
Abstract
Genome editing technologies hold great promise for numerous applications including the understanding of cellular and disease mechanisms and the development of gene and cellular therapies. Achieving high editing frequencies is critical to these research areas and to achieve the overall goal of being able to manipulate any target with any desired genetic outcome. However, gene editing technologies sometimes suffer from low editing efficiencies due to several challenges. This is often the case for emerging gene editing technologies, which require assistance for translation into broader applications. Enrichment strategies can support this goal by selecting gene edited cells from non-edited cells. In this review, we elucidate the different enrichment strategies, their many applications in non-clinical and clinical settings, and the remaining need for novel strategies to further improve genome research and gene and cellular therapy studies.
Similar content being viewed by others

Introduction
The development and widespread use of gene editing technologies such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), Meganucleases, and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems and derivatives hereof have provided great opportunities for site-specific genome editing [1, 2]. Especially the simplicity of the CRISPR/Cas system has made it the preferred choice for genome editing. The RNA-guided type II CRISPR/Cas system consists of a Cas9 nuclease, which is guided to a specific target site by a chimeric single guide RNA (sgRNA) [1]. Site-specific genome editing is achieved by introducing a nuclease-induced double-strand break (DSB) to the DNA and relying on the endogenous cellular repair mechanisms to alter the genome [3]. The most prevalent repair mechanism is the error-prone non-homologous end joining (NHEJ), which is active throughout the cell cycle by direct ligation of DNA ends, often resulting in small insertions or deletions (INDELs) at the site of the break due to end-processing during repair [4]. If a DSB lies between two homologous repeat sequences in the same direction, the single-strand annealing (SSA) pathway can anneal these sequences and mediate deletion of one repeat and the intervening sequence to seal the DSB [5]. In contrast, homology-directed repair (HDR) uses an exogenous DNA template for targeted integration of transgenes facilitated by homology arms identical to the DSB flanking sequence [6].
The bottlenecks of genome editing
Despite the great potential of gene editing technologies, not all exhibit high activity and efficiency. Gene editing efficiency can vary widely between genomic target loci and among cell types. Particularly, integration of transgenes into the genome relying on HDR-mediated integration (“knock in”) is an inefficient process suffering from low editing rates compared to gene disruptions mediated by NHEJ (“knock out”). HDR-mediated site-specific integration can in some scenarios be achieved with high efficiency in cell types and loci particularly permissive to gene editing [7], but is generally restricted to < 30% of cells obtaining a targeted integration in most cases [8,9,10]. Furthermore, HDR activity is largely restricted to the S and G2 mitotic phases of the cell cycle, where homologous sister chromatids are present for natural DNA repair. Therefore, HDR can display limited efficiency in quiescent or slowly cycling cells [11].
Depending on the desired edit and target locus, numerous factors and challenges can impair genome modification efficiencies, some of which include: (1) inefficient delivery of the gene editing system [12, 13], (2) toxicity caused by delivery modality and exposure to gene editing components [14,15,16], (3) restriction to a narrow target sequence window with inefficient nuclease target sites, which could be caused by complex target sequences with repetitive elements, unusual GC content, or a dense chromatin state [17, 18], and (4) quiescent cells without an active endogenous repair machinery [11, 19]. Consequently, suboptimal gene editing efficiencies may hamper the use of gene editing technologies for some applications [1, 20].
More recent genome editing technologies like base editing [21, 22] and prime editing [23] do not rely on the formation of a DSB, and are thus not restricted by inactive endogenous repair machinery. However, other challenges for these technologies persist. Base editing has recently shown great editing efficiencies and a potential for treating various monogenic diseases [24, 25]. Unfortunately, base editing is limited to point mutations only, and the purity of the editing outcome with base editors is a concern when there is more than one target base in the editing window, thereby limiting the potential of base editors [26, 27]. Reducing the size of the editing window with newer base editors to increase specificity might in turn reduce editing efficiency and limit the genomic sites that can be targeted because of PAM constraints. Instead, enrichment of base edited cells may be an alternative to improve editing efficiencies.
Prime editing exhibits a high rate of precise editing to unwanted INDEL formation [28]. However, prime editing efficiencies can be very low, suffer from impure edits, is restricted to small edits, and often requires extensive optimization [20, 28, 29]. Any approach that can substantially increase editing outcomes is critical for its forthcoming use.
Other gene editing technologies for the integration of larger DNA sequences into the genome without reliance on endogenous repair pathways have emerged. These include CRISPR-associated transposon (CAST) systems, which are transposons that have co-opted Cas proteins for precise RNA-guided DNA insertion. CAST-mediated editing has been demonstrated in prokaryotes [30,31,32,33] and recently in mammalian cells albeit with very low integration efficiencies (< 0.1%) [34], which can be increased with enrichment [35]. Similarly, Cas nucleases combined with site-specific recombinases have enabled integration of larger DNA segments into human cellular genomes. The platform termed Programmable Addition via Site-specific Targeting Elements (PASTE) facilitated multiplexed insertions of large DNA cargo at multiple genomic loci in both human cell lines, primary human T cells, and non-dividing primary human hepatocytes with high precision. [36] Another approach based on a twin prime editing strategy (TwinPE) yielded 9% efficiency of correcting a large sequence inversion associated with Hunter syndrome in human cells [37].
Despite great potential of these novel DSB-free gene editing technologies, further improvements are required to generalize these genome editing modalities and achieve robust editing across all desired target sites. Figure 1A summarizes some of the bottlenecks affecting genome editing efficiencies including cell type, cell and gene state, and the selected genome editing tool.

Enrichment rationale and applications. A Different conditions and choice of gene editing technology influence gene editing efficiencies. These are ordered based on their approximate impact on genome editing efficiency, but can be subject to high variability. B Applications for enrichment of gene edited cells include engineering of cell and animal models, engineering and isolation of single cell clones, editing of hard-to-edit cell types, making new gene editing tools more applicable, and facilitating the use of gene editing in gene and cellular therapies
Improving gene editing efficiencies
Various strategies have been developed to improve the yield and accuracy of correctly edited cells. These include approaches to improve activity and/or specificity by engineered Cas enzymes [38,39,40,41,42,43,44], sgRNA design optimizations [45, 46], improved design and delivery of the nuclease and HDR templates [7, 47, 48], manipulation of repair pathways to adjust NHEJ:HDR ratio [49, 50], cell cycle control [19, 51,52,53], retargeting undesired INDELs by recursive editing for a subsequent opportunity to perform the desired HDR-mediated integration [54], introducing non-cleavable Cas9 target sequences (CTSs) in the HDR template to facilitate Cas9-mediated nuclear import of the template [55], or recruitment of the HDR template to the target site by direct fusion to the nuclease [56]. Several strategies have been investigated, which are reviewed extensively elsewhere [20].
Selection of gene edited cells
A different overall strategy to improve the frequency of gene edited cells in a population aims to select edited cells from unedited cells. There are various means to do this based on negative or positive selection and using physical or biological separation methods. Some approaches may enable close to 100% selection efficiency while others merely enable a small enrichment. Enrichment may find various uses within basic biological studies as well as in clinical applications. CRISPR/Cas has democratized the generation of genetically engineered cell lines for studies of genotype–phenotype relationships, but generating a clonal cell line with the desired genotype may require labor-intensive screening of hundreds of clones to identify a correct one. Enrichment may facilitate the direct identification and isolation of very infrequent genotypes in a population or at least vastly increase the likelihood of identifying a correct clone, thereby reducing the labor intensity [57, 58].
Enrichment strategies can also prove valuable in a clinical setting. For some applications, large inter-patient variability in gene editing efficiency can be a limiting factor. Enrichment of correctly gene edited cells for therapies could reduce this issue and potentially assure a product for all patients. In other cases, gene editing efficiencies are not high enough to provide a therapeutic effect. One example is hematopoietic stem cell (HSC) therapies where edited and unedited cells compete during engraftment and hematopoiesis. For some therapies like x-linked severe combined immunodeficiency (X-SCID), there is a large survival benefit during lymphopoiesis for correctly edited cells, which means that a very small fraction of edited HSC is believed to suffice to provide a therapeutic benefit. For other hematopoietic disorders like chronic granulomatous disease (CGD), such enhanced survival advantage does not exist, and low editing rates would not provide a therapeutic effect [59].
New treatment modalities for Cystic Fibrosis also investigate genome editing strategies. Previous studies have shown that the presence of 10–25% CFTR-expressing cells is sufficient to restore CFTR function [60, 61]. Implementing enrichment of CRISPR/Cas edited cells increased the frequency of CFTR edited cells from 15 to 80%, thereby greatly exceeding the desired minimum editing level [60].
Cellular immunotherapies have revolutionized treatment of especially difficult-to-treat CD19 + hematological malignancies, and anti-CD19 Chimeric Antigen Receptor (CAR) T cell therapies are commercially available from several companies. Here, the patient’s own T cells are engineered to express the CAR using lentiviral vectors, and notably, the inter-trial and inter-patient variabilities are significant regarding CAR expressing cells. Different clinical trials have reported 5 to > 90% of T cells expressing the CAR following manufacturing [62,63,64,65,66], and with a desired release criteria of > 10–20% CAR T cells, improvements are necessary [67, 68]. This might prove particularly important when using CRISPR/Cas approaches to insert the CAR gene as efficiencies may be inferior to lentiviral delivery. The possibility to enrich engineered CAR T cells before infusion into patients might generate more successful CAR T cell therapies and potentially facilitate the transition into allogeneic cell products [69]. Furthermore, for such kind of cell therapy with in vitro expansion of the engineered cells, there might be a benefit in manufacturing costs since undesired therapeutically irrelevant cells are not included during the expansion process.
Strategies to enrich gene edited cells would facilitate the further use of programmable nucleases for many applications including engineering of cell and animal models, engineering and isolation of single cell clones, engineering hard-to-edit cell types, making new gene editing tools more applicable, and facilitate the use of gene editing in gene and cellular therapies (Fig. 1B). In this review we describe different enrichment strategies developed for the selection and isolation of gene modified cells from unmodified cells.
Enrichment of transfection- or transduction-positive cells
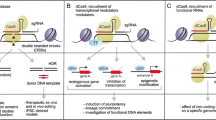
The first hurdle to overcome for genome editing is the delivery of the genome editing system into cells. A simple obvious approach is to enrich for cells that have efficiently taken up the genome editing system. This can be done for example through linkage with a selectable reporter such as fluorescent, antibiotic, or antigenic reporters, which allows for selection of cells efficiently transfected with the genome editing system. Introducing a reporter can be achieved by simple co-transfection of the reporter only allowing for enrichment of transfection-competent cells or by coupling the selectable reporter to the sgRNA or the nuclease for example via a self-cleaving 2A sequence (T2A) allowing for enrichment of nuclease- or sgRNA- expressing cells (Fig. 2A) [70,71,72,73,74,75,76,77,78,79,80,81].

Different enrichment strategies. A Enrichment of transfection-positive cells with selectable reporter genes. B Enrichment of nuclease-active cells using surrogate reporters based on NHEJ-, SSA-, or HDR-mediated restoration of a reporter gene. C Enrichment by co-targeting. D Co-integration of a selectable reporter gene to enrich for HDR-mediated transgene integration events. The reporter gene can subsequently stay permanently in the genome, be excised, or become silent due to induced, transient reporter expression. Constructs are not to scale. GOI (gene of interest)
This strategy proved especially valuable for multiplexed gene editing in primary CD34 + HSPCs. Here lentiviral sgRNA vectors containing fluorescent markers allowed for traceable and selectable multiplexed editing. Robust double knock outs of cell surface molecules CD45 and CD44 with an efficiency of ~ 70% was achieved in HSPCs. Multiplexed knockouts were also demonstrated for STAG1 and STAG2 genes as well as for the AHR and LSD1 or RCOR1 genes resulting in marked CD34+ HSPC expansion [82].
Enrichment of nuclease-active cells using exogenous surrogate reporters
Another way of not only selecting for transfected cells, but also nuclease-active cells involves the use of surrogate reporters that report on NHEJ [83,84,85,86,87,88], SSA [84, 85, 89,90,91,92], or HDR activity [83, 84, 90, 92, 93]. The use of surrogate reporter systems to enrich for genome edited cells is based on the premise that engineered nucleases able to edit the co-delivered surrogate reporter have a higher probability of also editing the genomic target site, since cells proficient for genome editing at one locus are more likely to be proficient for editing at another locus, called co-targeting or co-selection [94, 95]. These surrogate reporter systems show improved enrichment of gene edited cells compared to simple selection of transfection-positive cells [96, 97]. Various exogenous surrogate reporters based on NHEJ-, SSA-, or HDR-mediated editing by engineered nucleases have been developed for the enrichment of genome editing events. An overview of the three surrogate reporter types are presented in Fig. 2B.
NHEJ-based exogenous surrogate reporters
NHEJ-based exogenous surrogate reporters for enrichment of gene edited cells are based on a nuclease target sequence inserted to shift the reading frame of a selectable reporter gene. Only new INDEL events can restore the reading frame and the expression of the selectable reporter. Kim et al. were the first to develop an NHEJ-based surrogate reporter system with a frameshifted GFP reporter to enrich for gene edited cells demonstrating up to 39-fold enrichment of INDELs (from 0.62 to 24%) in the TP53 gene in the sorted GFP + cell population versus the unsorted population [98]. Enrichment of gene edited cells was also achieved using similar surrogate reporter systems relying on enrichment by magnetic selection of a truncated mouse MHC class I molecule (H2-kk) antigen or antibiotic selection using a hygromycin resistance gene (hygroR) as selectable frame-shifted genes [99]. Even though high numbers of reporter-positive cells can be observed with this strategy, perhaps due to excess reporter plasmids in the cells [97], NHEJ-mediated INDEL formation occurs with an uncontrollable outcome, which means that on average only 1/3 of the surrogate reporters would generate an in-frame selection gene. Additional changes have been implemented to increase the sensitivity of the reporter to 2/3 by including two different out-of-frame reporter genes (3n + 1 and 3n + 2). The new NHEJ-based surrogate reporter system was also developed to enrich for CRISPR/Cas-mediated genome editing events by both FACS, immunomagnetic selection, and antibiotic selection. Up to 11-fold enrichment (from 2.8 to 31%) of nuclease-induced mutations at the target site was observed compared to the unselected cell population [96]. Similar enrichment efficiencies have been achieved in other studies using this and similar surrogate reporters. [100,101,102,103].
Despite the ease of using surrogate reporters, a new reporter needs to be cloned for each nuclease target site to be edited, since the most efficient enrichment is achieved if the nuclease targets the same sequence as the genomic sequence. To solve this issue, NHEJ-based surrogate reporters have been developed with the target sequence flanked by restriction sites allowing for easy exchange of the target sequence [97]. More advanced approaches have also been developed that include 17 target sites in a row generating a single reporter able to enrich for edits at any of the 17 target sites [104]. However, this strategy increases the challenge of arranging all target sites so that a premature stop codon does not occur in the reporter, which would otherwise compromise the functionality of the reporter as it is required to be able to be turned on upon the right frameshifting INDELs. Nonetheless, this surrogate reporter achieved enrichment so 80% of reporter-positive cells contained the desired edit compared to < 10% in the reporter negative population [104].
SSA-based exogenous surrogate reporters
Unlike the NHEJ-based surrogate reporter systems, avoidance of in-frame premature stop codons at the target sequence is not required for the SSA-based surrogate reporter system, thus simplifying its in silico design and broadening its applications. Instead, the selection reporter is disrupted by a nuclease target site flanked by direct repeats (DRs), which allows for restoration of the reporter upon SSA-mediated intramolecular repair of the DSB introduced at the nuclease target site by deleting one of the DRs along the region in between. Several SSA-based surrogate reporter systems have been described using both fluorescence [91, 103] and antibiotic resistance [103, 105], achieving increased INDEL rates from 8.7 to 97.9% in the reporter positive population [91].
A dual surrogate reporter system containing two different reporter cassettes was designed to also act as repair template for HDR, thereby potentially allowing enrichment of both INDELs and integration events. One reporter cassette can function as surrogate reporter for nuclease-activity and enrichment and the second reporter for knock-in and screening of biallelically targeted cells based on dual antibiotic selection yielding 6.7-fold enrichment (from 2.70 to 18.18%) of biallelic integrations compared to the use of only one reporter [8]. Comparison of NHEJ- and SSA-based surrogate reporter systems in one study revealed superior enrichment when utilizing an SSA-based surrogate reporter system achieving up to 34.8-fold enrichment (from 2.1 to 72.7%) of INDELs compared to non-selected cells with an optimal DR length of 200 bp [103]. However, one study has demonstrated the opposite, that the NHEJ-based reporter is most efficient [106].
HDR-based exogenous surrogate reporters
HDR-based exogenous surrogate reporters enrich for cells that have both nuclease activity and an active HDR machinery, thereby potentially enriching for integration events. Different HDR-based surrogate reporter systems have been described, but all rely on restoration of a reporter gene by HDR-mediated repair of a DSB [107,108,109,110]. One system contains a truncated N-terminal part of a reporter gene (puromycin resistance (PuroR) or eGFP) followed by the nuclease target site and next a full length reporter gene with a stop codon instead of a start codon. Upon nuclease-induced DSB formation at the nuclease target site, the full-length reporter can be repaired by recombination with the N-terminal homologous part, thereby replacing the stop codon with a start codon and mediating reporter expression [107]. Similarly, disrupting the reporter by a nuclease expression cassette flanked by nuclease target sites mediated restoration of the reporter gene and simultaneous self-inactivation upon removal of the nuclease expression cassette to restore the reporter gene by HDR [108]. Another system restores a truncated puromycin resistance gene by intra-molecular HDR using a “universal” sgRNA target site not present in the human genome for introducing the DSB in the surrogate reporter. This identifies cells with HDR activity and a potential simultaneous integration in the genome if another sgRNA and matching HDR template was supplied. HDR-mediated editing of precise point mutations was increased up to 20.7-fold (from 2.22 to 45.93%) and HDR-mediated integration of an eGFP gene was enriched up to 8.9-fold (from 1.34 to 11.93%) with only 50 bp homology arms [109].
Most surrogate reporters enrich for both transfection-positive cells and nuclease-active cells if an additional reporter is included for assessing transfection efficiency (dual surrogate reporters). NHEJ and SSA-based reporters are best at enriching for knock out events, but all reporters can also be used to enrich for HDR events, since they enrich for nuclease activity [97]. However, these strategies are not able to directly enrich for specific editing events since they are only able to enrich for nuclease activity.
The biggest advantage of these episomal surrogate reporters is that they are transiently transfected and do not intentionally alter the genome, thereby constituting a scarless enrichment strategy. On the other hand, relying on plasmid surrogate reporters can potentially be a limitation since random integration of plasmid DNA into the genome can occur [111]. Furthermore, the introduction of a DSB both in the episomal reporter and in a genomic locus of interest, increases the risk of interference between the two sites and integration of the episomal reporter into the genomic locus. Absence of such unintended outcomes related to this enrichment strategy should be verified especially for clinically relevant applications. Another potential issue with surrogate reporters is an overestimation of editing efficiencies if relying on reporter expression for example to screen sgRNA efficiencies due to differences in chromatin state at the genomic loci versus the targeting site in the reporter [112].
Enrichment of nuclease-active cells using endogenous reporters
The premise of co-targeting is also the foundation of another group of enrichment strategies. Contrary to the use of exogenous surrogate reporters, these strategies rely on inconsequential mutations made to endogenous genes to create a selectable phenotype (for example drug resistance). Hence, enrichment of cells is facilitated through a modification at a second unrelated endogenous locus (Fig. 2C). Avoiding the use of exogenous surrogate reporters makes this category of enrichment strategies more compatible with therapeutic applications, depending on the selection edit made to the genome, since no exogenous reporter DNA is introduced into the cells [113]. This co-targeting strategy was originally described in C. elegans [114, 115] and later applied to mammalian cells as well.
Moriarity et al. used this strategy to co-target the HPRT gene in CD34+ HSPCs. Cells lacking endogenous HPRT expression become resistant to the cytotoxic drug 6-thioguanine (6-TG) thereby allowing for enrichment of cells with a simultaneous knock out of either the CCR5 or ARTEMIS gene by NHEJ. Up to 64.1% of the 6-TG resistant HSPCs presented were co-edited at the desired target site. [116] Another study applied the same co-targeting strategy and achieved enrichment of NHEJ-mediated AAVS1 editing events to over 80% with co-targeting [117]. However, since HPRT is X-linked, 6-TG resistant cells arise from modification of only a single active allele, which may not provide an adequate selection pressure if biallelic co-targeting knockouts are desired [116, 118].Another endogenous co-targeting strategy for enrichment of NHEJ or HDR modifications at a second locus utilizes mutations in the Na+ /K+ ATPase gene ATP1A1 that renders cells resistant to the inhibitor Ouabain (a cardiac glycoside). The authors achieved successful enrichment for CRISPR-induced INDELs and HDR events in both cell lines and in CD34+ HSPCs [113]. This strategy was further developed for induced pluripotent stem cells (iPSCs). Here they improved INDEL rates, whereas HDR rates were improved only at some loci, which could be attributed to differences in chromatin accessibility [119].
Other selectable targets for co-targeted enrichment of human primary cells include disruption of the SLC35F2 gene making hPSCs insensitive to the anti-cancer drug YM155 [100], disruption of the HBEGF gene encoding the receptor for Diphtheria Toxin (DT) making iPSCs and primary human T cells insensitive to DT, which improved INDEL formation up to 14-fold (from ~ 5 to ~ 70%) and HDR-mediated integration at a second locus more than 35-fold (from ~ 0.2 to 6%) [120], disruption of the B2M gene followed by negative selection by for example FACS [118], or disruption of a pre-introduced temperature-sensitive (ts) mutation in the essential TAF1 gene in cell lines rendering only edited cells temperature-resistant [121].
In a few cases a co-targeting-based enrichment strategy has been based on integration of a selection cassette at one genomic locus by HDR, thereby also enriching for HDR-mediated integration of another repair template at another independent locus. This has achieved up to 50-fold increased integration frequency at a second locus [122, 123].
These endogenous co-targeting strategies are in most cases more therapeutically applicable since no exogenous material is required to be introduced to the target cell population and the strategy might thereby be more suitable for use in primary cells. However, introducing two simultaneous DSBs to the genome can cause chromosomal translocations which is a primary driver of many cancers [124, 125]. Additionally, modifying an endogenous gene to create a selectable phenotype can also be problematic in some cases if it alters an essential or required gene function. Assuring that all desired target cells express the endogenous selection gene and checking for random escape from the selectable mutation should also be considered to confirm the enrichment potential of the strategy.
Enrichment following reporter integration
A straightforward strategy to directly enrich for a specific genomic modification is to introduce a selectable reporter gene into the target locus possibly along a desired gene or cDNA to be integrated. This strategy has been applied in different variations. An overview can be seen in Fig. 2D.
Reporter integration with permanent expression
Several studies have integrated reporter genes at specific genomic loci to track genome modification outcomes or to track endogenous genes and protein activity, localization, and dynamics [126,127,128,129,130,131,132,133,134]. This approach can be used for targeted disruption of a gene by integration of a reporter gene into the open reading frame of the target gene. The reporter gene enables direct tracking of cells with the disrupted target gene. It also enables enrichment for the integration of a target gene’s cDNA by utilizing a reporter tagged to this cDNA. Some reporter genes derived from endogenous genes can be clinically relevant since no foreign protein is introduced into cells. Especially, truncated signaling-inert membrane proteins, including tNGFR (truncated nerve growth factor receptor) and tEGFR (truncated epidermal growth factor receptor), have been used as reporters in clinical trials and shown to be safe [135,136,137,138]. However, constitutive overexpression and enrichment of a reporter like tEGFR in HSPCs would preclude patients from receiving anti-EGFR antibodies (e.g. cetuximab) for cancer treatment should the need arise. Thus, considering future clinical implications and choosing suitable reporter genes is important. Another way to circumvent this challenge could be to utilize an enrichment strategy relying on transiently expressed reporter genes, thereby allowing treatment with anti-reporter antibodies when expression of the reporter gene is silenced [139]. Integration of two different reporter genes at the HBB locus in HSPCs constituted a strategy for enrichment of cells with a biallelic targeted integration in more than 85% of cells. Furthermore, integrating an HBB cDNA correcting the sickle cell disease mutation followed by a clinically relevant EF1a-tNGFR cassette for enrichment of anti-sickling HSPCs, confirmed the potential to enrich functionally corrected HSPCs which expressed mRNA from the integrated anti-sickling cDNA driven by the endogenous promoter [140,141,142]. Similar results were obtained from HDR-mediated integrations at other loci where 99%, 92%, and 100% of reporter-positive HSPCs had at least monoallelic targeted integration at CCR5, IL2RG, and RUNX1 respectively [143].
Integrating various cDNAs from genes of interest fused to a fluorescent reporter through an internal ribosome entry site (IRES) has also been demonstrated to be greatly enriched upon selection based on the reporter expression [144]. Also, including an additional reporter gene in the HDR template outside the homology arms further allows for exclusion of cells with random integration events and cells with only episomal HDR template expression by negative selection [145].
Vaidyanathan et al. developed a gene-corrected airway stem cell therapy against Cystic Fibrosis (CF) by targeted replacement of full-length CFTR and enrichment by co-integration of a tCD19 reporter. Enrichment of modified cells by immunomagnetic selection obtained 60–80% tCD19 + upper airway basal stem cells (UABCs) and human bronchial epithelial cells (HBECs) from 11 different CF donors. Integration of the full-length CFTR cDNA and tCD19 enrichment cassette was confirmed into at least one allele per tCD19+ cell and the corrected airway basal stem cells were able to differentiate to produce epithelial sheets with restored CFTR-mediated chloride transport at an average of 70–80% of the levels seen for non-CF controls [60].
So far, the most promising cellular therapies include chimeric antigen receptor (CAR) T cell therapies, which are rapidly emerging as very promising cellular therapies especially for use as cancer immunotherapies, and site-specific gene editing technologies like the CRISPR/Cas system are increasingly used for next-generation engineering and production of CAR T cells [146]. A certain level of site-specific CAR integration is required to meet clinical release criteria to assure proficiency. Therefore, enrichment strategies have also been applied to specifically enrich for the engineering of CAR T cells. Integration of a multi-epitope molecule harboring a CD34 epitope and two CD20 mimotopes (RQR8) along a CD19-targeting CAR into the CD52 locus resulted in 60% of genome-edited T cells being CAR+ /CD20+ /CD34+ /CD52-, which could be increased to > 95% after CD34-based positive selection. A dual functionality of the RQR8 as both a selectable reporter and as a suicide switch sensitive to rituximab (anti-CD20) further advances this type of enrichment strategy for use in CAR T cell engineering [147]. Reporter genes such as tEGFR has also been coupled to CD19-targeting CARs for enrichment of CAR T cells [148]. A more refined approach may be to incorporate a selectable gene fragment within the CAR coding sequence itself. Strep-tag II and NGFR sequences have been introduced within the CAR N-terminus, enabling enrichment of CAR T cells to > 90% purity [69, 148].
Abrogating the need for a reporter gene has also been demonstrated by choosing essential genes as integration sites [149]. Here, the reading frame of the essential gene from the location of the target site to the stop codon is included in the repair template to restore the reading frame of the essential gene upon integration. This partial cDNA is followed by the desired transgene to be expressed, and only cells with restored expression of the target gene survives the editing process. Targeting the GAPDH locus achieved > 90% transgene integration efficiency into the GAPDH locus in primary cells. Furthermore, this strategy also reduces undesired INDELs and incorrect integrations since precise HDR is required for survival of edited cells [149].
Integration of an excisable reporter
Permanent expression of a selectable reporter gene integrated along a gene of interest to enrich for gene edited cells may interfere with neighboring genes, may be immunogenic, or perturb cell function and homeostasis, which can be especially troublesome in a therapeutic context. To solve this issue, a common strategy is to first integrate a GOI and reporter gene for positive selection of cells with an integration, then subsequently excise the reporter gene using recombinases or transposon systems and a negative selection marker. Only cells with successful removal of the enrichment cassette are included in the final enrichment step. The excision step can be performed by either Flp/FRT or Cre/loxP recombinase systems or an excision-only piggyBac transposon system by surrounding the enrichment cassette with relevant excision target sites [57]. This combined enrichment and excision strategy aims to minimize the impact at the target site since only the intended edit remains.
The Cre/loxP recombinase system can excise fragments flanked by two loxP sites, leaving a single 34 bp loxP “scar” behind [150]. Numerous enrichment strategies have relied on subsequent excision of the enrichment cassette following integration [131, 151,152,153]. A few studies even demonstrated that expression of the integration transgene increased following excision of the co-integrated enrichment cassette [152, 154].
Since both the Cre/loxP and the Flp/FRT systems leave behind a footprint in the form of a single loxP or FRT site after excision, an alternative method is the piggyBac transposon system which mediates “scarless” excision by removing transgenes flanked by piggyBac-specific inverted terminal repeats (ITRs) if the genetic sequence already contains a simple TTAA site, otherwise this will be left behind [155]. Furthermore, cytotoxicity and genotoxicity caused by prolonged expression of the Cre recombinase is well documented. The same has not been documented for the piggyBac transposase [156, 157]. The piggyBac transposon system has been the preferred choice for excision of enrichment cassettes following enrichment and has been described in numerous studies [122, 145, 157,158,159,160].
With the ambition to modify the genome as little as possible, these strategies do require multiple genomic manipulations and enrichment processes which in return can introduce additional risks of cellular- and genotoxicities. PiggyBac excision from heterochromatic regions has also been demonstrated to be far less efficient [157, 161]. Thus, if an integration, enrichment, and excision strategy is performed at a non-expressed genetic locus, then the transposon can be difficult to remove [157]. Another approach could be to excise the enrichment cassette by CRISPR/Cas-mediated excision. However, additive off-target risks and genomic translocations are associated with multiple DSBs. [153, 162]
Reporter integration with transient expression
As an alternative to enrichment cassette excision following integration, we have recently developed a strategy for enrichment of CRISPR/Cas-mediated transgene integration by transient CRISPR activation (CRISPRa) of an otherwise silent reporter gene. CRISPRa is a fusion complex consisting of catalytically deactivated Cas9 (dCas9) fused to transcriptional activators, for example the tripartite transactivator VP64-p65-Rta (VPR). The CRISPRa complex is directed to the region immediately upstream of the transcriptional start site (TSS) of a target gene by a sgRNA where it activates expression of the target gene [163, 164]. An enrichment cassette consisting of a silent miniCMV promoter driving a therapeutically relevant reporter gene is integrated along a GOI enabling a short transient burst in reporter expression allowing enrichment of cells with targeted integration. Up to 3.6-fold enrichment (from 17.7 to 66.8%) of cells with transgene integration was achieved at various loci (HBB, AAVS1, CCR5) using various reporter genes (tNGFR, tEGFR, tCD19, tCD8) in both primary human T cells and HSPCs. Furthermore, enriched CAR T cells displayed improved cytotoxicity over non-enriched cells [139]. This transient enrichment strategy constitutes a novel strategy for enrichment without the risks associated with permanent reporter expression or excision of reporter genes. A similar approach based on transient reporter expression by a Tet-On-driven system instead of CRISPRa has also been developed [165]. However, several Tet-On systems remain compromised by low inducibility and leaky promoter expression. Leaky expression was also observed in our strategy, so additional optimizations may be required for the further use of this type of enrichment strategy [139, 166].
Despite being superior due to the direct enrichment approach, these strategies also face challenges related to the more extensive genome modifications or promoter interference between transgene and reporter promoters. However, all enrichment strategies described in this review are dependent on strong reporter expression for selection to occur. One excision strategy was unable to counter-select for reporter-excised cells after piggyBac transfection, most likely due to silencing of the cassette since a transcriptionally inactive genomic locus was targeted, demonstrating the importance of reporter expression [160]. A comparison of different promoters driving reporter expression did not demonstrate any difference between the percentages of alleles that underwent HDR between any of the different promoter constructs tested, concluding that the promoter choice only affects expression levels and enrichment possibility [142].
Specialized enrichment strategies
Enrichment of biallelic editing
Genome modification at low frequencies often occur on only one allele, but for applications such as correction of disease-causing genetic mutations, gene therapies, and development of transgenic cellular or animal models it is highly desired to be able to enrich for cells with biallelic genome modifications. Since biallelic editing efficiencies are even lower than monoallelic editing efficiencies on a per-cell basis, especially for HDR-mediated editing, screening for cells with biallelic editing can be very cumbersome. Instead, specialized enrichment strategies for the enrichment of biallelic genome modifications have been developed (Table 1).
A co-targeting approach to impair sensitivity towards Diphtheria Toxin (DT) has been used for enrichment of biallelic integration events at another locus [120]. Sequential targeting of each allele of a gene of interest to directly integrate different reporters allows for enrichment of cells with integrations at both alleles. However, this approach is only able to enrich for cells where different reporter HDR templates are integrated at each allele, thereby overlooking cases where the same HDR template is integrated at both alleles (Fig. 3). Nonetheless, implementing a dual reporter system containing different reporter genes has resulted in highly efficient enrichment of biallelic integration events compared to using only one reporter for enrichment [8, 167]. Enrichment of biallelic editing using two fluorescent reporters were obtained in human iPSCs with either regular CRISPR/Cas HDR-mediated integration [145] or with two opposite-strand nicks by Cas nickase (nCas9) followed by HDR-mediated integration [160], and obtained in human primary T cells and HSPCs as well [143].Furthermore, Bak et al. demonstrated both biallelic, targeted integration of fluorescent reporters for a number of loci (ASXL1, RUNX1, HBB, CCR5) as well as di-genic editing, targeted integration at two loci simultaneously (HBB and IL2RG, HHB and AAVS1), yielding ~ 10% double positive HSPCs in both cases. Combining both di-genic and biallelic targeted integration, they succeeded in simultaneously targeting both alleles of ASXL1 or HBB and both alleles of RUNX1 with on-targeted integration at both alleles at both loci in 78% of HSPC clones. This approach allows for a total of six simultaneous genetic modifications: two endogenous genes inactivated (both alleles of each gene) plus the addition of four different transgenes. Furthermore, multi-genic HDR-mediated targeted integration was demonstrated in HSPCs targeting three (HBB/CCR5/IL2RG or RUNX1/HBB/ASXL1) or four (HBB/CCR5/ASXL1/RUNX1) loci at once showing successful triple- and quadruple-positive cells with 78% of HSPC clones obtaining the targeted tri-genic integrations at most alleles. These studies demonstrate tracking and enrichment of targeted integration events with both wild type and/or knock out alleles. Integrating a cDNA along the fluorescent reporter thereby allows for widespread applications [143]. This multiplexed HDR approach has been applied to model severe combined immunodeficiency (SCID) with biallelic knockouts of relevant genes and subsequently for gene correction of RAG2-SCID patient-derived CD34 + HSPCs by biallelic integration of a complete RAG2 cDNA. Cells were sorted based on one of the reporters and revealed successful cDNA integration at ~ 50% of all targeted alleles [9]. Other specific heterozygous mutations have also been modeled in HSPCs with this cDNA and fluorescent reporter integration approach [168, 169].

HDR editing outcomes. Constructs with different transgenes, for example specific cDNA variants, can be coupled to different reporter genes to allow for enrichment of different editing events and discrimination between heterozygous and homozygous alleles if different cDNA variants or transgenes are combined. The different possible editing outcomes are depicted. Cells with a desired genotype can be enriched by FACS
Enrichment of base editing
As an alternative to standard gene editing approaches that require formation of a DSB, base editors and prime editors, introducing small nucleotide changes independently of HDR, are emerging tools for creating specific genome modifications. The lack of requirement for DSBs and HDR pathway activity results in reduced INDEL rates and potentially higher editing efficiencies in a broader range of cellular contexts. Although base editing efficiencies can be really high, some loci and some cell types can still be challenging to edit, which has led to the development of several strategies for enrichment of base edited cells. Various surrogate reporters to reveal base editing activity has been described based on disruption of a premature stop codon [170,171,172,173], resolving a disruptive codon [174], inducing restoration of reporter expression by base excision repair [175], formation of a start codon for reporter expression [171], conversion of one fluorescence to another by a single base edit (BFP to GFP conversion) [176,177,178], or formation of an endogenous selectable phenotype for co-selection of base editing events [120]. An overview of base editing enrichment strategies is combined in Table 2.
Several strategies describe a BFP to GFP conversion upon a C-to-T substitution allowing for enrichment of cells with base editing activity. These strategies have shown up to 45-fold increase (in one experiment from ~ 1 to ~ 45%) in the desired base edit and reaching up to 85% editing (in another experiment from 20 to 85% corresponding to a 4.25-fold increase) in primary cells at multiple independent loci with increased multiplexed base editing frequencies as well [176,177,178].
A system able to enrich for both adenine and cytosine base edits utilizes a split fluorescent gene disrupted by the last intron of the mouse Vim gene that can be restored by correcting a splice donor site by either adenine or cytosine base editors as it allows variable target sequences corresponding to different PAMs and editing windows due to the intronic sequence which can be varied without restrictions. Up to 4.8-fold enrichment and up to 2.9-fold enrichment was achieved on five independent base editing target sites for cytosine and adenine base editors, respectively, based on the less efficient but more precise nuclease deficient Cas9 (dCas9) compared to a nicking Cas9 (nCas9) [179].
Some of the caveats of all base editing approaches are the potential formation of INDELs when using a nicking Cas9 (nCas9) and the potential editing of non-target, bystander bases that are located within the editing window of the sgRNA protospacer. Consequently, this could limit the application of base editing reporters where formation of INDELs or conversion of non-target bases result in mutations disturbing the specificity of the reporter. Thereby, caution should be used when designing these base editing reporters.
Enrichment of prime editing
Prime editing enables the introduction of short insertions, deletions, and nucleotide substitutions into the genome without requiring a DSB. Prime editors consist of an nCas9 fused to a reverse transcriptase (RT) enzyme. The RT extends the nicked DNA strand using a primer binding site (PBS) and an RNA template embedded in the 3’ terminus of a prime editing sgRNA (pegRNA). pegRNA design is complex since several PBS and RT template combinations are functional in a broad range of cell types and extensive optimization can be required [23]. Despite the great flexibility to modify the genome in almost any possible way, editing efficiency of prime editing is generally low achieving editing rates of around 10–30% [29]. To circumvent these limitations different enrichment approaches have been implemented. An overview of prime editing enrichment strategies is provided in Table 3.
Several of the base editing enrichment strategies could be repurposed for enrichment of prime editing events, which in one study was demonstrated to be less efficient as expected but still succeeded in generating reporter positive cells for enrichment [171]. Also strategies based on transfection-positive cells [28, 180] or conversion of fluorescence-based reporters could be used for enrichment of prime edited cells [181, 182].
Simon et al. were the first to develop a prime editor activity reporter (PEAR), relying on a surrogate reporter-based approach to enrich for prime editing based on their previous flexible base editing reporter [179]. The reporter contains an inactive splice site activated by prime editing enabling expression of GFP. Enrichment of nine different endogenous targets with single-nucleotide substitutions, insertions, or deletions achieved co-targeting of the surrogate reporter and an endogenous target editing frequency reaching up to 82% and achieved up to 7.8-fold enrichment of prime editing at the FANCF gene [183].
Next, Levesque et al. developed a prime editing enrichment strategy based on co-targeting the endogenous ATP1A1 gene encoding Na/K ATPase, also repurposed from a previous enrichment strategy. Various loci and modifications were co-selected for in K562 and HeLa S3 cells, and the frequencies of alleles harboring precise modifications markedly increased after enrichment at all tested loci. Furthermore, the enrichment strategy was advanced by identifying multiple ATP1A1 selectable mutations that allows for sequential enrichment steps by increasing the dose of Ouabain at each step. Successive rounds of enrichment yielded highly modified cells with multiple modifications reaching cells with two different modifications on 88% and 58% of alleles at the MTOR locus, respectively [184]. An overview of prime editing enrichment strategies is combined in Table 2.
For both base editing and prime editing, enrichment can alter the purity of the editing product, defined as % intended editing events / % all editing events, as the incidence of unintended edits like INDELs or incorporation of the sgRNA scaffold at the target site will also be enriched. A compromise is to use the less effective dCas9 to avoid nicks resulting in INDEL formation [179, 183, 184]. One base editing enrichment strategy using dCas9 demonstrated up to 30.1-fold increase in product purity compared to using a nCas9 and was able to enrich for base edited cells to achieve the same level of desired modifications as with the dCas9 [179].
In vivo enrichment
Several of the enrichment reporters described for CAR integrations could also be utilized for depletion of CAR-positive cells in vivo as a safety switch to rapidly remove the immunotherapeutic cells if toxicity is observed [147]. However, only a few studies describe enrichment of gene modified cells in vivo. Selection of gene modified cells in vivo has been reported based on a mutant O6‐methylguanine DNA methyltransferase (MGMTP140K) gene that confers resistance to O6‐BG/BCNU (O6‐Benzylguanine/Carmustine) given at doses lower than those used for cancer chemotherapy [185,186,187]. Efficient selection of genome modified HSCs was demonstrated using both gammaretrovirus and HIV-derived lentivirus vectors in both macaque and baboon nonhuman primate models. Genome modified cells were stable more than 14 months after the last drug treatment, resulting in increased frequencies of transgene-expressing cells from 11.3 to 76.9% for granulocytes, from 15.3 to 49.0% for lymphocytes, from 5.6% to 15.2% for erythrocytes, and from 6.7 to 64.0% for platelets [188]. Recently, MGMTP140K-mediated in vivo selection of prime edited cells was reported for correction of the sickle cell disease (SCD) mutation by introducing a T > A conversion. A helper‐dependent adenoviral vector (HDAd5/35++) was used for in vivo delivery of prime editors to target mobilized HSCs in the peripheral blood to correct the SCD mutation in a SCD mouse model (CD46/Townes mice). A MGMTP140K selection cassette was included for selection of transduced cells to confer resistance to O6‐BG/BCNU. Transduced cells were enriched for at days 6, 19, and 33 following vector injection to select for cells with the T > A conversion. The conversion efficiency reached an average of 43.6% at week 16 and was consistently detected in differentiated progeny cells [189]. Enrichment of base editing based on bacterial toxin resistance (DT resistance) was also demonstrated in vivo in a humanized mouse model expressing hHBEGF under the liver cell-specific albumin promoter. The base editing system and sgRNAs for hHBEGF and another endogenous target were delivered using adenoviral vectors (AdV8) and edited cells were enriched by DT treatment two weeks later. This approach resulted in 2.5-fold increase in base editing efficiency at the mouse Pcsk9 gene compared to control mice [120].
Discussion and future enrichment strategies
Despite several advancements for genome editing technologies, improvement of quality, safety, and efficiency is still required, particularly for clinical use. Many genome editing technologies are relatively new and their precise mechanisms of action are still being unraveled. While continuously subject to optimization, genome editing frequencies can still be so low that some kind of enrichment strategy is warranted. Enrichment of edited cells might help elucidate and consequently avoid some of the unintended outcomes of genome modification, as improvements to detection methods for gene editing outcomes will also help the engineering of safer and more precise genome modification technologies. It is possible that the combined efforts in improving the gene editing technologies themselves and improving enrichment strategies will aid future enhancements of gene editing efficiencies.
An ideal enrichment strategy would entail a direct selection of precisely edited cells while still being completely “scarless” and only having the desired precise modification at the specified target site. Both unedited cells and cells with unintended edits at on-target or off-target loci should be sorted away. No unintended genomic impact or off-target editing should be introduced, no adverse effects related to the editing or enrichment process should occur, and gentle and scalable selection applicable in all cellular contexts should be possible.
An enrichment strategy able to deselect cells or kill cells with unintended editing outcomes or abnormal DNA damage responses would be advantageous, yet still sought-after. One strategy approaches this by targeted integration into an essential gene, however unedited cells still remains [149]. Other strategies to approach this could include inducible reporter genes that would only be expressed if a specific repair mechanism is active in the cell or a fusion molecule (e.g. dCas9 fusion complex) scanning the genome and inducing cell death if an unintended edit was identified. Development of such strategies would enable implementation of a safety switch in all gene and cellular therapy products allowing for improved safety and control of therapeutic outcomes. Another strategy specifically for in vivo enrichment could be to integrate a unique receptor that signals cell division or cell survival followed by supplying a ligand specific for only that receptor in vivo after editing.
As an alternative to selectable reporter genes for enrichment, smaller structures like RNA aptamers could replace reporter genes to create less of an impact on the cellular genome when integrated due to their small size and simplicity. Fluorescent light‐up aptamers (FLAPs) are RNA sequences that can bind nontoxic cell‐permeable small‐molecule fluorogens thereby aiding visualization and allowing for selection of gene modified cells [190]. The most frequently used fluorogens are derived from 4-hydroxybenzlidene imidazolinone (HBI), the fluorogen moiety in GFP able to bind various FLAPs coupled to mRNA for detection purposes [191]. No enrichment strategy has so far been developed based on FLAPs, which could be due to associated challenges including relatively low brightness, limited thermostability or photostability, scaffold requirement, and detection challenges. However, as FLAPs have been developed in a multitude of colors they pose an interesting approach for enrichment of multiple simultaneous edits if potential challenges can be overcome.
Enrichment of gene modified cells provides a huge potential in basic life science research where engineered cell lines are extensively used. Development and engineering of specific cell lines can in some cases be very inefficient, especially introducing multiple edits for example to study co-dependence of specific proteins can be challenging. In addition, it can be very cumbersome to screen hundreds of clonal cells before finding a clone with the desired edit(s). Enrichment strategies can drastically reduce the number of clones to be screened or completely abrogate it and thereby simplify the engineering process. If the enrichment process is efficient enough, this might enable working with a pool of cells instead of a clone. Several enrichment strategies, spread across all the categories mentioned in this review, have also been specifically developed and used for enrichment of gene edited cells from other organisms and plants [17, 106, 192,193,194,195].
Human induced pluripotent stem cells (hiPSCs) are another example of a technology spread across a large number of research areas. iPSCs are used in many research areas including disease modeling, developmental biology, drug screening and development, regenerative medicine, and for generating patient-specific differentiated cells for personalized therapy. Yet, obtaining efficient genetic engineering of iPSCs and maintaining quality is challenging due to infrequent HDR, time-consuming clonal expansion, and low cell viability upon manipulation resulting in generally low editing efficiencies [196]. As a solution, a large number of studies have described enrichment of modified iPSCs [77, 78, 80, 100, 119, 120, 123, 126, 127, 131, 145, 149, 158,159,160]. Efficient CRISPR/Cas-mediated editing of iPSCs would prove a valuable step towards new therapies against a huge number of diseases and enrichment strategies might help facilitate this goal.
Enrichment of various cell types have already been applied to the clinic. Examples include CD34 enrichment of HSPCs which can boost graft function after allogenic hematopoietic stem cell transplantation and deplete T cells from the donor graft to limit graft versus host disease (GvHD) [197,198,199], or CD3+ T cell enrichment before viral transduction for CAR T cell therapies [200]. The most common enrichment methods rely on fluorescent activated cell sorting (FACS) or immunomagnetic enrichment / magnetic activated cell sorting (MACS) through direct (primary antibody-conjugated microbeads) or indirect (primary antibody plus secondary antibody-conjugated microbeads) sorting. FACS has the highest level of purity, however suffer from low throughput and yield compared to immunomagnetic enrichment, which in one study resulted in only 7–9% cell loss compared to ~ 70% cell loss for FACS [201]. Furthermore, immunomagnetic enrichment is cheaper and faster for low proportion samples and faster for enrichment of multiple samples due to parallel handling. During FACS, cells are subjected to strong lasers and hydrostatic pressure which might influence cell viability. Nonetheless, cell viability remains high (> 83%) with both methods, however slightly higher for immunomagnetically enriched cells [201]. Devices for clinical grade enrichment in a closed and sterile system have been developed including the CliniMACS Prodigy device (Miltenyi) and the MACSQuant Tyto (Miltenyi) for FACS and immunomagnetic selection, respectively [202], as well as the CellCelector (Sautorius) based on imaging. Both the CliniMACS Prodigy and the MACSQuant Tyto have been extensively used in clinical trials [198, 199, 203,204,205]. Future enrichment devices might be developed based on levitation of cells between magnets where the position of the cell depends on density, so different cells can be separated using this technique. Similar approaches could be based on cellular weight [206].
Enrichment of gene modified cells have also been used in a number of clinical trials. Neomycin resistance (NeoR) has been used for enrichment of cells transduced with a retroviral vectors, however strong immunogenicity of transduced cells was observed in one trial [197, 207]. Instead tNGFR has been used to magnetically immune-select transduced cells based on tNGFR to enrich for gene modified cells before transplantation into patients in a number of clinical trials [136, 138, 207]. Other clinical studies focusing on the ability to eliminate reactive T cells in the case of graft GvHD have enriched for genetically engineered donor lymphocytes expressing the herpes-simplex thymidine kinase suicide gene fused to CD34 or truncated CD34 (tCD34) before infusion into patients. Enrichment of CD34-TK75- or tCD34-TK75-positive cells yielded an almost pure infusion product and also allowed for tracking of modified cells in the patients. Circulating modified T cells persisted in the patients for over 12 months [205, 208]. Similarly, the use of inducible caspase 9 (iCasp9) as a suicide marker for a safety switch in T cell therapy have been demonstrated by transducing peripheral blood mononuclear cells (PBMCs) with a retroviral vector containing iCasp9 coupled to truncated CD19 (tCD19) through a 2A self-cleaving peptide. The cells were enriched for CD19 by immunomagnetic selection before infusion. iCasp9-transduced T cells were readily detectable 4 weeks post-infusion in all patients and remained at high level (11% of T cells) in one patient alive at 3.6 years [198, 204]. These trials demonstrate the potential to use clinically relevant selection markers for enrichment before infusion and for tracking modified cells post-infusion in patients.
Conclusion
Enrichment strategies constitute helpful tools to enhance all types of gene modification efficiencies and can be considered in many laboratory and clinical contexts, as they are able to offer high frequencies of genome edited cells, up to 100%, depending on the strategy. Most of the strategies mentioned in this review employ surrogate reporters or selectable transgenes for enrichment, of which the latter option has tried to transition into more clinically appropriate strategies, however not without limitations. Thus, the best enrichment choice depends on the individual situation since each enrichment strategy has both advantages and disadvantages. Nevertheless, enrichment strategies still play an important part in making gene editing technologies applicable, especially when new, exciting, but non-optimized technologies emerge.
Availability of data and materials
Not applicable.
Abbreviations
- 6-TG:
-
6-Thioguanine
- BCNU:
-
N,N′-bis(2-chloroethyl)-N-nitroso-urea (Carmustine)
- CAR:
-
Chimeric antigen receptor
- Cas:
-
CRISPR-associated
- CAST:
-
CRISPR-associated transposon
- CF:
-
Cystic fibrosis
- CGD:
-
Chronic granulomatous disease
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- CRISPRa:
-
CRISPR activation
- CTS:
-
Cas9 target sequence
- dCas9:
-
Deficient Cas9
- DR:
-
Direct repeats (DRs)
- DSB:
-
Double strand break
- DT:
-
Diphtheria toxin
- FACS:
-
Fluorescent activated cell sorting
- FLAP:
-
Fluorescent light‐up aptamer
- GOI:
-
Gene of interest
- GvHD:
-
Graft versus host disease
- HBEC:
-
Human bronchial epithelial cell
- HBI:
-
4-Hydroxybenzlidene imidazolinone
- HDR:
-
Homology-directed repair
- hiPSC:
-
Human induced pluripotent stem cell
- HSC:
-
Hematopoietic stem cell
- HSPC:
-
Hematopoietic stem and progenitor cell
- HygroR:
-
Hygromycin resistance
- iCasp9:
-
Inducible caspase 9
- INDEL:
-
Insertion and deletions
- iPSC:
-
Induced pluripotent stem cell
- iPSC:
-
Induced pluripotent stem cell
- IRES:
-
Internal ribosome entry site
- ITR:
-
Inverted terminal repeat
- MACS:
-
Magnetic activated cell sorting
- MGMT:
-
O6‐methylguanine DNA methyltransferase
- nCas9:
-
Cas9 nickase
- NeoR:
-
Neomycin resistance
- NHEJ:
-
Non-homologous end joining
- O6‐BG:
-
O6‐benzylguanine
- PASTE:
-
Programmable addition via site-specific targeting elements
- PBMC:
-
Peripheral blood mononuclear cell
- PBS:
-
Primer binding site
- PEAR:
-
Prime editor activity reporter
- pegRNA:
-
Prime editing sgRNA
- PuroR:
-
Puromycin resistance
- RT:
-
Reverse transcriptase
- SCD:
-
Sickle cell disease
- SCID:
-
Severe combined immunodeficiency
- sgRNA:
-
Single guide RNA
- SSA:
-
Single-strand annealing
- TALEN:
-
Transcription activator-like effector nucleases
- tCD19:
-
Truncated CD19
- tEGFR:
-
Truncated epidermal growth factor receptor
- tNGFR:
-
Truncated nerve growth factor receptor
- TSS:
-
Transcriptional start site
- TwinPE:
-
Twin prime editing
- UABC:
-
Upper airway basal stem cell
- VPR:
-
VP64-p65-Rta
- X-SCID:
-
X-linked severe combined immunodeficiency
- ZFN:
-
Zinc finger nuclease
References
Bak RO, Gomez-Ospina N, Porteus MH. Gene editing on center stage. Trends Genet. 2018;34:600–11.
Hendel A, Bak RO. Editorial: CRISPR and beyond: cutting-edge technologies for gene correction in therapeutic applications. Front Genome Ed. 2023;5:1203864.
Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–36.
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211.
Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18:495–506.
Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5: a012740.
Ferrari S, Jacob A, Cesana D, Laugel M, Beretta S, Varesi A, Unali G, Conti A, Canarutto D, Albano L, et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells. Cell Stem Cell. 2022;29:1428-1444.e1429.
Wu Y, Xu K, Ren C, Li X, Lv H, Han F, Wei Z, Wang X, Zhang Z. Enhanced CRISPR/Cas9-mediated biallelic genome targeting with dual surrogate reporter-integrated donors. FEBS Lett. 2017;591:903–13.
Iancu O, Allen D, Knop O, Zehavi Y, Breier D, Arbiv A, Lev A, Lee YN, Beider K, Nagler A, et al. Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol Ther Nucleic Acids. 2023;31:105–21.
Tasca F, Brescia M, Wang Q, Liu J, Janssen JM, Szuhai K, Gonçalves M. Large-scale genome editing based on high-capacity adenovectors and CRISPR-Cas9 nucleases rescues full-length dystrophin synthesis in DMD muscle cells. Nucleic Acids Res. 2022;50:7761–82.
Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, Passegué E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–85.
Lino CA, Harper JC, Carney JP, Timlin JA. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 2018;25:1234–57.
van Haasteren J, Li J, Scheideler OJ, Murthy N, Schaffer DV. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat Biotechnol. 2020;38:845–55.
Álvarez MM, Biayna J, Supek F. TP53-dependent toxicity of CRISPR/Cas9 cuts is differential across genomic loci and can confound genetic screening. Nat Commun. 2022;13:4520.
Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24:927–30.
Dorset SR, Bak RO. The p53 challenge of hematopoietic stem cell gene editing. Mol Therapy Methods Clin Dev. 2023;30:83–9.
Antonova E, Glazova O, Gaponova A, Eremyan A, Zvereva S, Grebenkina N, Volkova N, Volchkov P. Successful CRISPR/Cas9 mediated homologous recombination in a chicken cell line. F1000Res. 2018;7:238.
Liu X, Homma A, Sayadi J, Yang S, Ohashi J, Takumi T. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci Rep. 2016;6:19675.
Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3: e04766.
Wang JY, Doudna JA. CRISPR technology: a decade of genome editing is only the beginning. Science. 2023;379: eadd8643.
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–4.
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–71.
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–57.
Rothgangl T, Dennis MK, Lin PJC, Oka R, Witzigmann D, Villiger L, Qi W, Hruzova M, Kissling L, Lenggenhager D, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat Biotechnol. 2021;39:949–57.
Arbab M, Matuszek Z, Kray KM, Du A, Newby GA, Blatnik AJ, Raguram A, Richter MF, Zhao KT, Levy JM, et al. Base editing rescue of spinal muscular atrophy in cells and in mice. Science. 2023. https://doi.org/10.1126/science.adg6518.
Arbab M, Shen MW, Mok B, Wilson C, Matuszek Ż, Cassa CA, Liu DR. Determinants of base editing outcomes from target library analysis and machine learning. Cell. 2020;182:463-480.e430.
Lavrov AV, Varenikov GG, Skoblov MY. Genome scale analysis of pathogenic variants targetable for single base editing. BMC Med Genomics. 2020;13:80.
Schene IF, Joore IP, Oka R, Mokry M, van Vugt AHM, van Boxtel R, van der Doef HPJ, van der Laan LJW, Verstegen MMA, van Hasselt PM, et al. Prime editing for functional repair in patient-derived disease models. Nat Commun. 2020;11:5352.
Gao Z, Ravendran S, Mikkelsen NS, Haldrup J, Cai H, Ding X, Paludan SR, Thomsen MK, Mikkelsen JG, Bak RO. A truncated reverse transcriptase enhances prime editing by split AAV vectors. Mol Ther. 2022;30:2942–51.
Tou CJ, Orr B, Kleinstiver BP: Cut-and-paste DNA insertion with engineered type V-K CRISPR-associated transposases. bioRxiv 2022:2022.2001.2007.475005.
Klompe SE, Vo PLH, Halpin-Healy TS, Sternberg SH. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature. 2019;571:219–25.
Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, Zhang F. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365:48–53.
Vo PLH, Ronda C, Klompe SE, Chen EE, Acree C, Wang HH, Sternberg SH. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat Biotechnol. 2021;39:480–9.
Tou CJ, Orr B, Kleinstiver BP. Precise cut-and-paste DNA insertion using engineered type V-K CRISPR-associated transposases. Nat Biotechnol. 2023. https://doi.org/10.1038/s41587-022-01574-x.
Lampe GD, King RT, Halpin-Healy TS, Klompe SE, Hogan MI, Vo PLH, Tang S, Chavez A, Sternberg SH. Targeted DNA integration in human cells without double-strand breaks using CRISPR-associated transposases. Nat Biotechnol. 2023. https://doi.org/10.1038/s41587-023-01748-1.
Yarnall MTN, Ioannidi EI, Schmitt-Ulms C, Krajeski RN, Lim J, Villiger L, Zhou W, Jiang K, Garushyants SK, Roberts N, et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat Biotechnol. 2022. https://doi.org/10.1038/s41587-022-01527-4.
Anzalone AV, Gao XD, Podracky CJ, Nelson AT, Koblan LW, Raguram A, Levy JM, Mercer JAM, Liu DR. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat Biotechnol. 2022;40:731–40.
Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan S, Camarena J, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med. 2018;24:1216–24.
Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, Lorenzin F, Prandi D, Romanel A, Demichelis F, et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol. 2018;36:265–71.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5.
Zhang L, Zuris JA, Viswanathan R, Edelstein JN, Turk R, Thommandru B, Rube HT, Glenn SE, Collingwood MA, Bode NM, et al. AsCas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat Commun. 2021;12:3908.
Xu X, Chemparathy A, Zeng L, Kempton HR, Shang S, Nakamura M, Qi LS. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol Cell. 2021;81:4333-4345.e4334.
Pedrazzoli E, Bianchi A, Umbach A, Amistadi S, Brusson M, Frati G, Ciciani M, Badowska KA, Arosio D, Miccio A, et al. An optimized SpCas9 high-fidelity variant for direct protein delivery. Mol Ther. 2023. https://doi.org/10.1016/j.ymthe.2023.03.007.
Kim YH, Kim N, Okafor I, Choi S, Min S, Lee J, Bae SM, Choi K, Choi J, Harihar V, et al. Sniper2L is a high-fidelity Cas9 variant with high activity. Nat Chem Biol. 2023. https://doi.org/10.1038/s41589-023-01279-5.
Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–9.
Yin H, Song CQ, Suresh S, Wu Q, Walsh S, Rhym LH, Mintzer E, Bolukbasi MF, Zhu LJ, Kauffman K, et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol. 2017;35:1179–87.
Yao X, Zhang M, Wang X, Ying W, Hu X, Dai P, Meng F, Shi L, Sun Y, Yao N, et al. Tild-CRISPR allows for efficient and precise gene knockin in mouse and human cells. Dev Cell. 2018;45:526-536.e525.
Shui S, Wang S, Liu J. Systematic investigation of the effects of multiple SV40 nuclear localization signal fusion on the genome editing activity of purified SpCas9. Bioengineering (Basel). 2022. https://doi.org/10.3390/bioengineering9020083.
Liu M, Rehman S, Tang X, Gu K, Fan Q, Chen D, Ma W. Methodologies for improving HDR efficiency. Front Genet. 2018;9:691.
Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–8.
Lomova A, Clark DN, Campo-Fernandez B, Flores-Bjurstrom C, Kaufman ML, Fitz-Gibbon S, Wang X, Miyahira EY, Brown D, DeWitt MA, et al. Improving gene editing outcomes in human hematopoietic stem and progenitor cells by temporal control of DNA repair. Stem Cells. 2019;37:284–94.
Shin JJ, Schroder MS, Caiado F, Wyman SK, Bray NL, Bordi M, Dewitt MA, Vu JT, Kim WT, Hockemeyer D, et al. Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. 2020;32: 108093.
Wienert B, Nguyen DN, Guenther A, Feng SJ, Locke MN, Wyman SK, Shin J, Kazane KR, Gregory GL, Carter MAM, et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat Commun. 2020;11:2109.
Möller L, Aird EJ, Schröder MS, Kobel L, Kissling L, van de Venn L, Corn JE. Recursive Editing improves homology-directed repair through retargeting of undesired outcomes. Nat Commun. 2022;13:4550.
Shy BR, Vykunta V, Ha A, Roth TL, Talbot A, Nguyen DN, Chen YY, Blaeschke F, Vedova S, Mamedov MR, et al. Hybrid ssDNA repair templates enable high yield genome engineering in primary cells for disease modeling and cell therapy manufacturing. bioRxiv. 2021:2021.2009.2002.458799.
Ling X, Xie B, Gao X, Chang L, Zheng W, Chen H, Huang Y, Tan L, Li M, Liu T. Improving the efficiency of precise genome editing with site-specific Cas9-oligonucleotide conjugates. Sci Adv. 2020;6: eaaz0051.
Ren C, Xu K, Segal DJ, Zhang Z. Strategies for the enrichment and selection of genetically modified cells. Trends Biotechnol. 2019;37:56–71.
Reuven N, Shaul Y. Selecting for CRISPR-edited knock-in cells. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms231911919.
Jensen TI, Axelgaard E, Bak RO. Therapeutic gene editing in haematological disorders with CRISPR/Cas9. Br J Haematol. 2019;185:821–35.
Vaidyanathan S, Baik R, Chen L, Bravo DT, Suarez CJ, Abazari SM, Salahudeen AA, Dudek AM, Teran CA, Davis TH, et al. Targeted replacement of full-length CFTR in human airway stem cells by CRISPR/Cas9 for pan-mutation correction in the endogenous locus. Mol Ther. 2021. https://doi.org/10.1016/j.ymthe.2021.03.023.
Vaidyanathan S, Salahudeen AA, Sellers ZM, Bravo DT, Choi SS, Batish A, Le W, Baik R, de la Sean O, Kaushik MP, et al. High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell. 2020;26:161-171.e164.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6: 224ra225.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–9.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7: 303ra139.
Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9:1183–97.
Zhang C, He J, Liu L, Wang J, Wang S, Liu L, Ge J, Gao L, Gao L, Kong P, et al. Novel CD19 chimeric antigen receptor T cells manufactured next-day for acute lymphoblastic leukemia. Blood Cancer J. 2022;12:96.
Talleur AC, Qudeimat A, Métais JY, Langfitt D, Mamcarz E, Crawford JC, Huang S, Cheng C, Hurley C, Madden R, et al. Preferential expansion of CD8+ CD19-CAR T cells postinfusion and the role of disease burden on outcome in pediatric B-ALL. Blood Adv. 2022;6:5737–49.
Casucci M, Falcone L, Camisa B, Norelli M, Porcellini S, Stornaiuolo A, Ciceri F, Traversari C, Bordignon C, Bonini C, Bondanza A. Extracellular NGFR spacers allow efficient tracking and enrichment of fully functional CAR-T Cells co-expressing a suicide gene. Front Immunol. 2018;9:507.
Fehse B, Uhde A, Fehse N, Eckert HG, Clausen J, Rüger R, Koch S, Ostertag W, Zander AR, Stockschläder M. Selective immunoaffinity-based enrichment of CD34+ cells transduced with retroviral vectors containing an intracytoplasmatically truncated version of the human low-affinity nerve growth factor receptor (deltaLNGFR) gene. Hum Gene Ther. 1997;8:1815–24.
Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–4.
Duda K, Lonowski LA, Kofoed-Nielsen M, Ibarra A, Delay CM, Kang Q, Yang Z, Pruett-Miller SM, Bennett EP, Wandall HH, et al. High-efficiency genome editing via 2A-coupled co-expression of fluorescent proteins and zinc finger nucleases or CRISPR/Cas9 nickase pairs. Nucleic Acids Res. 2014;42: e84.
Grav LM, Lee JS, Gerling S, Kallehauge TB, Hansen AH, Kol S, Lee GM, Pedersen LE, Kildegaard HF. One-step generation of triple knockout CHO cell lines using CRISPR/Cas9 and fluorescent enrichment. Biotechnol J. 2015;10:1446–56.
Gu Y, Hou W, Xu C, Li S, Shih JW, Xia N. The enhancement of RNAi against HIV in vitro and in vivo using H-2K(k) protein as a sorting method. J Virol Methods. 2012;182:9–17.
Lee K, Mackley VA, Rao A, Chong AT, Dewitt MA, Corn JE, Murthy N. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. Elife. 2017. https://doi.org/10.7554/eLife.25312.
Li K, Wang G, Andersen T, Zhou P, Pu WT. Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS ONE. 2014;9: e105779.
Nasri M, Mir P, Dannenmann B, Amend D, Skroblyn T, Xu Y, Schulze-Osthoff K, Klimiankou M, Welte K, Skokowa J. Fluorescent labeling of CRISPR/Cas9 RNP for gene knockout in HSPCs and iPSCs reveals an essential role for GADD45b in stress response. Blood Adv. 2019;3:63–71.
Ramachandran H, Martins S, Kontarakis Z, Krutmann J, Rossi A. Fast but not furious: a streamlined selection method for genome-edited cells. Life Sci Alliance. 2021. https://doi.org/10.26508/lsa.202101051.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Steyer B, Bu Q, Cory E, Jiang K, Duong S, Sinha D, Steltzer S, Gamm D, Chang Q, Saha K. Scarless genome editing of human pluripotent stem cells via transient puromycin selection. Stem Cell Rep. 2018;10:642–54.
Wei Q, Croy BA, Etches RJ. Selection of genetically modified chicken blastodermal cells by magnetic-activated cell sorting. Poult Sci. 2001;80:1671–8.
Bäckström A, Yudovich D, Žemaitis K, Nilsén Falck L, Subramaniam A, Larsson J. Combinatorial gene targeting in primary human hematopoietic stem and progenitor cells. Sci Rep. 2022;12:18169.
Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, Scharenberg AM. Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods. 2011;8:671–6.
Kuhar R, Gwiazda KS, Humbert O, Mandt T, Pangallo J, Brault M, Khan I, Maizels N, Rawlings DJ, Scharenberg AM, Certo MT. Novel fluorescent genome editing reporters for monitoring DNA repair pathway utilization at endonuclease-induced breaks. Nucleic Acids Res. 2014;42: e4.
Sun N, Bao Z, Xiong X, Zhao H. SunnyTALEN: a second-generation TALEN system for human genome editing. Biotechnol Bioeng. 2014;111:683–91.
Kumar A, Birnbaum MD, Moorthy BT, Singh J, Palovcak A, Patel DM, Zhang F. Insertion/deletion-activated frame-shift fluorescence protein is a sensitive reporter for genomic DNA editing. BMC Genomics. 2019;20:609.
Zhang H, Zhou Y, Wang Y, Zhao Y, Qiu Y, Zhang X, Yue D, Zhou Z, Wei W. A surrogate reporter system for multiplexable evaluation of CRISPR/Cas9 in targeted mutagenesis. Sci Rep. 2018;8:1042.
Zhang H, Zhang X, Fan C, Xie Q, Xu C, Zhao Q, Liu Y, Wu X, Zhang H. A novel sgRNA selection system for CRISPR-Cas9 in mammalian cells. Biochem Biophys Res Commun. 2016;471:528–32.
Xu K, Ren C, Liu Z, Zhang T, Zhang T, Li D, Wang L, Yan Q, Guo L, Shen J, Zhang Z. Efficient genome engineering in eukaryotes using Cas9 from Streptococcus thermophilus. Cell Mol Life Sci. 2015;72:383–99.
Yang Y, Liu S, Cheng Y, Nie L, Lv C, Wang G, Zhang Y, Hao L. Highly efficient and rapid detection of the cleavage activity of Cas9/gRNA via a fluorescent reporter. Appl Biochem Biotechnol. 2016;180:655–67.
Zhou Y, Liu Y, Hussmann D, Brøgger P, Al-Saaidi RA, Tan S, Lin L, Petersen TS, Zhou GQ, Bross P, et al. Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system. Cell Mol Life Sci. 2016;73:2543–63.
Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–16.
Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–16.
Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–88.
Ramakrishna S, Cho SW, Kim S, Song M, Gopalappa R, Kim JS, Kim H. Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. Nat Commun. 2014;5:3378.
Niccheri F, Pecori R, Conticello SG. An efficient method to enrich for knock-out and knock-in cellular clones using the CRISPR/Cas9 system. Cell Mol Life Sci. 2017;74:3413–23.
Kim H, Um E, Cho SR, Jung C, Kim H, Kim JS. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat Methods. 2011;8:941–3.
Kim H, Kim MS, Wee G, Lee CI, Kim H, Kim JS. Magnetic separation and antibiotics selection enable enrichment of cells with ZFN/TALEN-induced mutations. PLoS ONE. 2013;8: e56476.
Kim KT, Park JC, Jang HK, Lee H, Park S, Kim J, Kwon OS, Go YH, Jin Y, Kim W, et al. Safe scarless cassette-free selection of genome-edited human pluripotent stem cells using temporary drug resistance. Biomaterials. 2020;262: 120295.
Koo OJ, Park SJ, Lee C, Kang JT, Kim S, Moon JH, Choi JY, Kim H, Jang G, Kim JS, et al. Production of mutated porcine embryos using zinc finger nucleases and a reporter-based cell enrichment system. Asian-Australas J Anim Sci. 2014;27:324–9.
Kim YH, Ramakrishna S, Kim H, Kim JS. Enrichment of cells with TALEN-induced mutations using surrogate reporters. Methods. 2014;69:108–17.
Ren C, Xu K, Liu Z, Shen J, Han F, Chen Z, Zhang Z. Dual-reporter surrogate systems for efficient enrichment of genetically modified cells. Cell Mol Life Sci. 2015;72:2763–72.
Liu WH, Völse K, Senft D, Jeremias I. A reporter system for enriching CRISPR/Cas9 knockout cells in technically challenging settings like patient models. Sci Rep. 2021;11:12649.
Yasuda H, Kim E, Reza AM, Kim JH. A highly efficient method for enriching TALEN or CRISPR/Cas9-edited mutant cells. J Genet Genomics. 2016;43:705–8.
He Z, Shi X, Liu M, Sun G, Proudfoot C, Whitelaw CB, Lillico SG, Chen Y. Comparison of surrogate reporter systems for enrichment of cells with mutations induced by genome editors. J Biotechnol. 2016;221:49–54.
Flemr M, Bühler M. Single-step generation of conditional knockout mouse embryonic stem cells. Cell Rep. 2015;12:709–16.
Zhang C, Xu K, Hu L, Wang L, Zhang T, Ren C, Zhang Z. A suicidal zinc finger nuclease expression coupled with a surrogate reporter for efficient genome engineering. Biotechnol Lett. 2015;37:299–305.
Yan N, Sun Y, Fang Y, Deng J, Mu L, Xu K, Mymryk JS, Zhang Z. A universal surrogate reporter for efficient enrichment of CRISPR/Cas9-mediated homology-directed repair in mammalian cells. Mol Ther Nucleic Acids. 2020;19:775–89.
Wen Y, Liao G, Pritchard T, Zhao TT, Connelly JP, Pruett-Miller SM, Blanc V, Davidson NO, Madison BB. A stable but reversible integrated surrogate reporter for assaying CRISPR/Cas9-stimulated homology-directed repair. J Biol Chem. 2017;292:6148–62.
Peng R, Lin G, Li J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 2016;283:1218–31.
Verkuijl SA, Rots MG. The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr Opin Biotechnol. 2019;55:68–73.
Agudelo D, Duringer A, Bozoyan L, Huard CC, Carter S, Loehr J, Synodinou D, Drouin M, Salsman J, Dellaire G, et al. Marker-free coselection for CRISPR-driven genome editing in human cells. Nat Methods. 2017;14:615.
Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 2014;198:837–46.
Kim H, Ishidate T, Ghanta KS, Seth M, Conte D Jr, Shirayama M, Mello CC. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 2014;197:1069–80.
Moriarity BS, Rahrmann EP, Beckmann DA, Conboy CB, Watson AL, Carlson DF, Olson ER, Hyland KA, Fahrenkrug SC, McIvor RS, Largaespada DA. Simple and efficient methods for enrichment and isolation of endonuclease modified cells. PLoS ONE. 2014;9: e96114.
Liao S, Tammaro M, Yan H. Enriching CRISPR-Cas9 targeted cells by co-targeting the HPRT gene. Nucleic Acids Res. 2015;43: e134.
Hansen M, Cai X, Bowen S, Largaespada DA, Li MV. Flow assisted mutation enrichment (FAME): a highly efficacious and efficient method to enrich Double Knockouts (DKO) after gene editing. PLoS ONE. 2021;16: e0247375.
Liu JT, Corbett JL, Heslop JA, Duncan SA. Enhanced genome editing in human iPSCs with CRISPR-CAS9 by co-targeting ATP1a1. PeerJ. 2020;8: e9060.
Li S, Akrap N, Cerboni S, Porritt MJ, Wimberger S, Lundin A, Möller C, Firth M, Gordon E, Lazovic B, et al. Universal toxin-based selection for precise genome engineering in human cells. Nat Commun. 2021;12:497.
Reuven N, Adler J, Myers N, Shaul Y. CRISPR co-editing strategy for scarless homology-directed genome editing. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22073741.
Shy BR, MacDougall MS, Clarke R, Merrill BJ. Co-incident insertion enables high efficiency genome engineering in mouse embryonic stem cells. Nucleic Acids Res. 2016;44:7997–8010.
Mitzelfelt KA, McDermott-Roe C, Grzybowski MN, Marquez M, Kuo CT, Riedel M, Lai S, Choi MJ, Kolander KD, Helbling D, et al. Efficient precision genome editing in iPSCs via genetic co-targeting with selection. Stem Cell Rep. 2017;8:491–9.
Brunet E, Jasin M. Induction of chromosomal translocations with CRISPR-Cas9 and other nucleases: understanding the repair mechanisms that give rise to translocations. Adv Exp Med Biol. 2018;1044:15–25.
Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372:143–9.
Haupt A, Grancharova T, Arakaki J, Fuqua MA, Roberts B, Gunawardane RN. Endogenous protein tagging in human induced pluripotent stem cells using CRISPR/Cas9. J Vis Exp. 2018. https://doi.org/10.3791/58130.
Roberts B, Haupt A, Tucker A, Grancharova T, Arakaki J, Fuqua MA, Nelson A, Hookway C, Ludmann SA, Mueller IA, et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol Biol Cell. 2017;28:2854–74.
Merkle FT, Neuhausser WM, Santos D, Valen E, Gagnon JA, Maas K, Sandoe J, Schier AF, Eggan K. Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep. 2015;11:875–83.
Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, Huang B. Versatile protein tagging in cells with split fluorescent protein. Nat Commun. 2016;7:11046.
Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci U S A. 2016;113:E3501-3508.
Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–7.
Cho NH, Cheveralls KC, Brunner AD, Kim K, Michaelis AC, Raghavan P, Kobayashi H, Savy L, Li JY, Canaj H, et al. OpenCell: endogenous tagging for the cartography of human cellular organization. Science. 2022;375: eabi6983.
Wang S, Li Y, Zhong L, Wu K, Zhang R, Kang T, Wu S, Wu Y. Efficient gene editing through an intronic selection marker in cells. Cell Mol Life Sci. 2022;79:111.
Dever DP, Scharenberg SG, Camarena J, Kildebeck EJ, Clark JT, Martin RM, Bak RO, Tang Y, Dohse M, Birgmeier JA, et al. CRISPR/Cas9 genome engineering in engraftable human brain-derived neural stem cells. iScience. 2019;15:524–35.
Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, Dinauer M, Sadat M, Aiuti A, Deola S, et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med. 2003;9:367–9.
Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, Turchetto L, Colombi S, Bernardi M, Peccatori J, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009;10:489–500.
Bordignon C, Bonini C, Verzeletti S, Nobili N, Maggioni D, Traversari C, Giavazzi R, Servida P, Zappone E, Benazzi E, et al. Transfer of the HSV-tk gene into donor peripheral blood lymphocytes for in vivo modulation of donor anti-tumor immunity after allogeneic bone marrow transplantation. Hum Gene Ther. 1995;6:813–9.
Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, Bordignon C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–24.
Mikkelsen NS, Hernandez SS, Jensen TI, Schneller JL, Bak RO. Enrichment of transgene integrations by transient CRISPR activation of a silent reporter gene. Mol Ther Methods Clin Dev. 2023;29:1–16.
Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–9.
Bak RO, Dever DP, Porteus MH. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc. 2018;13:358–76.
Charlesworth CT, Camarena J, Cromer MK, Vaidyanathan S, Bak RO, Carte JM, Potter J, Dever DP, Porteus MH. Priming human repopulating hematopoietic stem and progenitor cells for Cas9/sgRNA gene targeting. Mol Ther Nucleic Acids. 2018;12:89–104.
Bak RO, Dever DP, Reinisch A, Cruz Hernandez D, Majeti R, Porteus MH. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. Elife. 2017;6: e27873.
Zhang L, Huang R, Lu L, Fu R, Guo G, Gu Y, Liu Z, He L, Malissen M, Liang Y. Gene knock-in by CRISPR/Cas9 and cell sorting in macrophage and T cell lines. J Vis Exp. 2021. https://doi.org/10.3791/62328.
Arias-Fuenzalida J, Jarazo J, Qing X, Walter J, Gomez-Giro G, Nickels SL, Zaehres H, Schöler HR, Schwamborn JC. FACS-assisted CRISPR-Cas9 genome editing facilitates Parkinson’s disease modeling. Stem Cell Rep. 2017;9:1423–31.
Wagner DL, Koehl U, Chmielewski M, Scheid C, Stripecke R. Review: sustainable clinical development of CAR-T Cells—switching from viral transduction towards CRISPR-Cas gene editing. Front Immunol. 2022;13: 865424.
Mosti L, Langner LM, Chmielewski KO, Arbuthnot P, Alzubi J, Cathomen T. Targeted multi-epitope switching enables straightforward positive/negative selection of CAR T cells. Gene Ther. 2021. https://doi.org/10.1038/s41434-021-00220-6.
Liu L, Sommermeyer D, Cabanov A, Kosasih P, Hill T, Riddell SR. Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nat Biotechnol. 2016;34:430–4.
Allen AG, Khan SQ, Margulies CM, Viswanathan R, Lele S, Blaha L, Scott SN, Izzo KM, Gerew A, Pattali R, et al. A highly efficient transgene knock-in technology in clinically relevant cell types. Nat Biotechnol. 2023. https://doi.org/10.1038/s41587-023-01779-8.
Van Duyne GD. Cre recombinase. Microbiol Spectr. 2015;3: Mdna3-0014-2014.
Ho TT, Zhou N, Huang J, Koirala P, Xu M, Fung R, Wu F, Mo YY. Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 2015;43: e17.
Zhu Z, Verma N, González F, Shi ZD, Huangfu D. A CRISPR/Cas-mediated selection-free knockin strategy in human embryonic stem cells. Stem Cell Rep. 2015;4:1103–11.
Xi L, Schmidt JC, Zaug AJ, Ascarrunz DR, Cech TR. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol. 2015;16:231.
Pham CT, MacIvor DM, Hug BA, Heusel JW, Ley TJ. Long-range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci U S A. 1996;93:13090–5.
Yusa K. piggyBac Transposon. Microbiol Spectr. 2015;3:Mdna3-0028–2014.
Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci U S A. 2001;98:9209–14.
Yusa K. Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat Protoc. 2013;8:2061–78.
Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordóñez A, Hannan NR, Rouhani FJ, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–4.
Wang G, Yang L, Grishin D, Rios X, Ye LY, Hu Y, Li K, Zhang D, Church GM, Pu WT. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat Protoc. 2017;12:88–103.
Eggenschwiler R, Moslem M, Fráguas MS, Galla M, Papp O, Naujock M, Fonfara I, Gensch I, Wähner A, Beh-Pajooh A, et al. Improved bi-allelic modification of a transcriptionally silent locus in patient-derived iPSC by Cas9 nickase. Sci Rep. 2016;6:38198.
Li MA, Pettitt SJ, Eckert S, Ning Z, Rice S, Cadiñanos J, Yusa K, Conte N, Bradley A. The piggyBac transposon displays local and distant reintegration preferences and can cause mutations at noncanonical integration sites. Mol Cell Biol. 2013;33:1317–30.
Kühn R, Chu VT. Pop in, pop out: a novel gene-targeting strategy for use with CRISPR-Cas9. Genome Biol. 2015;16:244.
Bendixen L, Jensen TI, Bak RO. CRISPR-Cas-mediated transcriptional modulation: the therapeutic promises of CRISPRa and CRISPRi. Mol Ther. 2023. https://doi.org/10.1016/j.ymthe.2023.03.024.
Jensen TI, Mikkelsen NS, Gao Z, Foßelteder J, Pabst G, Axelgaard E, Laustsen A, König S, Reinisch A, Bak RO. Targeted regulation of transcription in primary cells using CRISPRa and CRISPRi. Genome Res. 2021;31:2120–30.
Genovese P, Ferrari S, Lombardo Angelo L, Naldini L, Fiumara M. Selection by means of artificial transactivators. WO: OSPEDALE San Raffaele SRL Fond Telethon; 2020.
Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, Koyama D, Goto T, Hanajiri R, Nishida T, et al. A tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res. 2016;4:658–68.
Li X, Sun B, Qian H, Ma J, Paolino M, Zhang Z. A high-efficiency and versatile CRISPR/Cas9-mediated HDR-based biallelic editing system. J Zhejiang Univ Sci B. 2022;23:141–52.
Sconocchia T, Foßelteder J, Köhnke T, Majeti R, Reinisch A. Engineering oncogenic heterozygous gain-of-function mutations in human hematopoietic stem and progenitor cells. J Vis Exp. 2023. https://doi.org/10.3791/64558.
Foßelteder J, Pabst G, Sconocchia T, Schlacher A, Auinger L, Kashofer K, Beham-Schmid C, Trajanoski S, Waskow C, Schöll W, et al. Human gene-engineered calreticulin mutant stem cells recapitulate MPN hallmarks and identify targetable vulnerabilities. Leukemia. 2023;37:843–53.
Ma Y, Zhang J, Yin W, Zhang Z, Song Y, Chang X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods. 2016;13:1029–35.
Katti A, Foronda M, Zimmerman J, Diaz B, Zafra MP, Goswami S, Dow LE. GO: a functional reporter system to identify and enrich base editing activity. Nucleic Acids Res. 2020;48:2841–52.
Wang P, Xu L, Gao Y, Han R. BEON: a functional fluorescence reporter for quantification and enrichment of adenine base-editing activity. Mol Ther. 2020;28:1696–705.
Brookhouser N, Nguyen T, Tekel SJ, Standage-Beier K, Wang X, Brafman DA. A Cas9-mediated adenosine transient reporter enables enrichment of ABE-targeted cells. BMC Biol. 2020;18:193.
Martin AS, Salamango DJ, Serebrenik AA, Shaban NM, Brown WL, Harris RS. A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci Rep. 2019;9:497.
St Martin A, Salamango D, Serebrenik A, Shaban N, Brown WL, Donati F, Munagala U, Conticello SG, Harris RS. A fluorescent reporter for quantification and enrichment of DNA editing by APOBEC-Cas9 or cleavage by Cas9 in living cells. Nucleic Acids Res. 2018;46: e84.
Coelho MA, Li S, Pane LS, Firth M, Ciotta G, Wrigley JD, Cuomo ME, Maresca M, Taylor BJM. BE-FLARE: a fluorescent reporter of base editing activity reveals editing characteristics of APOBEC3A and APOBEC3B. BMC Biol. 2018;16:150.
Standage-Beier K, Tekel SJ, Brookhouser N, Schwarz G, Nguyen T, Wang X, Brafman DA. A transient reporter for editing enrichment (TREE) in human cells. Nucleic Acids Res. 2019;47: e120.
Brookhouser N, Tekel SJ, Standage-Beier K, Nguyen T, Schwarz G, Wang X, Brafman DA. BIG-TREE: base-edited isogenic hPSC line generation using a transient reporter for editing enrichment. Stem Cell Rep. 2020;14:184–91.
Tálas A, Simon DA, Kulcsár PI, Varga É, Krausz SL, Welker E. BEAR reveals that increased fidelity variants can successfully reduce the mismatch tolerance of adenine but not cytosine base editors. Nat Commun. 2021;12:6353.
Adikusuma F, Lushington C, Arudkumar J, Godahewa GI, Chey YCJ, Gierus L, Piltz S, Geiger A, Jain Y, Reti D, et al. Optimized nickase- and nuclease-based prime editing in human and mouse cells. Nucleic Acids Res. 2021;49:10785–95.
Sürün D, Schneider A, Mircetic J, Neumann K, Lansing F, Paszkowski-Rogacz M, Hänchen V, Lee-Kirsch MA, Buchholz F. Efficient generation and correction of mutations in human iPS cells utilizing mRNAs of CRISPR base editors and prime editors. Genes (Basel). 2020. https://doi.org/10.3390/genes11050511.
Lin Q, Zong Y, Xue C, Wang S, Jin S, Zhu Z, Wang Y, Anzalone AV, Raguram A, Doman JL, et al. Prime genome editing in rice and wheat. Nat Biotechnol. 2020;38:582–5.
Simon DA, Tálas A, Kulcsár PI, Biczók Z, Krausz SL, Várady G, Welker E. PEAR, a flexible fluorescent reporter for the identification and enrichment of successfully prime edited cells. Elife. 2022. https://doi.org/10.7554/eLife.69504.
Levesque S, Mayorga D, Fiset JP, Goupil C, Duringer A, Loiselle A, Bouchard E, Agudelo D, Doyon Y. Marker-free co-selection for successive rounds of prime editing in human cells. Nat Commun. 2022;13:5909.
Neff T, Horn PA, Peterson LJ, Thomasson BM, Thompson J, Williams DA, Schmidt M, Georges GE, von Kalle C, Kiem HP. Methylguanine methyltransferase-mediated in vivo selection and chemoprotection of allogeneic stem cells in a large-animal model. J Clin Invest. 2003;112:1581–8.
Davis BM, Reese JS, Koç ON, Lee K, Schupp JE, Gerson SL. Selection for G156A O6-methylguanine DNA methyltransferase gene-transduced hematopoietic progenitors and protection from lethality in mice treated with O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1997;57:5093–9.
Chen Y, Schroeder JA, Gao C, Li J, Hu J, Shi Q. In vivo enrichment of genetically manipulated platelets for murine hemophilia B gene therapy. J Cell Physiol. 2021;236:354–65.
Beard BC, Trobridge GD, Ironside C, McCune JS, Adair JE, Kiem HP. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J Clin Invest. 2010;120:2345–54.
Li C, Georgakopoulou A, Newby GA, Chen PJ, Everette KA, Paschoudi K, Vlachaki E, Gil S, Anderson AK, Koob T, et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood. 2023. https://doi.org/10.1182/blood.2022018252.
Neubacher S, Hennig S. RNA structure and cellular applications of fluorescent light-up aptamers. Angew Chem Int Ed Engl. 2019;58:1266–79.
Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333:642–6.
Ewen-Campen B, Perrimon N. ovo(D) Co-selection: a method for enriching CRISPR/Cas9-edited alleles in drosophila. G3 (Bethesda). 2018;8:2749–56.
Challagulla A, Shi S, Nair K, ONeil TE, Morris KR, Wise TG, Cahill DM, Tizard ML, Doran TJ, Jenkins KA. Marker counter-selection via CRISPR/Cas9 co-targeting for efficient generation of genome edited avian cell lines and germ cells. Anim Biotechnol. 2021; https://doi.org/10.1080/10495398.2021.1885428.
Piñero-Lambea C, Garcia-Ramallo E, Miravet-Verde S, Burgos R, Scarpa M, Serrano L, Lluch-Senar M. SURE editing: combining oligo-recombineering and programmable insertion/deletion of selection markers to efficiently edit the Mycoplasma pneumoniae genome. Nucleic Acids Res. 2022;50: e127.
Shimatani Z, Kashojiya S, Takayama M, Terada R, Arazoe T, Ishii H, Teramura H, Yamamoto T, Komatsu H, Miura K, et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol. 2017;35:441–3.
Li XL, Li GH, Fu J, Fu YW, Zhang L, Chen W, Arakaki C, Zhang JP, Wen W, Zhao M, et al. Highly efficient genome editing via CRISPR-Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018;46:10195–215.
Dunbar CE, Cottler-Fox M, O’Shaughnessy JA, Doren S, Carter C, Berenson R, Brown S, Moen RC, Greenblatt J, Stewart FM, et al. Retrovirally marked CD34-enriched peripheral blood and bone marrow cells contribute to long-term engraftment after autologous transplantation. Blood. 1995;85:3048–57.
Zhang P, Raju J, Ullah MA, Au R, Varelias A, Gartlan KH, Olver SD, Samson LD, Sturgeon E, Zomerdijk N, et al. Phase I trial of inducible caspase 9 T cells in adult stem cell transplant demonstrates massive clonotypic proliferative potential and long-term persistence of transgenic T cells. Clin Cancer Res. 2019;25:1749–55.
Adair JE, Chandrasekaran D, Sghia-Hughes G, Haworth KG, Woolfrey AE, Burroughs LM, Choi GY, Becker PS, Kiem HP. Novel lineage depletion preserves autologous blood stem cells for gene therapy of Fanconi anemia complementation group A. Haematologica. 2018;103:1806–14.
Zhou X, Tu S, Wang C, Huang R, Deng L, Song C, Yue C, He Y, Yang J, Liang Z, et al. Phase I trial of fourth-generation anti-CD19 chimeric antigen receptor T cells against relapsed or refractory B cell non-hodgkin lymphomas. Front Immunol. 2020;11: 564099.
Sutermaster BA, Darling EM. Considerations for high-yield, high-throughput cell enrichment: fluorescence versus magnetic sorting. Sci Rep. 2019;9:227.
Pello OM, Lanzarot D, Colorado M, Amunarriz C, Insunza A, Álvarez-Rodríguez L, Díez de Velasco M, Sainz-Sainz N, Arroyo JL. Optimal large-scale CD34+ enrichment from a leukapheresis collection using the clinimacs prodigy platform. Clin Case Rep. 2020;8:2650–3.
Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, Krueger W, Worden AA, Kadan MJ, Yim S, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med. 2020;26:1569–75.
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83.
Eissenberg LG, Rettig MP, Ritchey JK, Prior JL, Schwarz SW, Frye J, White BS, Fulton RS, Ghobadi A, Cooper ML, et al. [(18)F]FHBG PET/CT imaging of CD34-TK75 transduced donor T cells in relapsed allogeneic stem cell transplant patients: safety and feasibility. Mol Ther. 2015;23:1110–22.
Durmus NG, Tekin HC, Guven S, Sridhar K, Arslan Yildiz A, Calibasi G, Ghiran I, Davis RW, Steinmetz LM, Demirci U. Magnetic levitation of single cells. Proc Natl Acad Sci U S A. 2015;112:E3661-3668.
Verzeletti S, Bonini C, Marktel S, Nobili N, Ciceri F, Traversari C, Bordignon C. Herpes simplex virus thymidine kinase gene transfer for controlled graft-versus-host disease and graft-versus-leukemia: clinical follow-up and improved new vectors. Hum Gene Ther. 1998;9:2243–51.
Zhan H, Gilmour K, Chan L, Farzaneh F, McNicol AM, Xu JH, Adams S, Fehse B, Veys P, Thrasher A, et al. Production and first-in-man use of T cells engineered to express a HSVTK-CD34 sort-suicide gene. PLoS ONE. 2013;8: e77106.
Acknowledgements
Figures were created using Biorender.com.
Funding
Funding in the Bak Lab is supported by grants from the Danish health authorities (SST) (4-1612-391/1), the EU Commission in the form of an ERC Starting Grant (project 101041231, Horizon Europe Pillar I) and a grant from the Horizon Research and Innovation Actions (project 101057438, Horizon Europe Pillar II), a Lundbeck Foundation Fellowship (R238-2016-3349), the Independent Research Fund Denmark (0134-00113B, 0242-00009B, and 9144-00001B), an AIAS-COFUND (Marie Curie) fellowship from Aarhus Institute of Advanced Studies (AIAS) co-funded by Aarhus University’s Research Foundation and the European Union’s seventh Framework Program under grant agreement no 609033, the Novo Nordisk Foundation (NNF19OC0058238 and NNF17OC0028894), Innovation Fund Denmark (8056-00010B), the Carlsberg Foundation (CF20-0424 and CF17-0129), Slagtermester Max Wørzner og Hustru Inger Wørzners Mindelegat, the AP Møller Foundation, the Riisfort Foundation, the Agnes and Poul Friis’ Foundation, and a Genome Engineer Innovation Grant from Synthego.
Author information
Authors and Affiliations
Contributions
NSM took the lead in writing the manuscript with input and supervision from ROB.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
ROB holds equity in Graphite Bio and UNIKUM Tx and is part-time employee of UNIKUM Therapeutics. NSM declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mikkelsen, N.S., Bak, R.O. Enrichment strategies to enhance genome editing. J Biomed Sci 30, 51 (2023). https://doi.org/10.1186/s12929-023-00943-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-023-00943-1