M, Mihai C, Riemekasten G, Sfikakis PP, Kowal-Bielecka O, Riccardi A, Distler O. EUSTAR collaborators. Progressive interstitial lung disease in patients with systemic sclerosis-associated interstitial lung disease in the EUSTAR database. Ann Rheum Dis. 2021;80(2):219–27. https://doi.org/10.1136/annrheumdis-2020-217455.
Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, Elicker BM, Wolters PJ, Koth LL, King TE Jr, Collard HR. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723–8. https://doi.org/10.1378/chest.13-1474.
Cottin V, Schmidt A, Catella L, Porte F, Fernandez-Montoya C, Le Lay K, Bénard S. Burden of idiopathic pulmonary fibrosis progression: a 5-Year longitudinal Follow-Up study. PLoS ONE. 2017;12(1):e0166462. https://doi.org/10.1371/journal.pone.0166462.
Brown AW, Fischer CP, Shlobin OA, Buhr RG, Ahmad S, Weir NA, Nathan SD. Outcomes after hospitalization in idiopathic pulmonary fibrosis: a cohort study. Chest. 2015;147(1):173–9.
Wälscher J, Witt S, Schwarzkopf L, Kreuter M. Hospitalisation patterns of patients with interstitial lung disease in the light of comorbidities and medical treatment - a German claims data analysis. Respir Res. 2020;21(1):73.
Torrisi SE, Kahn N, Wälscher J, Polke M, Lee JS, Molyneaux PL, Sambataro FM, Heussel CP, Vancheri C, Kreuter M. Outcomes and incidence of PF-ILD according to different definitions in a Real-World setting. Front Pharmacol. 2021;12:790204. https://doi.org/10.3389/fphar.2021.790204.
Ma X, Zhu L, Kurche JS, Xiao H, Dai H, Wang C. Global and regional burden of interstitial lung disease and pulmonary sarcoidosis from 1990 to 2019: results from the Global Burden of Disease study 2019. Thorax. 2022;77(6):596–605. https://doi.org/10.1136/thoraxjnl-2020-216732.
Kreuter M, Koegler H, Trampisch M, Geier S, Richeldi L. Differing severities of acute exacerbations of idiopathic pulmonary fibrosis (IPF): insights from the INPULSIS® trials. Respir Res. 2019;20(1):71. https://doi.org/10.1186/s12931-019-1037-7.
Hariri LP, Mino-Kenudson M, Shea B, Digumarthy S, Onozato M, Yagi Y, Fraire AE, Matsubara O, Mark EJ. Distinct histopathology of acute onset or abrupt exacerbation of hypersensitivity pneumonitis. Hum Pathol. 2012;43(5):660–8. https://doi.org/10.1016/j.humpath.2011.06.001.
Kreuter M, Bendstrup E, Kondoh Y, et al. On behalf of the INBUILD trial investigators Acute exacerbations in patients with Progressive Fibrosing interstitial lung diseases: data from the INBUILD Trial.B94. LEARNING FROM REGISTRIES AND CLINICAL TRIALS IN ILD. American Thoracic Society; 2022. pp. A3428–8.
Kreuter M, Behr J, Bonella F, Costabel U, Gerber A, Hamer OW, Heussel CP, Jonigk D, Krause A, Koschel D, Leuschner G, Markart P, Nowak D, Pfeifer M, Prasse A, Wälscher J, Winter H, Kabitz HJ. S1-Leitlinie Interdisziplinäre Diagnostik Interstitieller Lungenerkrankungen Im Erwachsenenalter [Consensus guideline on the interdisciplinary diagnosis of interstitial lung diseases]. Pneumologie. 2023;77(5):269–302. https://doi.org/10.1055/a-2017-8971. German.
Torrisi SE, Ley B, Kreuter M, Wijsenbeek M, Vittinghoff E, Collard HR, Vancheri C. The added value of comorbidities in predicting survival in idiopathic pulmonary fibrosis: a multiCenter observational study. Eur Respir J. 2019;53(3):1801587. https://doi.org/10.1183/13993003.01587-2018.
Acknowledgements
The registry is located at the Translational Lung Research Center Heidelberg (TLRC), a member of the German Center for Lung Research (DZL), and supported by an unrestricted grant from InterMune (now Roche). Financial support is independent of the scientific concept of the registry, data analysis, interpretation, or publication other than an acknowledgement of financial support. We would like to thank Phillen Maqhuzu and Larissa Schwarzkopf from Helmholtz Center Munich for their support regarding database and statistics. Furthermore, we would like to thank Eva Brammen from Chrestos Concept GmbH & Co. KG for support regarding the database, tables and figures.
Funding
This study was supported by an unrestricted grant from InterMune (now Roche). Financial support is independent of the scientific concept of the registry, data analysis, interpretation, or publication other than an acknowledgement of financial support.
Author information
Authors and Affiliations
Contributions
MK and PM were responsible for the study design. HJK, LH, PH, AE, CW, DS, CS, MJ, SV, MH, AH, MP, JB, RK, AG, MP, PH, VS, and CL had contributions to the conception of the work and were involved in recruiting the patients and documentation in the full analysis set. PM and LS were responsible for the statistical analyses. KB was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was conducted ethically in accordance with the World Medical Association Declaration of Helsinki and was approved by the Ethics Committee of the Medical Faculty of the University of Heidelberg, Germany (S-525/2013) as well as by all local ethics committees of the participating centres. Informed consent to participate in the study was obtained from all participants.
Consent for publication
Not applicable.
Conflict of interest
KB received payment for lectures from Boehringer Ingelheim and a grant from Sarkoidose-Netzwerk e.V. LH received fees for consulting and lectures from Boehringer-Ingelheim, GSK, AstraZeneca, Pfizer and BMS. PH received fees for lectures from Galapagos, GSK, Boehringer Ingelheim, AstraZeneca, Roche. DS reports fees for lectures or consultations from AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Janssen, MSD, Sanofi, all outside the submitted work. CS reports fees for lectures GSK, AstraZeneca, Boehringer Ingelheim, Berlin Chemie. MJ reports fees for lectures or consultations from AstraZeneca, Bencard, Boehringer Ingelheim, GSK, HAL Allergy, Sanofi, all outside the submitted work. SV reports fees from Berlin Chemie, GSK, Lilly, Pfizer, and Boehringer-Ingelheim. LS reports consulting fees from Galapagos. JB reports personal fees for lectures and consulting from Astra-Zeneca, Biogen, Boehringer-Ingelheim, BMS, Ferrer, Novartis, Roche, and Sanofi. AG reports grants, leture payments and/or consulting fees from Boehringer Ingelheim, Roche, Lung Therapeutics and Pieris. MP has received payment or honoraria for lectures, presentations or educational events from Boehringer Ingelheim and AstraZeneca. VS received support for attending meetings from CSL Behring. PM received fees for consulting and lectures from Boehringer-Ingelheim and Roche. MK reports grants, consulting fees, or payment for lectures from Boehringer Ingelheim, Galapagos, AstraZeneca, BMS and Roche. All other authors have nothing to disclose. The registry was supported by an unrestricted grant by Roche.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
^Deceased: Rainer Karpavicius
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Buschulte, K., Kabitz, HJ., Hagmeyer, L. et al. Disease trajectories in interstitial lung diseases – data from the EXCITING-ILD registry. Respir Res 25, 113 (2024). https://doi.org/10.1186/s12931-024-02731-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-024-02731-3