
Abstract
Background
This study aims to analyze the pathogenic gene in a Chinese family with non-syndromic hearing loss and identify a novel mutation site in the TNC gene.
Methods
A five-generation Chinese family from Anhui Province, presenting with autosomal dominant non-syndromic hearing loss, was recruited for this study. By analyzing the family history, conducting clinical examinations, and performing genetic analysis, we have thoroughly investigated potential pathogenic factors in this family. The peripheral blood samples were obtained from 20 family members, and the pathogenic genes were identified through whole exome sequencing. Subsequently, the mutation of gene locus was confirmed using Sanger sequencing. The conservation of TNC mutation sites was assessed using Clustal Omega software. We utilized functional prediction software including dbscSNV_AdaBoost, dbscSNV_RandomForest, NNSplice, NetGene2, and Mutation Taster to accurately predict the pathogenicity of these mutations. Furthermore, exon deletions were validated through RT-PCR analysis.
Results
The family exhibited autosomal dominant, progressive, post-lingual, non-syndromic hearing loss. A novel synonymous variant (c.5247A > T, p.Gly1749Gly) in TNC was identified in affected members. This variant is situated at the exon–intron junction boundary towards the end of exon 18. Notably, glycine residue at position 1749 is highly conserved across various species. Bioinformatics analysis indicates that this synonymous mutation leads to the disruption of the 5' end donor splicing site in the 18th intron of the TNC gene. Meanwhile, verification experiments have demonstrated that this synonymous mutation disrupts the splicing process of exon 18, leading to complete exon 18 skipping and direct splicing between exons 17 and 19.
Conclusion
This novel splice-altering variant (c.5247A > T, p.Gly1749Gly) in exon 18 of the TNC gene disrupts normal gene splicing and causes hearing loss among HBD families.
Similar content being viewed by others


Background
Hearing loss significantly impacts patients' quality of life, which is mainly caused by a combination of genetic and environmental factors, with approximately 60% of cases attributed to genetics [1]. Hereditary hearing loss can be classified into syndromic hearing loss and non-syndromic hearing loss based on hearing symptoms, with non-syndromic hearing loss (NSHL) accounting for around 70% of hereditary hearing loss [2,3,4]. The non-syndromic hearing loss can be categorized into autosomal recessive inheritance, autosomal dominant inheritance, X-chromosome linked inheritance, and mitochondrial maternal inheritance, based on different modes of transmission. With the rapid development of molecular sequencing technology, especially whole exome sequencing (WES), more genes related to autosomal dominant non-syndromic hearing loss (ADNSHL) have been discovered. To date, more than 79 loci for ADNSHL have been mapped to chromosomal regions and 63 genes for ADNSHL have been identified (http:// hereditaryhearingloss.org).
The TNC gene (NM_002160.2, NP_002151.2), located on chromosome 9q31.3-q34.3 and spanning 28.54 Mb, is responsible for DFNA56 and encodes the multifunctional hexoscan protein Tenascin-C [5]. Tenascin-C is an extracellular matrix (ECM) glycoprotein found in the basement membrane and spiral layer of the cochlear bone, consisting of a tenascin assembly (TA) domain, a linear array of epidermal growth factor-like (EGFL) repeats, a series of fibronectin type III (FN-III) domains, and a globular domain at the terminal [6]. The FN-III domain is the crucial functional component of tenascin-C, playing a significant role in auditory development and repair of hearing injuries [7]. The reporting of missense and synonymous mutations has consistently increased in recent years. Numerous studies have shown that synonymous mutations can contribute to diseases through various mechanisms, such as influencing gene splicing [8], mRNA stability [9], and protein translation rate [10]. Since 2013, TNC has been identified as a novel pathogenic gene associated with non-syndromic hearing loss [11]. Reports of TNC mutations in non-syndromic hearing loss are emerging. However, there have been no reports on synonymous mutations in TNC causing non-syndromic hearing loss. In this study, a five-generation Chinese family (family HBD) from Anhui Province was included, which exhibited autosomal dominant non-syndromic hearing loss. Whole exome sequencing and Sanger verification identified a novel splice-altering variant in the TNC, c.5247A > T (p.Gly1749Gly), within family HBD.
Methods
Family members and clinical data
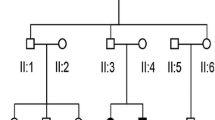
The family, referred to here as HBD, is a five-generation Chinese family from Anhui Province with autosomal dominant non-syndromic hearing loss (Fig. 1). The proband is a 23-year-old male with a family history of hearing loss. This family has autosomal dominant, progressive, post-lingual, non-syndromic sensorineural hearing loss, and these features are consistent with ADNSHL. A total of 20 family members participated in the study, including 12 males and 8 females. Among them were 5 patients with non-syndromic hearing loss, all of whom had moderate or higher hearing loss and did not use hearing aids or undergo surgery. Informed consent has been obtained from all participating members of the HBD family or their guardians and approved by the hospital Ethics Committee (No. YYLL20230039).

Pedigree of the Chinese family HBD. Filled symbols represent affected individuals, and empty symbols represent unaffected individuals. The square represents males and the circle represents females. The arrow indicates the proband. WT: wild-type; M: mutant
All participants underwent medical history collection, physical examination, intelligence assessment, hearing examination and genetic testing. The basic data included age of onset, disease progression, trauma history, noise exposure, utilization of ototoxic drugs, hearing aid usage, presence of tinnitus and vertigo. The physical examination included assessing the skin condition, hair quality, limb functionality, iris appearance, inner canthal distance measurement, outer ear structure and inner ear health. The Wechsler Adult Intelligence Scale-Revised in China (WAIS-RC) is used for intelligence assessment, including the evaluation of full intelligence quotient (FIQ), verbal intelligence quotient (VIQ), and performance intelligence quotient (PIQ).
The hearing examination includes pure tone audiometry (PTA), auditory brainstem response (ABR), acoustic immittance measurement, tinnitus detection, distortion product otoacoustic emissions (DPOAE), computed tomography (CT) scan of the temporal bone and vestibular system. In order to determine the degree of hearing loss in patients, PTA is employed to assess the average air conduction thresholds at frequencies of 250, 500, 1000, 2000, 4000 and 8000 Hz. To conduct ABR testing, the skin should be cleansed using an alcohol swab, followed by placement of two electrodes on the midline of the forehead and vertex. Subsequently, the patient should assume a supine position. A click stimulus with an intensity level of 70 dB was selected for testing at a rate of 21.1 stimuli per second. ABR testing is conducted on both ears of each participant to automatically measure amplitude and latency values for waves I, III, and V, as well as interpeak latencies for waves I-III, III-V, and I-V in order to assess potential lesions along the auditory pathway from cochlea to brainstem. DPOAE is an objective indicator of cochlear and outer hair cell functionality. Examinations were conducted using the AccuScreen 42D automatic otoacoustic emission tester (Madsen, Denmark) in a low-noise environment with background noise levels ≤ 40 dB. The tested frequency range included 1.5, 2.0, 3.0, 4.0, 5.0, and 6.0 kHz frequencies. Signal amplitude and signal-to-noise ratio were recorded at each frequency to calculate the pass rate. The severity of hearing loss was defined as mild (26–40 dB), moderate (41–55 dB), moderately severe (56–70 dB), severe (71–90 dB), or profound (> 90 dB) [11]. Furthermore, high resolution computed tomography (HRCT) was also performed on five affected individuals.
DNA extraction
The peripheral blood samples of 20 participants from the HBD family were collected, and DNA was extracted from the peripheral blood using QIAamp DNA Blood Midi KIT (Qiagen, Dusseldorf, Germany), the purity and concentration of the extracted DNA were determined using a Nanodrop2000 ultramicrospectrophotometer.
Screening for mutations in common hearing loss gene
The Genetic Deafness Microarray was utilized to identify common deafness genes in families III:3, III:4, III:5, III:6, IV:5, IV:6, IV:7, IV:8. These included GJB2, GJB3, mitochondrial MT-RNR1, and SLC26A4 [12].
Whole exome sequencing
The IDT xGen Exome Research Panel v1.0 kit was used to capture exon regions, including the DNA coding region and adjacent shear region of genes associated with hereditary hearing loss. Dian Diagnostics Group Co., Ltd., completed the sequencing process using the NovaSeq 6000 platform (Illumina, San Diego, CA, USA).
The initial sequencing data obtained were processed according to the following criteria: 1) filtering out sequences with low quality; 2) removing sequencing adapters and primer sequences; 3) excluding regions with poor base quality to eliminate any chaotic sequences. This was followed by aligning against the GRCh37/hg19 reference sequence using Burrow-Wheeler Aligner (BWA) software [13]. Subsequently, duplicate sequences were removed using the Picard tool [14]. Next, Genome Analysis ToolKit (GATK) software is employed for mutation site detection, followed by annotation of these sites using snpEFF software [15]. The database includes 1000 Genome, NHLBI Exome Sequencing Project, ClinVar, and dbNSFP. The prediction tool for identifying harmful genetic mutations includes MutationTaster [16], dbscSNV_AdaBoost [17], dbscSNV_RandomForest [18], NNSplice [19], and NetGene2 [20].
Sanger sequencing
Following whole exome sequencing, candidate genes were isolated and subjected to Sanger sequencing analysis. Through PCR amplification and sequencing, the mutation can be detected and identified. The primers for TNC gene mutation sites have been designed and synthesized as follows: 5'-ATTCCTTGGAGTCAGCTA-3' and 5'-CCAGTAGGTCTTGTTCAA-3'. Subsequently, these primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd.
Evolutionary conservation analysis
To determine the phylogenetic conservation of the variant, Clustal Omega software were used to align the sequence of Homo sapiens (human) with a range of different mammals such as Macaca fascicularis (monkey), Pan troglodytes (chimpanzee), Halichoerus grypus (seal), Marmota monax (marmot), Ursus arctos (bear), and Delphinus delphis (delphinus) [21].
Splicing site mutation validation
In order to investigate the impact of the identified variant (c.5247A > T, p.Gly1749Gly) on splice sites, we performed RT-PCR amplification of exons 17–20 of the TNC gene and subsequently using agarose gel electrophoresis. RNA extraction was performed on fresh blood samples obtained from individuals III:3, III:4, III:5, III:6, IV:5, IV:6, IV:7, and IV:8 utilizing a spin column-based blood total RNA purification kit (Sangon Biotech, Shanghai, PR China). Following this step, the obtained total RNA underwent reverse transcription using the iScript Reverse Transcription Supermix for RT-qPCR Kit (Bio-Rad, USA). Using cDNA as a template, the exons 17–20 sequences were amplified using the following primer sequences: forward 5'-AAGCCGAACCGGAAGTTGACAACC-3' and reverse 5'-CTTTCTCGCCTGTGTAGGAGATGA-3'. Subsequently, electrophoresis was conducted on a 2% agarose gel at 95 V for 30 min, and the results were visualized under UV illumination.
The real-time fluorescence quantitative PCR method was employed to quantify the expression levels of bands amplified, with all reactions being performed in triplicate.
Results
Clinical features
The HBD family, which consisted of 7 patients with hearing loss, including 2 deceased individuals (I:1, II:1), ultimately comprised 5 affected and 15 unaffected individuals. The age at onset of hearing impairment ranged from 6 to 16 years. The hearing examination and clinical data of the affected members in HBD showed progressive, post-lingual, non-syndromic sensorineural hearing loss. Notably, there is no history of trauma, noise exposure, or ototoxic drug use. The Affected members showed no abnormalities during the physical examination and intelligence assessment. However, slight speech expression abnormalities were observed in the proband (IV:7) and their sister (IV:8) (Table 1).
The affected individuals initially had low frequency hearing loss, which gradually worsened as they aged (Fig. 2). No vertigo or tinnitus was reported in affected members of HBD. The ABR test results were normal for the affected members, but DPOAE testing revealed cochlear dysfunction. High resolution CT scans showed normal structure in the middle and inner ear (Table 1).

Pure-tone audiograms of the affected members in HBD. dB, decibels; Hz, Hertz; y, years
WES identifies a causative variant in TNC
To identify the genetic cause of hearing loss, a common genetic test for deafness is conducted utilizing a genetic deafness chip. The findings do not reveal any variants in GJB2, GJB3, SLC26A4, and mitochondrial MT-RNR1. Consequently, WES was performed on four affected individuals (III:3, III:5, IV:7, IV:8) and three unaffected individuals (II:2, III:6, IV:6) in HBD. WES analysis successfully identifies a novel synonymous variant (c.5247A > T, p.Gly1749Gly) located in exon 18 of the TNC gene among the four affected family members. Subsequent Sanger sequencing confirms that this variant in TNC (c.5247A > T) co-segregates in the family (Fig. 3). This variant was detected in individuals IV:7, IV:8, III:3, and III:5, all of whom were diagnosed with hearing loss (Fig. 3A-D). The variant was not detected in individuals II:2, III:6, and IV:6, who exhibited normal hearing abilities (Fig. 3E-G). Furthermore, this variant was not present in the 1000 Genomes database, NHLBI Exome Sequencing Project, and ClinVar. In addition to the TNC gene mutation, WES testing of the proband revealed mutations in genes MYO3A (c.610G > A, c.4484G > A), CDH23 (c.574G > C), and DMXL2 (c.364 + 8G > A). However, these mutations did not correspond with the family members' inherited deafness, and none of the other affected family members had these mutations, so we decided to exclude them.

The sanger sequence results of the variant c.5247A > T in HBD family. Arrows indicate the position of the nucleotide changes identified
Pathogenicity and evolutionary conservation analysis
The pathogenicity analysis for this variant was determined based on various tools, including dbscSNV (likely pathogenic) and Mutation Taster (disease causing) in Table 2. The impact of this variant on splicing was assessed using various computational tools, including NNSplice and NetGene2. Predictions from these tools suggest that this synonymous variant leads to the disruption of the 5' end donor splicing site in the 18th intron of the TNC gene (Table 2). This variant is located at the nucleotide end of exon 18, which corresponds to the boundary of the exon–intron junction point, specifically within the 13th FN-III domain in the translated protein (Fig. 4). Mutations occurring at this site have the potential to disrupt normal splicing and alter downstream splicing events (Table 2). Notably, glycine residue at position 1749 is highly conserved across a range of different species (such as monkey, chimpanzee, seal, marmot, bear, and dolphin), as depicted in Fig. 5.

Gene mapping of HBD family with non-syndromic hearing loss. A Structure of TNC gene. B Structure of TNC translated protein. Mutations of c.5247A > T (p.Gly1749Gly) identified in TNC gene is located in exon 18, which corresponds to the13th FN-III domain in the translated protein

Evolutionary conservation of the Gly1749 residue of TNC across a range of species. Protein alignment shows high conservation for the position 1749 in the TNC protein among different species
Splicing site mutation analysis
In order to validate the impact of this splice-altering variant, we performed RT-PCR amplification of exons 17–20 sequences to confirm aberrantly spliced RNA presence in affected members. Agarose electrophoresis revealed that the four affected members showed two distinct bands on RT-PCR, 660 bp and 537 bp respectively, whereas normal individuals displayed only a single band (Fig. 6A). The results demonstrate that this splice-altering variant affects the splicing process of the TNC gene, causing exon skipping in exon 18. Furthermore, this observation was further supported by quantifying expression levels across different bands (Fig. 6B).

Splicing Site Mutation Analysis for c.5247A > T in TNC. A Agarose gel showing the result of the RT-PCR. B Relative expression of bands
Discussion
In this study, a novel splice-altering variant (c.5247A > T, p.Gly1749Gly) in the TNC was identified in a Chinese family with non-syndromic hearing loss. The glycine at position 1749 is conserved across a range of different species. Although this mutation does not result in an amino acid change, it has the potential to disrupt normal splicing and alter downstream splicing events, potentially impacting the function or structure of the gene.
Tenascin-C is a member of extracellular matrix glycoprotein, which exists in the basement membrane and spiral layer of cochlear bone, and is one of the main components of the basement membrane of mammals [22]. The TNC plays a role in regulating fluid and ion transport between endolymphatic and exolymph. Any mutation in the TNC may disrupt basement membrane ion homeostasis [23]. The hearing loss resulting from TNC mutation is classified as low-frequency non-syndromic hearing loss. Below 1000 Hz, the hearing loss is severe, gradually decreasing and returning to normal as the frequency increases. Therefore, it is crucial for patients to undergo effective early diagnosis before the progression of hearing loss to severe hearing loss [24, 25].
Since the discovery in 2013 that TNC mutation is implicated in non-syndromic hearing loss [26], TNC has been included in the non-syndromic hearing loss gene database. However, due to the rarity of TNC mutation, there remains a scarcity of reports on non-syndromic hearing loss attributed to TNC mutation. In 2013, Zhang et al. initially identified through genetic linkage analysis that two missense mutations (c.5317G > A, p.V1773M and c.5368A > T, p.T1796S) in the TNC gene were associated with autosomal dominant hearing loss, thereby confirming TNC as a novel gene implicated in hearing loss [26]. The report indicates that the TNC mutations (c.5317G > A, p.V1773M and c.5368A > T, p.T1796S) at these two loci are both located in exon 19, corresponding to the 13th FN-III domain. These two mutations occur at the connecting edges of two conserved domains within the FN-III structure, thereby inducing conformational changes in this region and affecting the molecular binding ability of tenascin-C. Furthermore, a case report analysis conducted in Portugal demonstrated that a 20-year-old female patient exhibited progressive bilateral hearing loss, which was ultimately attributed to a missense mutation (c.1337G > A, p.R446Q) in the TNC gene [24].
The disease was observed in a total of 7 individuals within this family. Following thorough hearing examinations and medical history investigations, no abnormalities were detected in skin and hair pigmentation, iris heterochromia, inner canthal distance, upper limb structure or megacolon presence. Based on these findings, it was determined that this family was non-syndromic hearing loss. None of the patients in this family had a history of trauma, noise exposure, or ototoxic drug use. The patients exhibit a genetic predisposition, with a male-to-female ratio of 2:5. Both genders are susceptible to developing the disease. The inheritance pattern followed Mendelian autosomal dominant characteristics. Therefore, this family is diagnosed with autosomal dominant non-syndromic hearing loss. In this study, we conducted a screening of four common deafness genes (GJB2, GJB3, mitochondrial MT-RNR1, and SLC26A4) in the Chinese population to exclude mutations in these commonly associated deafness gene loci. Subsequently, through whole exome sequencing, we identified a novel mutation in the TNC which was further confirmed by Sanger sequencing. Notably, this mutation co-segregated with the observed phenotype of familial hearing loss. To establish controls for comparison, three individuals with normal hearing from HBD family were selected and no mutations were detected at TNC. Therefore, it can be concluded that the TNC mutation is responsible for causing hearing loss in HBD families.
The N-terminal EGF domain of tenascin-C is capable of binding and activating the EGFR located on the surface of muscle stem cells, thereby facilitating the proliferation of muscle stem cells [27]. The current evidence demonstrates that tenascin-C has the ability to both promote and inhibit the growth of various neurons and neurites [28]. Considering the primary role of the tenascin-C in the human body, it is hypothesized that tenascin-C primarily functions in repairing the basement membrane in the cochlea. Following a mutation in TNC, the repair mechanism for inner ear damage becomes impaired, leading to a gradual accumulation of hearing impairment [26]. Therefore, the hearing impairment in affected individuals within this family is initially characterized by low-frequency hearing loss, which progressively worsens with age.
The amino acid sequence remains unchanged by synonymous mutations, and it was initially believed that a substantial number of these mutations lacked biological significance. However, numerous studies have demonstrated that synonymous mutations can induce disease by impacting precursor mRNA splicing, mRNA structural integrity and stability, as well as protein translation efficiency and speed. The most prevalent mechanism involves interference with precursor mRNA splicing through modulation of splicing enhancer or silencer elements, or the generation of an occult donor/receptor site, resulting in aberrant protein encoding or production of unstable abnormal mRNA [29]. Nucleotide changes near the splicing site are widely believed to cause protein dysfunction [30]. In this study, we have identified a mutation site located at the terminal nucleotide position of exon 18, precisely at the boundary of the exon–intron junction point. Mutations occurring at this specific site can significantly disrupt RNA splicing. Species conservation analysis revealed that this site was highly conserved in various species. When combined with the mutation predictions from dbscSNV, Mutation Taster, NNSplice, and NetGene2, it is speculated that the mutation occurring at this site may disrupt normal splicing and alter downstream splicing events, thereby potentially impacting protein function. Concurrently, the experimental findings demonstrated that the mutation impeded exon 18 splicing, resulting in complete exon 18 skipping and direct splicing of exons 17 and 19. Currently, the mutation site has not yet been identified in the 1000 Genomes database, NHLBI Exome Sequencing Project, and ClinVar databases. Additionally, no reports have been made regarding synonymous mutations in the TNC leading to non-syndromic hearing loss. The identification of this mutation would be beneficial for diagnosing TNC-related hearing loss.
The study still has limitations as we did not perform cDNA sequencing of exons 17–19, thus unable to determine the specific changes in protein levels. Additionally, we are unsure about the exact impact of this splice-altering variant on protein structure and function. Therefore, in future research, we will focus on addressing these shortcomings.
Conclusions
In conclusion, our study has identified a novel splice-altering mutation (c.5247A > T, p.Gly1749Gly) in the TNC that is responsible for autosomal dominant non-syndromic hearing loss in HBD families. This represents the first reported instance of a pathogenic splice-altering mutation in the TNC within a non-syndromic hearing loss population, thereby broadening the range of pathogenic loci associated with the genetic database for non-syndromic hearing loss.
Availability of data and materials
Sequence data that support the findings of this study have been deposited in the NCBI ClinVar repository with the primary accession code VCV002775398.1.
Abbreviations
- NSHL:
-
Non-syndromic hearing loss
- ADNSHL:
-
Autosomal dominant non-syndromic hearing loss
- ECM:
-
Extracellular matrix
- EGFL:
-
Epidermal growth factor-like
- FN-III:
-
Fibronectin type III
- PTA:
-
Pure tone audiometry
- ABR:
-
Auditory brainstem response
- DPOAE:
-
Distortion product otoacoustic emissions
- CT:
-
Computed tomography
- HRCT:
-
High resolution computed tomography
- BWA:
-
Burrow-Wheeler Aligner
- GATK:
-
Genome Analysis ToolKit
- WES:
-
Whole exome sequencing
- PCR:
-
Polymerase chain reaction
- TA:
-
Tenascin assembly
- WAIS-RC:
-
Wechsler Adult Intelligence Scale-Revised in China
- FIQ:
-
Full intelligence quotient
- VIQ:
-
Verbal intelligence quotient
- PIQ:
-
Performance intelligence quotient
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
References
Jin X, Huang S, An L, Zhang C, Dai P, Gao H, Ma X. Variant analysis of 92 Chinese Han families with hearing loss. BMC Med Genomics. 2022;15(1):12.
Kim YR, Kim HM, Lee B, Baek JI, Lee KY, Park HJ, Kim UK. Identification of novel missense mutation related with non-syndromic sensorineural deafness, DFNA11 in korean family by NGS. Genes Genomics. 2023;45(2):225–30.
Liu Y, Tan M, Cai L, Lv L, Chen Q, Chen W, Yang H, Xu Y. Genetic profiles of non-syndromic severe-profound hearing loss in Chinese Hans by whole-exome sequencing. Gene. 2022;819:146258.
Zhang W, Song J, Tong B, Ma M, Guo L, Yuan Y, Yang J. Identification of a novel CNV at the EYA4 gene in a Chinese family with autosomal dominant non-syndromic hearing loss. BMC Med Genomics. 2022;15(1):113.
Gan NS, Oziębło D, Skarżyński H, Ołdak M. Monogenic causes of low-frequency non-syndromic hearing loss. Audiol Neurootol. 2023;28(5):327–37.
Shi M, Cao L, Ding D, Shi L, Hu Y, Qi G, Zhan L, Zhu Y, Yu W, Lv P, Yu N. Acute noise causes down-regulation of ECM protein expression in Guinea pig cochlea. Mol Biotechnol. 2023;65(5):774–85.
Meiners S, Mercado ML, Nur-e-Kamal MS, Geller HM. Tenascin-C contains domains that independently regulate neurite outgrowth and neurite guidance. J Neurosci. 1999;19(19):8443–53.
Deng LX, Yang Y, Yang J, Zhou LW, Wang K, Zhou JH. A presumed synonymous mutation of PKD2 caused autosomal dominant polycystic kidney disease in a Chinese family. Curr Med Sci. 2021;41(5):1029–36.
Simhadri VL, Hamasaki-Katagiri N, Lin BC, Hunt R, Jha S, Tseng SC, Wu A, Bentley AA, Zichel R, Lu Q, Zhu L, Freedberg DI, Monroe DM, Sauna ZE, Peters R, Komar AA, Kimchi-Sarfaty C. Single synonymous mutation in factor IX alters protein properties and underlies haemophilia B. J Med Genet. 2017;54(5):338–45.
Bertalovitz AC, Badhey MLO, McDonald TV. Synonymous nucleotide modification of the KCNH2 gene affects both mRNA characteristics and translation of the encoded hERG ion channel. J Biol Chem. 2018;293(31):12120–36.
Smith JR, Huang AR, Lin FR, Reed NS, Deal JA. The population attributable fraction of dementia from audiometric hearing loss among a nationally representative sample of community-dwelling older adults. J Gerontol A Biol Sci Med Sci. 2023;78(7):1300–6.
Duan S, Guo Y, Chen X, Li Y. Genetic mutations in patients with nonsyndromic hearing impairment of minority and Han Chinese ethnicities in Qinghai, China. J Int Med Res. 2021;49(4):3000605211000892.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92.
Steinhaus R, Proft S, Schuelke M, Cooper DN, Schwarz JM, Seelow D. MutationTaster2021. Nucleic Acids Res. 2021;49(1):446–51.
Moles-Fernández A, Duran-Lozano L, Montalban G, Bonache S, López-Perolio I, Menéndez M, Santamariña M, Behar R, Blanco A, Carrasco E, López-Fernández A, Stjepanovic N, Balmaña J, Capellá G, Pineda M, Vega A, Lázaro C, de la Hoya M, Diez O, Gutiérrez-Enríquez S. Computational tools for splicing defect prediction in breast/ovarian cancer genes: how efficient are they at predicting RNA alterations? Front Genet. 2018;9:366.
Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human non-synonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235–41.
Riepe TV, Khan M, Roosing S, Cremers FPM, ‘t Hoen PAC. Benchmarking deep learning splice prediction tools using functional splice assays. Hum Mutat. 2021;42(7):799–810.
Millat G, Lafont E, Nony S, Rouvet I, Bozon D. Functional characterization of putative novel splicing mutations in the cardiomyopathy-causing genes. DNA Cell Biol. 2015;34(7):489–96.
Sievers F, Higgins DG. The clustal omega multiple alignment package. Methods Mol Biol. 2021;2231:3–16.
Ishiyama A, Mowry SE, Lopez IA, Ishiyama G. Immunohistochemical distribution of basement membrane proteins in the human inner ear from older subjects. Hear Res. 2009;254(1–2):1–14.
Whitlon DS, Zhang X, Kusakabe M. Tenascin-C in the cochlea of the developing mouse. J Comp Neurol. 1999;406(3):361–74.
Sousa PCD, Gamboa I, Duarte D, et al. TNC gene mutation: a rare cause for early-onset sensorineural hearing loss. Allied Academies. 2018;8(2):167.
Bezdjian A, Bruijnzeel H, Pagel J, Daniel SJ, Thomeer HGXM. Low-frequency sensorineural hearing loss in familial hemophagocytic lymphohistiocytosis type 5. Ann Otol Rhinol Laryngol. 2018;127(6):409–13.
Zhao Y, Zhao F, Zong L, Zhang P, Guan L, Zhang J, Wang D, Wang J, Chai W, Lan L, Li Q, Han B, Yang L, Jin X, Yang W, Hu X, Wang X, Li N, Li Y, Petit C, Wang J, Wang HY, Wang Q. Exome sequencing and linkage analysis identified tenascin-C (TNC) as a novel causative gene in nonsyndromic hearing loss. PLoS ONE. 2013;8(7):e69549.
Swindle CS, Tran KT, Johnson TD, Banerjee P, Mayes AM, Griffith L, Wells A. Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J Cell Biol. 2001;154(2):459–68.
Kwiatkowska M, Reinhard J, Roll L, Kraft N, Dazert S, Faissner A, Volkenstein S. The expression pattern and inhibitory influence of Tenascin-C on the growth of spiral ganglion neurons suggest a regulatory role as boundary formation molecule in the postnatal mouse inner ear. Neuroscience. 2016;319:46–58.
Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 2003;12(3):205–16.
Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, Caspari R, Möslein G, Schulmann K, Propping P, Friedl W. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Hum Mutat. 2004;24(5):370–80.
Acknowledgements
We are grateful for all the staff at the medical records department for their help in data collection.
Funding
This study was supported by Wannan Medical College research project, China (No. 2021WYY0071) and First People's Hospital of Wuhu research project, China (No. 2020WYY0221).
Author information
Authors and Affiliations
Contributions
M.H. assisted with the statistical analysis and drafted the manuscript. M. H. performed with the statistical analysis, and revised the manuscript. Q. Z. and K. Y. collected the data and assisted the statistical analysis. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by the Ethics committee of First People's Hospital of Wuhu (No. YYLL20230039) and adhered to the Declaration of Helsinki’s ethical standards. All study participants or their legal guardians have signed written informed consent and agreed to participate in the study, as well as to allow their data and information to be utilized for scientific research purposes. We confrm that all methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
We confirm that all participants have signed written informed consent for publication of their genetic data, clinical details, and/or any accompanying images. For minor patient, parental signed written informed consent for publication of their child's genetic data, clinical details, and/or any accompanying images.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, M., Hu, M., Zhang, Q. et al. A novel splice-altering TNC variant (c.5247A > T, p.Gly1749Gly) in an Chinese family with autosomal dominant non-syndromic hearing loss. BMC Med Genomics 17, 189 (2024). https://doi.org/10.1186/s12920-024-01964-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01964-x