
Abstract
Background
Rhinovirus (RV) infection of airway epithelial cells triggers asthma exacerbations, during which airway smooth muscle (ASM) excessively contracts. Due to ASM contraction, airway epithelial cells become mechanically compressed. We previously reported that compressed human bronchial epithelial (HBE) cells are a source of endothelin-1 (ET-1) that causes ASM contraction. Here, we hypothesized that epithelial sensing of RV by TLR3 and epithelial compression induce ET-1 secretion through a TGF-β receptor (TGFβR)-dependent mechanism.
Methods
To test this, we used primary HBE cells well-differentiated in air–liquid interface culture and two mouse models (ovalbumin and house dust mite) of allergic airway disease (AAD). HBE cells were infected with RV-A16, treated with a TLR3 agonist (poly(I:C)), or exposed to compression. Thereafter, EDN1 (ET-1 protein-encoding gene) mRNA expression and secreted ET-1 protein were measured. We examined the role of TGFβR in ET-1 secretion using either a pharmacologic inhibitor of TGFβR or recombinant TGF-β1 protein. In the AAD mouse models, allergen-sensitized and allergen-challenged mice were subsequently infected with RV. We then measured ET-1 in bronchoalveolar lavage fluid (BALF) and airway hyperresponsiveness (AHR) following methacholine challenge.
Results
Our data reveal that RV infection induced EDN1 expression and ET-1 secretion in HBE cells, potentially mediated by TLR3. TGFβR activation was partially required for ET-1 secretion, which was induced by RV, poly(I:C), or compression. TGFβR activation alone was sufficient to increase ET-1 secretion. In AAD mouse models, RV induced ET-1 secretion in BALF, which positively correlated with AHR.
Conclusions
Our data provide evidence that RV infection increased epithelial-cell ET-1 secretion through a TGFβR-dependent mechanism, which contributes to bronchoconstriction during RV-induced asthma exacerbations.
Similar content being viewed by others

Introduction
Asthma is a chronic airway disease characterized by airway inflammation, progressive airway remodeling, and acute exacerbations [1, 2]. In patients with asthma, respiratory viral infections are common triggers of exacerbations, most frequently attributed to rhinovirus (RV) [3,4,5,6,7]. During an acute exacerbation, excessive contraction of airway smooth muscle (ASM) that narrows airways (bronchoconstriction) is a major pathological event contributing to the severity of asthma symptoms [1, 8, 9]. However, mechanisms underlying the link between RV infection of airway epithelial cells and bronchoconstriction remain poorly understood. It is unknown whether bronchoconstriction is directly caused by mediators secreted from infected airway epithelial cells, even prior to the recruitment and activation of inflammatory immune cells. To address this knowledge gap, we focused on endothelin-1 (ET-1) that is secreted from human bronchial epithelial (HBE) cells because ET-1 is a potent bronchoconstrictor that causes airway narrowing [10].
Since the identification of ET-1 in endothelial cells, its regulatory mechanisms and biological functions have been extensively studied in vascular endothelial cells in the context of pulmonary arterial hypertension (PAH) [11, 12]. However, the regulatory mechanisms of ET-1 expression may depend on the context of the disease and the specific cell types [12, 13]. In the lung, besides PAH, ET-1 is most well-recognized in the context of pulmonary fibrosis, where TGF-β plays a prominent role in the pathogenesis of the disease [12,13,14]. In patients with idiopathic pulmonary fibrosis (IPF), ET-1 concentration is increased in serum and bronchoalveolar lavage fluid (BALF), and expression of ET-1 is increased in bronchial epithelial cells and type II alveolar epithelial cells [15]. In an in vitro study using rat type II alveolar epithelial cells, ET-1 secretion is induced by TGF-β 1 [16], which could be a link between ET-1 and IPF. Like in IPF, in patients with asthma, both TGF-β1 and ET-1 are increased in serum and BALF [14, 17,18,19]. The expression of TGF-β1 and ET-1 in bronchial epithelial cells is positively correlated with disease severity, airway remodeling, and airflow obstruction [20,21,22]. However, mechanisms underlying increased ET-1 expression, as well as the role of TGF-β1 in ET-1 expression in HBE cells, remain unknown. Using an in vitro system that mimics the effect of mechanical compression on airway epithelial cells during bronchoconstriction, we previously demonstrated that mechanical compression significantly induces ET-1 secretion from HBE cells [23, 24]. Our data also revealed that this epithelial cell-derived ET-1 induces ASM contraction [24]. To extend our previous findings and identify molecular mechanisms behind increased ET-1 expression in bronchial epithelial cells, we hypothesized that RV infection induces ET-1 expression and results in ET-1 secretion. To examine mechanisms of RV-induced ET-1 secretion from bronchial epithelial cells, we used primary HBE cells differentiated in air–liquid interface (ALI) culture. Then, to determine RV-induced ET-1 in BALF and its correlation with airway hyperresponsiveness (AHR), we used mouse models (ovalbumin and house dust mite) of allergic airway disease (AAD).
Materials and methods
Culture of primary human bronchial epithelial cells
Primary HBE cells at passage 1 were obtained from the Cystic Fibrosis Center Tissue Procurement and Cell Culture Core, under the protocol (No. 03-1396) approved by the Biomedical Institutional Review Board at the University of North Carolina, Chapel Hill. As previously published [24,25,26,27], primary HBE cells at passage 2 were cultured from donors with no history of lung disease and differentiated in ALI culture. HBE cells were cultured and maintained using a 1:1 mixture of Dulbecco’s modified Eagle’s medium (DMEM, Corning Inc., Corning, NY) and bronchial epithelial cell growth basal medium (BEBM, Lonza, Basel, Switzerland), supplemented with bronchial epithelial growth medium (BEGM) SingleQuot kit (Lonza, Basel, Switzerland), nystatin (20 units/ml, Sigma Aldrich, St. Louis, MO), all-trans-retinoic acid (50 nM, Sigma Aldrich, St. Louis, MO), and bovine serum albumin (1.5 µg/ml, Sigma Aldrich, St. Louis, MO). Upon reaching confluence after 5–7 days in submerged culture, media from the apical side was removed to establish the air–liquid interface (ALI) and cells cultured for an additional 14–16 days, to achieve well-differentiated phenotypes as presented previously [26]. Prior to infection with human rhinovirus A16 (RV-A16) or exposure to stimuli, cells were maintained for 20 h in minimal medium, which is depleted of hydrocortisone, epidermal growth factor, and bovine pituitary extract. For each experimental condition, differentiated HBE cells were used in duplicate (ie. two transwells per control or any treatment condition for the cells from each donor).
Infection of HBE cells by RV-A16
RV-A16 was grown by infection of H1-HeLa cells (CRL-1958) and purified by ultracentrifugation (200,000 × g, for two hours, at 10 °C) through 30% (w/v) sucrose cushion as described [28, 29]. Virus pellet was resuspended in PBS containing 0.01% BSA. Well-differentiated HBE cells were apically infected with RV-A16 at 1 \(\times\) 105 ~ 107 plaque-forming units (PFU) per transwell with 1.1 cm2 surface area (106 PFU \(\approx\) multiplicity of infection (MOI) of 1) for four hours, as previously described [30, 31]. Uninfected cells were treated the same as infected cells, except for the presence of the virus in apical media.
In vitro exposure of HBE cells to poly(I:C), TGF-β1, or mechanical compression
Poly(I:C) (Invivogen, San Diego, CA) or recombinant human (rh) TGF-β1 (10 ng/ml, Cell Signaling Technologies, Danvers, MA) was spiked into the basolateral media of HBE cells in ALI cultures [26, 27]. PBS for poly(I:C) or sodium citrate for rhTGF-β1 was used as vehicle control. As previously described [23, 24, 32], HBE cells were subjected to mechanical compression at a magnitude of 30 cm H2O pressure for three hours. Time-matched controls were subjected to 0 cm H2O pressure. In experiments where a pharmacological inhibitor of TGF-β receptor 1, SB431542 was used (10 μM, Cell Signaling Technology), this was spiked to the basolateral medium at a final concentration of 10 μM, one hour prior to exposure to either stimulation. As a vehicle control for SB431542, 0.1% DMSO was used.
Real-time quantitative PCR analysis and enzyme-linked immunosorbent assay (ELISA)
As previously described [24, 33], we performed real-time RT-qPCR using the primers listed in Table 1, and then calculated fold-change for EDN1 normalized to GAPDH using the 2−ΔΔCT method; we quantified the amount of ET-1 protein in basolateral media from HBE cells or in BALF from mice using an ELISA kit (R&D Systems, Minneapolis, MN).
RV-induced exacerbation of allergic airways disease (AAD)
Measurements of lung function parameters, as well as bronchoalveolar lavage fluid supernatants, were collected from historical studies previously published [35,36,37,38,39], which are described below.
All animal models were reviewed and approved by the Animal Care and Ethics Committee at the University of Newcastle on protocols A-2016-605 and A-2020-014. BALB/c mice (at 6–8 wks old) obtained from Australian Bioresources (Mossvale, NSW Australia) were used in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
To induce AAD, mice were sensitized and challenged with either ovalbumin (OVA) or house dust mite (HDM) [35,36,37,38,39]. Non-sensitized and non-challenged mice received sterile endotoxin-free saline. Mock infection utilized UV-inactivated RV-A1.
OVA
In the first model of AAD, mice were sensitized with hen egg OVA (50 μg/200 μl in 1% alhydrogel) on day − 14 and day − 7 intraperitoneally and then challenged with low LPS OVA (50 μg/30 μl PBS) for 3 consecutive days (days − 2, − 1, and 0) intranasally. On day 0, 6 h after the final OVA challenge, mice were infected with 2.5 × 106 TCID50/ml of RV-A1, or PBS, intranasally.
HDM
In the second model of AAD, mice were sensitized with HDM (50 μg/50 μl in sterile saline) intranasally for three consecutive days. Twelve days after administration of the last sensitization dose, mice were challenged intranasally with HDM (5 μg/50 μl PBS) once daily for four consecutive days. At 24 h after final HDM challenge, mice were infected with RV-A1 (50 μl containing 1 × 107 TCID50/ml), or UV-inactivated RV intranasally.
Airway hyperresponsiveness
At 24 h after infection, mice were anesthetized for assessment of AHR and bronchoalveolar lavage fluid (BALF) samples were collected as previously described [37,38,39]. ET-1 protein in BALF was assessed by ELISA (R&D Systems) and correlated with previously published AHR data [37,38,39]. Airway resistance was expressed as a percentage change over baseline.
Collection of bronchoalveolar lavage fluid
Upon euthanasia, mouse tracheas were cannulated, and lower airways flushed with HBSS (HycloneTM, GE Life Sciences). Cells were pelleted from lavage fluids and supernatants were used for measuring ET-1, as described above. To determine the correlation between ET-1 concentration in BALF and airway resistance, values of these two parameters were matched for each mouse.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 software (San Diego, CA). In experiments comparing two groups, a two-tailed Student’s t-test was used. In experiments comparing three or more groups, a one-way ANOVA or a mixed model was fitted with Tukey's correction for multiple comparisons (in vitro) or Holm-Sidak’s correction for multiple comparisons (in vivo). For calculating the correlation between ET-1 concentration and airway resistance, a Pearson correlation coefficient was calculated. A p-value less than 0.05 was considered statistically significant.
Results
In well-differentiated HBE cells, RV infection and poly(I:C) induce EDN1 expression and ET-1 secretion
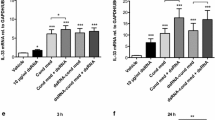
Because ET-1 causes ASM cell contraction, as a potent bronchoconstrictor, we hypothesize that ET-1 could be a potential link between viral infection and bronchoconstriction. To determine the role of RV in ET-1 secretion, we infected HBE cells in air–liquid interface culture with increasing doses of RV-A16 (at MOI of 0.1, 1, or 10). Following a previously published protocol [40], we confirmed the productive infection of HBE cells by measuring replication of RV RNA [40]. RV RNA detected at 4 h post-infection (hpi) reflected the input of RV at each dose (Fig. 1A). Compared to the level of RV RNA detected at 4 hpi in each of the three doses, RV RNA was significantly increased (up to 2 logs) by 24 hpi (Fig. 1A–C). The increased RV RNA then slightly decreased by 48 hpi. At these three time points, we then measured mRNA expression of EDN1 (the gene encoding ET-1 protein). At any of the three doses of RV, EDN1 mRNA expression at 4 hpi was not different from EDN1 mRNA expression of mock-infected cells as reflected by fold-changes of ~ 1 (Fig. 1D–F). At 24 hpi, infection with RV at MOI of 0.1 showed a modest but significant increase in EDN1 expression (1.3-fold) compared to 4 hpi (*p < 0.05, Fig. 1D). Infection with the next dose of RV (at MOI of 1) significantly increased EDN1 expression at 24 hpi by 2.0-fold compared to 4 hpi (**p < 0.01, Fig. 1E). The increased EDN1 expression detected at 24 hpi was transient, returning to baseline by 48 hpi. With the highest infectious dose of RV (at MOI of 10) we tested, we also detected increased EDN1 expression at 24 hpi by 2.4-fold compared to 4 hpi (p = 0.054, Fig. 1F). We then next measured secreted ET-1 protein in the basolateral media by ELISA at 24 hpi, to coincide with peak EDN1 expression induced by RV infection. RV infection (at MOI of 10) significantly induced basolateral secretion of ET-1 compared to mock-infected cells (*p < 0.01, Fig. 1G) and cells infected with RV at MOI of 0.1 (#p < 0.05, Fig. 1G).

Rhinovirus infection induces ET-1 production. RV-A16 infection increased RV RNA in a dose-dependent manner (A–C). *p < 0.05, **p < 0.01, ***p < 0.001 vs 4 h, #p < 0.05, ##p < 0.01 vs 24 h. RV-A16 infection induced EDN1 expression (D–F, *p < 0.05 and **p < 0.01 vs 4 h, #p < 0.05 vs 24 h) and ET-1 secretion (G, **p < 0.001 vs 0 MOI, #p < 0.05 vs 0.1 MOI) in a dose-dependent manner. Each data point represents the mean of duplicate transwells of differentiated HBE cells from each donor (mean ± SD, n = 3 distinct donors)
Given our dose-dependent data demonstrating that RV at either MOI of 1 or 10 led to the significant replication of RV and induction of EDN1 expression and ET-1 secretion (Fig. 1), we chose the dose of RV at MOI of 5, between 1 and 10, for the succeeding experiments. Having observed maximal RV-induced EDN1 expression and ET-1 secretion at 24 hpi, we used this time point in additional studies to investigate mechanisms of ET-1 secretion from HBE cells. Within the infected cells, RV replication generates double-stranded RNA, which then activates toll-like receptor 3 (TLR3) [41, 42]. To determine if TLR3 activation alone is sufficient to induce ET-1 secretion by HBE cells, we incubated HBE cells with a TLR3 agonist, poly(I:C) (at 0, 3 or 10 μg/ml) for 24 h [41, 43]. Compared to vehicle control, poly(I:C) (at 10 μg/ml) significantly induced EDN1 expression (***p < 0.001, Fig. 2A) and ET-1 secretion (***p < 0.001, Fig. 2B). Like RV infection, poly(I:C) treatment showed a dose-dependent induction of ET-1 secretion from HBE cells.

Poly(I:C), a TLR3 agonist, induces ET-1 production. Poly(I:C) induced EDN1 expression A and ET-1 secretion B in a dose-dependent manner. ***p < 0.001 vs vehicle control, ##p < 0.01 vs 3 μg/ml. Each data point represents the mean of duplicate transwells of differentiated HBE cells from each donor (mean ± SD, n = 5 distinct donors)
RV-induced ET-1 secretion depends on the activation of the TGF-β receptor
To elucidate signaling pathways that regulate ET-1 synthesis and secretion, we investigated the potential role of the TGF-β receptor. TGF-β receptor activation increases viral replication [44] in HBE cells and TGF-β1 induces ET-1 secretion in rat type II alveolar epithelial cells [16], suggesting that the TGF-β receptor activation is a mechanism of RV-induced ET-1 secretion. To determine the necessity of TGF-β receptor activation for RV-induced ET-1 secretion, we blocked the activity of TGF-β receptor using a pharmacological inhibitor of TGF-β receptor, SB431542 (denoted as SB). RV infection (at MOI of 5) significantly induced EDN1 expression and secretion in the absence of SB (Fig. 3A, B). Then, SB pretreatment significantly attenuated RV-induced EDN1 expression (#p < 0.05, Fig. 3A) and RV-induced ET-1 secretion (#p < 0.05, Fig. 3B). In the same vein, pretreatment with SB decreased poly(I:C)-induced EDN1 expression (Fig. 3C) and significantly attenuated poly(I:C)-induced ET-1 secretion (###p < 0.001, Fig. 3D).

RV-induced ET-1 secretion depends on the activation of the TGF-β receptor. Pretreatment with SB431542 attenuated RV-induced EDN1 expression (A) and ET-1 secretion (B). **p < 0.01 and ***p < 0.001 vs vehicle; #p < 0.05 vs RV alone. Pretreatment with SB431542 decreased poly(I:C)-induced EDN1 expression (C) and ET-1 secretion (D). ****p < 0.0001 vs vehicle; ###p < 0.001 vs poly(I:C) alone. Each data point represents the mean of duplicate transwells of differentiated HBE cells from each donor (mean ± SD; A, B: n = 4 distinct donors, C, D: n = 5 distinct donors)
TGF-β receptor activation is both necessary and sufficient to induce epithelial ET-1 secretion
Given our new data demonstrating that RV induced ET-1 secretion through the activation of TGF-β receptor, we tested whether a non-viral stimulus, mechanical compression, also induced ET-1 secretion through TGF-β receptor. Consistent with our previous findings [23, 24], mechanical compression significantly induced EDN1 expression (**p < 0.01, Fig. 4A) and ET-1 secretion (****p < 0.0001, Fig. 4B) above sham control. Pretreatment with SB significantly attenuated EDN1 expression (##p < 0.01, Fig. 4A) and ET-1 secretion (##p < 0.01, Fig. 4B), both of which were induced by mechanical compression. To determine if the activation of TGF-β receptor alone was sufficient to induce ET-1 secretion from HBE cells, we treated HBE cells with rhTGF-β1 protein (at 10 ng/ml) for 24 h. Compared to vehicle control, rhTGF-β1 treatment significantly increased EDN1 expression (*p < 0.05, Fig. 4C) and ET-1 secretion (*p < 0.05, Fig. 4D).

TGF-β receptor activation is necessary and sufficient to induce ET-1 secretion. Pretreatment with SB431542 attenuated compression-induced EDN1 expression A and ET-1 secretion (B). **p < 0.01, ****p < 0.0001 vs vehicle; ##p < 0.01 vs compression alone. rhTGF-β1 (10 ng/ml) induced EDN1 expression C and ET-1 secretion D *p < 0.05 vs vehicle. Each data point represents the mean of duplicate transwells of differentiated HBE cells from each donor (mean ± SD; A, B: n = 3 distinct donors, C, D: n = 6 distinct donors)
In mouse models of AAD, RV-induced exacerbations are accompanied by increased ET-1 in BALF, correlating with AHR
To investigate a mechanistic link between ET-1 secretion and RV-induced bronchoconstriction in vivo, we utilized data and samples from two well-established mouse models of AAD sensitized and challenged with experimental allergens, either OVA or HDM [36, 38, 39, 45]. From these models, we measured ET-1 protein in BALF collected after mock or RV infections. In both OVA-allergic (*p < 0.05, Fig. 5A) and HDM-allergic (*p < 0.05, Fig. 5B) mice, RV infection significantly increased ET-1 measured in BALF over allergen challenge alone. To extend these findings, we tested for associations between ET-1 concentration in BALF and AHR and found a positive correlation (r = 0.5334, p = 0.0275, Fig. 5C).

In mouse models of AAD, RV increased ET-1 concentration in BALF, correlating with AHR. In OVA-induced A or HDM-induced B model of AAD, RV infection increased ET-1 secretion in BALF collected at 24 hpi. *p < 0.05 vs OVA or HDM alone, ***p < 0.001 vs PBS + Mock; Mean ± SEM, n = 6 (A) and n = 12–15 (B). ET-1 concentration in BALF was correlated with AHR (C), n = 7–10 mice
Discussion
Despite extensive epidemiological data associating RV infection with asthma exacerbations, the mechanisms of virus-induced bronchoconstriction and relationship with airway hyperresponsiveness are not fully understood. To investigate a potential mechanistic link of RV infection to asthma exacerbations, in particular, bronchoconstriction provoked through the action of the infected airway epithelial cells, we focused on the epithelial cell secretion of ET-1, a potent bronchoconstrictor. Since the discovery of ET-1 production by endothelial cells, mechanisms regulating its production have been extensively studied in endothelial cells because of its association with PAH [12]. However, regulatory mechanisms of ET-1 production may be disease-dependent and cell type-specific [13]. In the lung, ET-1 is prominently expressed in bronchial epithelial cells, which could be a source of secreted ET-1 in lung diseases. In our previous studies, we identified HBE cells as a source of secreted ET-1 [23, 24], which then significantly induces ASM contraction [24]. We, therefore, sought to identify mechanisms by which bronchial epithelial cells synthesize and secrete ET-1 in the context of RV-induced bronchoconstriction. Our data reveal that ET-1 secretion was induced by RV infection, TLR3 activation, or mechanical compression through the activation of the TGF-β receptor and that TGF-β receptor activation itself was sufficient to induce ET-1 secretion. We then extended our in vitro findings to an in vivo system using two mouse models of RV-induced asthma exacerbation, in which AAD is established by sensitization and challenge with OVA or HDM followed by sequential infection with RV. In both of these AAD models, RV significantly increased levels of ET-1 detected in BALF. The increased ET-1 in BALF correlated with AHR, suggesting a mechanistic link between ET-1 and airway responsiveness.
In well-differentiated HBE cells cultured from non-diseased donors, RV RNA was increased in a time- and dose-dependent manner (Fig. 1A–C). The kinetics of increased RV RNA were similar to the temporal changes of cell-associated viral RNA, which have been previously reported in HBE cells from non-diseased donors [40]. Similar to the kinetics of RV replication, RV-induced ET-1 production was increased in a time- and dose-dependent manner (Fig. 1D–G). Our data also demonstrate that poly(I:C), an agonist of TLR3 and viral mimic, induced ET-1 production in a dose-dependent manner (Fig. 2A, B). These results suggest that viral replication and TLR3 activation may be a mechanism for increased ET-1 secretion from HBE cells infected with RV. The TLR3-ET-1 axis could be a general mechanism of induction of bronchoconstriction when airway epithelial cells are infected with respiratory RNA viruses that can activate TLR3.
We further dissected intracellular signaling pathways by which ET-1 production is increased in airway epithelial cells. In endothelial cells or inflammatory cells, ET-1 expression is induced by various cytokines, such as IL-1β, TNF-α, and TGF-β [46]. Among them, we speculated a potential link between TGF-β1 and ET-1. In the lung, besides pulmonary hypertension, ET-1 is most well-studied in the context of pulmonary fibrosis, where TGF-β plays a prominent role in the pathogenesis of the disease [12, 13]. In patients with IPF, ET-1 is increased in serum and BALF and cellular expression of ET-1 is increased in airway epithelial cells and type II alveolar epithelial cells [15]. An in vitro study using rat type II alveolar epithelial cells demonstrated that TGF-β1 induces ET-1 secretion [16]. Moreover, TGF-β receptor activation leads to the reduction of antiviral responses while promoting viral replication in HBE cells, suggesting a potential role for the TGF-β receptor in the modulation of cellular responses during RV infection [44]. However, in this earlier study, RV infection did not directly activate SMAD determined by the detection p-SMAD, despite the inhibitory effect of TGF-β receptor on the viral replication suggesting that the baseline activity of TGF-β receptor is sufficient for airway epithelial cells to respond to RV infection. Thus, we examined the role of TGF-β receptor signaling for ET-1 secretion by blocking TGF-β receptor activity using a pharmacological inhibitor of TGF-β receptor or by activating TGF-β receptor activity using TGF-β1. Pre-treatment with SB, a TGF-β receptor inhibitor, significantly attenuated EDN1 expression and ET-1 secretion, both of which were otherwise increased by RV infection or poly(I:C) treatment (Fig. 3). Like the effect of TGF-β receptor inhibition on ET-1 secretion that is induced by RV or poly(I:C), SB pretreatment also attenuated ET-1 secretion that is induced by mechanical compression (Fig. 4A, B). In our previous studies, we demonstrated the role of TGF-β pathways in mechanically compressed HBE cells. For example, RNA sequencing analysis revealed that TGF-β pathways are enriched in HBE cells by mechanical compression [47]; and TGF-β receptor signaling is partially required for compression-induced goblet cell hyperplasia [32]. Our new data here indicate that TGF-β receptor activation significantly contributed to compression-induced ET-1 secretion, which is a process relevant to bronchoconstriction as airway narrowing causes mechanical compression of HBE cells. Together, our in vitro data reveal that the activation of the TGF-β receptor on HBE cells is partially required for both viral and non-viral induction of ET-1 secretion from HBE cells. Thus, RV infection in combination with mechanical compression caused by bronchoconstriction may further augment secretion of ET-1 through the activation of TGF-β receptor, potentially leading to prolonged bronchoconstriction. Moreover, our data highlight the significance of crosstalk between RV infection and mechanical stimulation of airway epithelial cells in RV-induced asthma exacerbations. For example, a recent study demonstrated that mechanical compression suppresses antiviral innate immune responses from asthmatic airway epithelial cells following RV infection [48]. For a better understanding of virus-induced bronchoconstriction and its link with airway hyperresponsiveness, how these two factors, viral infection and mechanical compression, interact may be a major area of research for patients with RV-induced asthma exacerbations.
To determine the requirement of the TGF-β receptor in ET-1 secretion, we used SB431542 at 10 μM concentration that completely abolished the TGF-β receptor activity, as assessed by detection of p-SMAD2/3 in response to TGF-β1 (data not shown). However, complete inhibition of TGF-β receptor activity partially attenuated ET-1 synthesis or secretion that is induced by each of the three stimuli (Figs. 3, 4A, B). While blocking of TGF-β receptor activity led to the partial attenuation of increased ET-1 secretion, our data also indicate that TGF-β1 alone (Fig. 4C, D) was sufficient to induce ET-1 secretion. Our data further suggest that increased ET-1 secretion mediated by TGF-β receptor pathway could be a common mechanism for bronchoconstriction that is caused by infections with respiratory viruses (in addition to RV) that are known to induce TGF-β1, such as RSV [7, 49]. Moreover, comparative transcriptome analyses revealed that EDN1 is one of the 43 hub genes induced by SARS-CoV-2 and also induced by other respiratory viruses, including human influenza viruses [50], suggesting that ET-1 may play additional roles in pathogenic processes such as inflammation and pulmonary fibrosis. In addition to virus-induced exacerbations, TGF-β1-induced ET-1 secretion from bronchial epithelial cells may constitute a novel pathway leading to bronchoconstriction during an asthma exacerbation either subsequent to or independent of viral infections. An OVA-induced AAD model in SMAD2 overexpressing mice suggests the potential role of TGF-β1 receptor in linking augmented ET-1 expression in airway epithelial cells and AHR [51]. In patients with asthma, TGF-β1 is increased in the lung and the most abundant source of TGF-β1 could be either injured epithelial cells, myofibroblasts, or active eosinophils [52]. Among these potential cellular sources of TGF-β1, considering the substantial role of eosinophils in asthma, more studies are necessary to determine whether TGF-β1 secreted by eosinophils directly stimulate airway epithelial cells to produce ET-1, which then exacerbates bronchoconstriction in asthma.
Given our in vitro data demonstrating that RV infection of airway epithelial cells induces the secretion of ET-1, a potent bronchoconstrictor, we aimed to determine if increased ET-1 is linked to RV-induced bronchoconstriction in vivo. We utilized samples and data from two well-established mouse models of AAD using sensitization and challenge with experimental allergens, OVA or HDM [36, 38, 39, 45]. In both OVA and HDM AAD models, where we have previously observed that infection with RV-A1 induces acute exacerbations [36, 38, 39, 45], RV infection significantly increased ET-1 measured in BALF over allergen challenge alone (Fig. 5A, B). Furthermore, when we pooled data on AHR from previous studies to match our ET-1 measurements from BALF [38, 39, 45], we found a significant correlation between AHR as measured by airway resistance and ET-1 concentrations in BALF (Fig. 5C). These in vivo data extend our in vitro findings by linking ET-1 secretion to RV-induced bronchoconstriction and AHR. Because AHR is a characteristic feature of asthma, our data support the hypothesis that airway epithelial cell-expressed ET-1 contributes to airway narrowing during RV-induced exacerbations of asthma. While ET-1 could be one of several mediators of bronchoconstriction, including histamine and leukotrienes [53, 54], here we demonstrate that ET-1 secreted from airway epithelial cells could be a critical mediator for virus-induced bronchoconstriction.
Conclusions
In summary, our data reveal that RV infection of HBE cells induces secretion of ET-1 (a potent bronchoconstrictor) potentially through the sensing of double-stranded viral RNA by TLR3. Moreover, we demonstrate that both viral infection and mechanical compression induced ET-1 secretion from epithelial cells through the activation of the TGF-β receptor. Our data reveal that the activation of TGF-β receptor is not only required but also sufficient to induce ET-1 secretion in HBE cells. Given the effect of ET-1 on the contraction of ASM cells, this may be a novel mechanism mediating virus-induced bronchoconstriction. Mechanical compression of airway epithelial cells could further induce secretion of ET-1. In mouse models of AAD, increased ET-1 concentration in BALF correlated with AHR, both of which are characteristic features of asthma. Together the results from our in vitro and in vivo studies suggest that enhanced ET-1 secretion by airway epithelial cells may be a major driver of bronchoconstriction during RV-induced asthma exacerbations.
Availability of data and materials
All data generated or analyzed during this study are included in this manuscript.
Abbreviations
- AAD:
-
Allergic airway disease
- AHR:
-
Airway hyperresponsiveness
- ASM:
-
Airway smooth muscle
- BALF:
-
Bronchoalveolar lavage fluid
- ELISA:
-
Enzyme-linked immunosorbent assay
- ET-1:
-
Endothelin-1
- HBE cells:
-
Human bronchial epithelial cells
- HPI:
-
Hours post-infection
- HDM:
-
House dust mite
- MOI:
-
Multiplicity of infection
- OVA:
-
Ovalbumin
- Poly(I:C):
-
Polyinosinic:polycytidylic acid
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- RSV:
-
Respiratory syncytial virus
- RV:
-
Rhinovirus
- SARS-CoV:
-
Severe acute respiratory syndrome coronavirus
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- TGF-β:
-
Transforming growth factor beta
- TLR3:
-
Toll-like receptor 3
References
Kaminsky DA, Chapman DG. Asthma and lung mechanics. Compr Physiol. 2020;10(3):975–1007.
Israel E, Reddel HK. Severe and difficult-to-treat asthma in adults. N Engl J Med. 2017;377(10):965–76.
Gern JE, Busse WW. Relationship of viral infections to wheezing illnesses and asthma. Nat Rev Immunol. 2002;2(2):132–8.
Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178(7):667–72.
Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J Allergy Clin Immunol. 2017;140(4):895–906.
Tay H, Wark PA, Bartlett NW. Advances in the treatment of virus-induced asthma. Expert Rev Respir Med. 2016;10(6):629–41.
Mikhail I, Grayson MH. Asthma and viral infections: an intricate relationship. Ann Allergy Asthma Immunol. 2019;123(4):352–8.
Lo D, Kennedy JL, Kurten RC, Panettieri RA, Koziol-White CJ. Modulation of airway hyperresponsiveness by rhinovirus exposure. Respir Res. 2018;19(1):208.
Kennedy JL, Koziol-White CJ, Jeffus S, Rettiganti MR, Fisher P, Kurten M, et al. Effects of rhinovirus 39 infection on airway hyperresponsiveness to carbachol in human airways precision cut lung slices. J Allergy Clin Immunol. 2018;141(5):1887-90.e1.
Chalmers GW, Little SA, Patel KR, Thomson NC. Endothelin-1-induced bronchoconstriction in asthma. Am J Respir Crit Care Med. 1997;156(2 Pt 1):382–8.
Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–5.
Barton M, Yanagisawa M. Endothelin: 30 years from discovery to therapy. Hypertension. 2019;74(6):1232–65.
Park SH, Saleh D, Giaid A, Michel RP. Increased endothelin-1 in bleomycin-induced pulmonary fibrosis and the effect of an endothelin receptor antagonist. Am J Respir Crit Care Med. 1997;156(2 Pt 1):600–8.
Aschner Y, Downey GP. Transforming growth factor-beta: master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol. 2016;54(5):647–55.
Fagan KA, McMurtry IF, Rodman DM. Role of endothelin-1 in lung disease. Respir Res. 2001;2(2):90–101.
Jain R, Shaul PW, Borok Z, Willis BC. Endothelin-1 induces alveolar epithelial-mesenchymal transition through endothelin type A receptor-mediated production of TGF-beta1. Am J Respir Cell Mol Biol. 2007;37(1):38–47.
Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, Holgate ST, et al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156(2 Pt 1):642–7.
Mattoli S, Soloperto M, Marini M, Fasoli A. Levels of endothelin in the bronchoalveolar lavage fluid of patients with symptomatic asthma and reversible airflow obstruction. J Allergy Clin Immunol. 1991;88(3 Pt 1):376–84.
Aoki T, Kojima T, Ono A, Unishi G, Yoshijima S, Kameda-Hayashi N, et al. Circulating endothelin-1 levels in patients with bronchial asthma. Ann Allergy. 1994;73(4):365–9.
Vignola AM, Chanez P, Chiappara G, Merendino A, Pace E, Rizzo A, et al. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1997;156(2 Pt 1):591–9.
Pegorier S, Arouche N, Dombret MC, Aubier M, Pretolani M. Augmented epithelial endothelin-1 expression in refractory asthma. J Allergy Clin Immunol. 2007;120(6):1301–7.
Ackerman V, Carpi S, Bellini A, Vassalli G, Marini M, Mattoli S. Constitutive expression of endothelin in bronchial epithelial cells of patients with symptomatic and asymptomatic asthma and modulation by histamine and interleukin-1. J Allergy Clin Immunol. 1995;96(5 Pt 1):618–27.
Tschumperlin DJ, Shively JD, Kikuchi T, Drazen JM. Mechanical stress triggers selective release of fibrotic mediators from bronchial epithelium. Am J Respir Cell Mol Biol. 2003;28(2):142–9.
Lan B, Mitchel JA, O’Sullivan MJ, Park CY, Kim JH, Cole WC, et al. Airway epithelial compression promotes airway smooth muscle proliferation and contraction. Am J Physiol Lung Cell Mol Physiol. 2018;315(5):L645–52.
Mitchel JA, Antoniak S, Lee JH, Kim SH, McGill M, Kasahara DI, et al. IL-13 augments compressive stress-induced tissue factor expression in human airway epithelial cells. Am J Respir Cell Mol Biol. 2016;54(4):524–31.
O’Sullivan MJ, Mitchel JA, Mwase C, McGill M, Kanki P, Park JA. In well-differentiated primary human bronchial epithelial cells, TGF-beta1 and TGF-beta2 induce expression of furin. Am J Physiol Lung Cell Mol Physiol. 2021;320(2):L246–53.
Mitchel JA, Das A, O’Sullivan MJ, Stancil IT, DeCamp SJ, Koehler S, et al. In primary airway epithelial cells, the unjamming transition is distinct from the epithelial-to-mesenchymal transition. Nat Commun. 2020;11(1):5053.
Griggs TF, Bochkov YA, Nakagome K, Palmenberg AC, Gern JE. Production, purification, and capsid stability of rhinovirus C types. J Virol Methods. 2015;217:18–23.
Lee WM, Chen Y, Wang W, Mosser A. Growth of human rhinovirus in H1-HeLa cell suspension culture and purification of virions. Methods Mol Biol. 2015;1221:49–61.
Ashraf S, Brockman-Schneider R, Bochkov YA, Pasic TR, Gern JE. Biological characteristics and propagation of human rhinovirus-C in differentiated sinus epithelial cells. Virology. 2013;436(1):143–9.
Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3(1):69–80.
Park JA, Tschumperlin DJ. Chronic intermittent mechanical stress increases MUC5AC protein expression. Am J Respir Cell Mol Biol. 2009;41(4):459–66.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8.
Chu EK, Cheng J, Foley JS, Mecham BH, Owen CA, Haley KJ, et al. Induction of the plasminogen activator system by mechanical stimulation of human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2006;35(6):628–38.
Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14(2):199–204.
Williams TC, Loo SL, Nichol KS, Reid AT, Veerati PC, Esneau C, et al. IL-25 blockade augments antiviral immunity during respiratory virus infection. Commun Biol. 2022;5(1):415.
Hatchwell L, Girkin J, Dun MD, Morten M, Verrills N, Toop HD, et al. Salmeterol attenuates chemotactic responses in rhinovirus-induced exacerbation of allergic airways disease by modulating protein phosphatase 2A. J Allergy Clin Immunol. 2014;133(6):1720–7.
Hatchwell L, Collison A, Girkin J, Parsons K, Li J, Zhang J, et al. Toll-like receptor 7 governs interferon and inflammatory responses to rhinovirus and is suppressed by IL-5-induced lung eosinophilia. Thorax. 2015;70(9):854–61.
Girkin JL, Hatchwell LM, Collison AM, Starkey MR, Hansbro PM, Yagita H, et al. TRAIL signaling is proinflammatory and proviral in a murine model of rhinovirus 1B infection. Am J Physiol Lung Cell Mol Physiol. 2017;312(1):L89–99.
Amineva SP, Aminev AG, Gern JE, Palmenberg AC. Comparison of rhinovirus A infection in human primary epithelial and HeLa cells. J Gen Virol. 2011;92(Pt 11):2549–57.
Slater L, Bartlett NW, Haas JJ, Zhu J, Message SD, Walton RP, et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010;6(11): e1001178.
Holtzman MJ, Byers DE, Alexander-Brett J, Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol. 2014;14(10):686–98.
Marsh EK, Prestwich EC, Williams L, Hart AR, Muir CF, Parker LC, et al. Pellino-1 regulates the responses of the airway to viral infection. Front Cell Infect Microbiol. 2020;10:456.
Bedke N, Sammut D, Green B, Kehagia V, Dennison P, Jenkins G, et al. Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response. PLoS ONE. 2012;7(9): e44580.
Collison A, Hatchwell L, Verrills N, Wark PA, de Siqueira AP, Tooze M, et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat Med. 2013;19(2):232–7.
Stow LR, Jacobs ME, Wingo CS, Cain BD. Endothelin-1 gene regulation. Faseb J. 2011;25(1):16–28.
Kılıç A, Ameli A, Park JA, Kho AT, Tantisira K, Santolini M, et al. Mechanical forces induce an asthma gene signature in healthy airway epithelial cells. Sci Rep. 2020;10(1):966.
Veerati PC, Reid AT, Nichol KS, Wark PAB, Knight DA, Bartlett NW, et al. Mechanical forces suppress antiviral innate immune responses from asthmatic airway epithelial cells following rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2023.
Xia YC, Radwan A, Keenan CR, Langenbach SY, Li M, Radojicic D, et al. Glucocorticoid insensitivity in virally infected airway epithelial cells is dependent on transforming growth factor-beta activity. PLoS Pathog. 2017;13(1): e1006138.
Chandrashekar DS, Athar M, Manne U, Varambally S. Comparative transcriptome analyses reveal genes associated with SARS-CoV-2 infection of human lung epithelial cells. Sci Rep. 2021;11(1):16212.
Gregory LG, Jones CP, Mathie SA, Pegorier S, Lloyd CM. Endothelin-1 directs airway remodeling and hyper-reactivity in a murine asthma model. Allergy. 2013;68(12):1579–88.
Ojiaku CA, Yoo EJ, Panettieri RA. Transforming growth factor β1 function in airway remodeling and hyperresponsiveness. The missing link? Am J Respir Cell Mol Biol. 2017;56(4):432–42.
Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012;18(5):684–92.
Blease K, Lukacs NW, Hogaboam CM, Kunkel SL. Chemokines and their role in airway hyper-reactivity. Respir Res. 2000;1(1):54–61.
Acknowledgements
The authors thank Dr. Scott Randell and the Cystic Fibrosis Center Tissue Procurement and Cell Culture Core (The University of North Carolina, Chapel Hill) for providing primary human bronchial epithelial cells.
Funding
This research was funded in part by the NIH Grants (R01HL148152, P30ES000002, T32HL007118, P30DK065988, R56HL142890, U19AI104317, R01AI148707), Fujifilm Corporation.
Author information
Authors and Affiliations
Contributions
Conception and design: ABCD, JG, MJO, JEG, YAB, NWB, J-AP. Data acquisition: ABCD, JG, AM, AC, CM, MJO, T-KNP. Analysis and interpretation: ABCD, JG, AM, AC, CM, MJO, T-KNP, JM, CK-W, JEG, YAB, NWB, J-AP. Drafting the manuscript: ABCD, JG, CK-W, JEG, YAB, NWB, J-AP. All authors have reviewed the manuscript. All authors declare no competing interests related to data presented in this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Primary human bronchial epithelial (HBE) cells were obtained from the Cystic Fibrosis Center Tissue Procurement and Cell Culture Core, under the protocol (No. 03-1396) approved by the Biomedical Institutional Review Board at the University of North Carolina, Chapel Hill. All animal models were reviewed and approved by the Animal Care and Ethics Committee at the University of Newcastle on protocols A-2016-605 and A-2020-014. This manuscript does not contain any individual person’s data in any form.
Competing interests
JEG has received consulting fees from AstraZeneca and Meissa Vaccines Inc and has stock options in Meissa Vaccines Inc.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dy, A.B.C., Girkin, J., Marrocco, A. et al. Rhinovirus infection induces secretion of endothelin-1 from airway epithelial cells in both in vitro and in vivo models. Respir Res 24, 205 (2023). https://doi.org/10.1186/s12931-023-02510-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02510-6