
Abstract
Background
There is a high incidence of cervical cancer in Xinjiang. Genetic variation in human papillomavirus may increase its ability to invade, spread, and escape host immune response.
Methods
HPV16 genome was sequenced for 90 positive samples of HPV16 infection. Sequences of the E4, E5 and L2 genes were analysed to reveal sequence variation of HPV16 in Xinjiang and the distribution of variation among the positive samples of HPV16 infection.
Results
Eighty-one of the 90 samples of HPV16 infection showed variation in HPV16 E4 gene with 18 nucleotide variation sites, of which 8 sites were synonymous variations and 11 missense variations. 90 samples of HPV16 infection showed variation in HPV16 E5 and L2 genes with 16 nucleotide variation sites (6 synonymous, 11 missense variations) in the E5 gene and 100 nucleotide variation sites in L2 gene (37 synonymous, 67 missense variations). The frequency of HPV16 L2 gene missense variations G3377A, G3599A, G3703A, and G3757A was higher in the case groups than in the control groups.
Conclusions
Phylogenetic tree analysis showed that 87 samples were European strains, 3 cases were Asian strains, there were no other variations, and G4181A was related to Asian strains. HPV16 L2 gene missense variations G3377A, G3599A, G3703A, and G3757A were significantly more frequent in the case groups than in the control groups.
Similar content being viewed by others

Introduction
Cervical cancer (CC) is the fourth leading cause of death from malignancy among women, with 604,000 new cases and 342,000 deaths worldwide in 2020 according to the World Health Organization (WHO) [1,2,3]. The occurrence and development of cervical cancer is related to the economic and health status of the region, and the incidence and mortality rate of cervical cancer in Xinjiang, China, is still very high [4, 5].
Cancer etiology research in the past 25 years has revealed persistent infection of high-risk human papillomavirus (HR-HPV) as a main cause of cervical cancer development and progression [6,7,8]. The positive rate of HPV in Xinjiang is 14.02% with HPV52, HPV53, HPV16 and HPV18 being the most common types and HPV16 being the most pathogenic among all HPV types [9, 10]. HPV16 is the most dangerous and most preventable virus type. HPV16 is divided into four main variant lineages: A lineage contains EUR (A1-A3) and As (A4); B lineage contains AF-1, C lineage contains (AF-2) and D lineage contains NA (D1), AAII (D2) and AAI (D3). The HPV16 that infects women in Xinjiang, China, are mostly European strains in A lineage [11, 12].
The complete HPV16 genome is approximately 7.9 kb, consisting of six early genes (E1, E2, E4, E5, E6, and E7), two late genes (L1 and L2), and one long control region (LCR) [13]. The HPV16 E4 gene encodes the E4 protein, which is the protein with the highest HPV16 expression. The main amino acids are derived from the E4 ORF, which is contained in the E2 gene [14]. The E4 protein plays a role in viral transmission by enhancing viral replication and the excretion of virions [15, 16]. The HPV16 E5 gene encodes a transmembrane protein with 83 amino acids that serves as an innate immune evasion factor. The transmembrane protein is involved in immune surveillance and immune evasion, leading to persistent viral infection. The transmembrane protein also plays a central role in the regulation of the host immune system, and directly related to the initial stages of cervical cancer development [17, 18]. The HPV L2 gene encodes a minor capsid protein, which promotes retrograde transport of the viral genome, integrating the viral genome into the host gene during the intercellular phase, which may lead to irreversible changes in the cell [19, 20].
Because the HPV16 E4, E5 and L2 genes affect a series of processes of virus invasion, immune escape and transmission, the distribution of the HPV16 E4, E5 and L2 genes in Xinjiang is not yet clear. Therefore, in this study, we focused on the variation in the HPV16 E4, E5 and L2 genes and their distribution in the case group and control group.
Materials and methods
Collection of samples
A total of 90 patient samples were collected from Yili Friendship Hospital, Kashgar District People’s Hospital and Shihezi University Affiliated Hospital with HPV16-positive cervical cell samples. All samples had been collected from 2016 to 2017 and patients of age were from 30 to 60 years old. The diagnosis of cervical cancer was confirmed by pathological examination according to “Diagnosis and Treatment, Obstetrics and Gynaecology” and the FIGO stage (International Federation of Gynaecology and Obstetrics, 2009). The inclusion criteria for control groups were HPV16 positive and the absence of lesions or inflammation in the cervix. Informed consent was obtained from all patients. All patients had no history of long-term travel or residence, and samples were collected and stored in a -80 ℃ low-temperature refrigerator. The pathological information of the samples is shown in Table S1 of the supplementary materials.
HPV genotyping
The HPV genotyping (23 types) was performed with PCR-reverse dot blot hybridization technology (Shenzhen Co., Ltd., China). All of the detection procedures were conducted in accordance with the manufacturer’s instructions [12].
DNA extraction and PCR amplification of samples
A DNA extraction kit (Tiangen Biochemical Co., Ltd.) was used to extract DNA, which was stored in a -20 °C freezer. Using 1% agarose electrophoresis examined the quality of DNA samples; DNA samples were diluted to a working concentration of 10–20 ng/µL. Samples without DNA bands were re-extracted. The mixed reaction solution (40 µL) consisted of 20 µL of 2×Taq enzyme PCR SuperMix, 1 µL of forward primer (10µmol/L), 1 µL of reverse primer (10µmol/L), 2 µL of DNA sample, and 16 µL of ddH2O. The PCR cycling conditions were 94 °C for 5 min; 34 cycles of 94 °C for 30 s, 52 °C for 30 s, 72 °C for 1 min; and 72 °C for 5 min. Using 1% agarose electrophoresis examined the quality of PCR products, and samples with bright and regular bands at 650 bp were qualified for subsequent DNA sequencing. PCR products were stored in a -20 °C freezer. Information on the primers is shown in Table 1.
Sequencing
Sequencing was performed by Shanghai Sangon, The Beijing Genomics Institute and other sequencing companies, and the PCR product was purified by SAP (Promega) and EXO I (Epicentre): 0.5 U SAP and 4 U Exo I were added to 8 µl PCR products. The mixture was incubated at 37 °C for 60 min, followed by incubation at 75 °C for 15 min. Finally, the BIG-DYE Terminator V3.1 cycle sequencing kit from ABI Co. was used for sample sequencing on a DNA analyzer (ABI3130XL) after purification with alcohol. The sequencing primers were: 16B1228E-6 F, 16B1228E-7 F, 16B1228E-8 F, 16B1228E-9 F, 16B1228E-10 F (Table 1).
Phylogenetic analysis of HPV16 variants
The raw sequences were assembled by Molecular Evolutionary Genetics Analysis (MEGA) software and were aligned with the European prototype virus strain (GenBank: NC_001526.4) and the accession numbers of other HPV16 variants including European strains A1-A3: A1 (HQ644283.1, HQ644268.1, HQ644280.1, HQ644282.1), A2: (AF5.36179.1), A3: (HQ644236.1), Asian strains A4 (HQ644235.1, HQ644248.1, AF534061.1, HQ644251.1), African strains B (HQ644238.1, HQ644240.1, HQ644290.1, HQ644298.1) and C (AF472509.1, HQ644239.1, HQ644249.1, HQ644237.1), North American strains: D1 (HQ644257.1), Asian American strains: D2 (HQ644263.1, HQ644277.1, HQ644279.1, HQ644281.1), D3 (HQ644247.1, HQ644255.1, HQ644253.1, AF402678.1). Phylogenetic tree of HPV16 E4, E5, and L2 gene was constructed with MEGA [4, 21], using the Maximum likelihood method, the Bootstrap method (1000 replication), the Tamura 3-parameter model, Gamma distributed with Invariant sites (G + I) and showing only greater than 50% of the Bootstrap values, where the Bootstrap values were > 70% indicates good reliability.
Statistical analysis
The variation frequency of HPV16 E4, E5, and L2 gene variation was directly counted. SPSS 26.0 software was used to analyze the statistical results and the correlation between HPV16 E4, E5, and L2 single nucleotide variation and cervical cancer. P values < 0.05 is accepted as statistically significant in chi-square test.
Results
Sequence variation in HPV16 E4, E5 and L2 genes
HPV16 E4, E5 and L2 gene variation results: gene sequencing results of 90 DNA samples with polymorphic sites shown in Tables 2, 3 and 4.
Among the 90 HPV16-positive samples, 81 samples had HPV16 E4 gene sequence variations in 18 nucleotide sites with 8 synonymous and 11 missense. The common synonymous variations were nt2520 (T-C) (25/81, 30.86%), nt2546 (C-T/A) (76/81, 93.83%; 2/81, 2.47%), nt2585 (G-A) (23/81, 28.40%), and nt2660 (T-C) (19/81, 23.46%). The number of variation of missense variation sites did not exceed 1. The frequency of missense variation was much lower than that of synonymous variations, indicating that the E4 gene was relatively conserved and had a stabilizing effect on the spread of the virus.
All of the 90 HPV16-positive samples had HPV16 E5 sequence variations in 16 sites with 6 synonymous and 11 missense variations. As shown in Table 3, the sites with high synonymous variation in the E5 gene were nt3213 (A-T) (20/90, 22.22%); the sites with more than 1 missense variation were: C2995T, A3115C, T3122C, C3127A/G, and A3178G, leading to amino acid changes leucine to phenylalanine (L4F), isoleucine to leucine (I44L), valine to alanine (V46A), leucine to isoleucine/valine (L48I/V) and isoleucine to valine (I65V) respectively. The A3115C and A3178G variations had very high frequency, 75.56% and 100%, respectively, and the simultaneous occurance of these two variations may significantly change the structure of the E5 protein and indirectly change the ability of virus immune escape.
Among the 90 HPV16 positive samples, all samples had L2 gene sequence variation in 100 nucleotide sites with 37 synonymous and 67 missense variations. The sites with high synonymous variations were nt4074 (G-A) (90/90, 100%) and nt4602 (A-G) (15/90, 16.67%) (Table 4). The most common missense variations were G3622A, G3641A, G3658A, G3703A, G3757A, T4177C, A4362C/T and A4654C, which led the amino acid changes aspartic acid to asparagine (D84N), arginine to lysine (R90K), aspartic acid to asparagine (D96N), glutamic acid to lysine (E111K), aspartic acid to asparagine (D129N), serine to proline (S269P), leucine to phenylalanine (L330F), and isoleucine to leucine (I428L). Through the three gene variation sites of HPV16 E4, E5 and L2, it can be found that the frequency of missense variation of the E4 gene is lower than that of the E5 and L2 genes, indicating that the E4 gene is more conserved than these two genes.
Phylogenetic tree analysis of the nucleotide sequences of HPV16 E4, E5 and L2
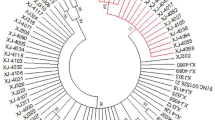
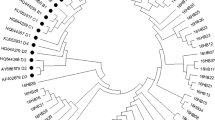
The Maximum likelihood method phylogenetic tree constructed with HPV16 E4, E5 and L2 gene sequences showed that 87 of the 90 HPV16 positive samples were European strains and 3 samples were Asian strains. No African, American or Asian-American strains were found. The Asian strains were associated with the missense variant G4181A, and the phylogenetic tree was shown in Fig. 1.

Phylogenetic tree analysis of HPV16 E4 E5 and L2 genes, the red dotted line is the reference virus strain
Genetic variation of genomic HPV16 E4, E5 and L2 in the case and control groups
Genetic variation of genomic HPV16 E4 in the case and control groups
The pathological information of the samples was statistical, including 47 cases in the control groups and 43 cases in the case groups. There were 6 synonymous variations and 1 missense variations in the control groups (non-cervical cancer group) (Table 5). In comparison, there were 7 synonymous variations and 10 missense variations in the case groups (cervical cancer group). The sequence variations did not differ significantly between the control groups and the case groups (P > 0.05) (Table 5). Most of the missense variations appeared in the case group, indicating the trend of E4 gene missense variations in the case group.
Genetic variation of genomic HPV16 E5 in the case and control groups
The control groups (non-cervical cancer group) had 3 synonymous variations and 8 missense variations. In comparison, the case groups (cervical cancer group) had 4 synonymous variations and 8 missense variations (Table 6). The statistical results showed that the synonymous variations A3213T was significantly higher than the control groups in the case groups, and the difference was statistically significant (P = 0.024). It is worth mentioning that the variation frequency of the missense variation A3115C in the case groups was 83.72%, which was higher than the frequency of variation in the control groups (68.09%), but the difference was not statistically significant (P > 0.05).
Genetic variation of genomic HPV16 L2 in the case and control groups
There were 24 synonymous variations and 44 missense variations in the control groups and 22 synonymous variations and 44 missense variations in the case groups (Table 7), among which the missense variations were G3377A (P = 0.036), G3599A (P = 0.004), G3703A (P = 0.038), and G3757A (P = 0.019). The frequency of variations in the case groups was significantly higher than that in the control groups, and the difference was statistically significant (P < 0.05). The amino acid changes were arginine to glutamine (R2Q), alanine to glutamic acid (G76E), glutamate to lysine (E111K), and aspartate to asparagine (D129N).
Discussion
Most of the research on HPV gene variation has focused on the E6 and E7 genes, while there are fewer studies on E4, E5, and L2 gene variations, and the understanding of HPV16 E4, E5 and L2 gene variations in Xinjiang is even more insufficient. The HPV16 E4, E5 and L2 genes dominate virion propagation, immune surveillance and escape, and the integration of the viral genome into the chromosomes of the nucleus [15, 18]; Sequence variation in E4, E5 and L2 genes, thus, will directly affect the ability of HPV virus to invade, immune, and spread.
We found that 87 cases (87/90, 96.67%) of the 90 HPV16 positive samples in Xinjiang were European strains, and the other 3 cases (3/90, 3.33%) were Asian strains. We speculated that the Asian strain was directly related to the L2 gene missense variation G4181A through bioinformatics comparison. The literature reported that the amino acid variations of HPV16 E4 protein include T22A, P36T, A43K, Q53R, L62I and L62P, which of these amino acid variations are related to the severity of cervical malignancy [22]. The E4 gene also encodes the E1^E4 protein, the first five amino acids of which are derived from E1 ORF, while the remaining amino acids are derived from E4 ORF. nine amino acid variations (A7V, A7P, L16I, D45E, L59I, L59T, Q66P, S72F, H75Q) were detected in the E1^E4 protein, and these were associated with the severity of cervical malignancy [23]. In this study, it was found that the amino acid variation of E4 protein (E1^E4 protein) : C7W, A10S (A7S), Y13C (Y10C), K40T (K37T), R43I (R40I), Q51H (Q48H), Q53H (Q50H), E67D (E64D), L79V (L76V), H82Y (H79Y) and T83R (T80R), which of amino acid variations had low frequency. Therefore, since the amino acid variations the frequency of E4 protein were much lower than that of E5 and L2 proteins, indicating that E4 gene was more conserved than E5 and L2 genes. Moreover, the missense variations of the E4 gene that was new variations were concentrated in the case group (cervical cancer). The HPV16 E4 gene is located in the central position of the E2 gene, which encodes the hinge domain in the E2 protein. The most common synonymous variations in the E4 gene, T2520C (30.86%), C2546T (93.83%) and G2585A (28.40%), are also missense variations in the hinge region of the E2 gene, which cause amino acid changes in the E2 protein and affect the viral replication process [14]. The reported that amino acid variations of E5 protein include F19I, V21A, C24S, L27P, P31L, I44L, L47S, L48A, V62A, I65V, I65L, and L73V [22, 24]. We found that the new amino acid variations of E5 protein include L4F, L16V, L28S, V46A, L48I, L48V, I65R, I65M and L81V, which L48I and L48V were the amino acid variations with high frequency. It was reported that a total of 17 amino acid variations with high frequency in L2 proteins, including D43E, S122P, V243I, T245A, L266F, L266V, S269P, L330F, D334N, T351P, T351S, T352P, T352A, S378VS378F, S384A, V385I, I420T, A424T, I428L and A443G. The amino acid variation I428L was present almost uniquely in Asia, and the frequency of S269P and L330F was higher than that of the reference amino acid, at position 330 phenylalanine of L2 protein was more common than the reference amino acid leucine in Europe, Asia, and North America [25]. We also found high-frequency variations of S269P and L330F in the L2 protein, which were consistent with the previous reports. In addition, the other high frequency amino acid variations have also been found in L2 protein, including D84N, R90K, D96N, E111K and D129N (Table 4). We found that L2 missense variations G3377A (R2Q), G3599A (G76E), G3703A (E111K), and G3757A (D129N), were in significantly higher frequency in the case groups (cervical cancer) than in the control groups(non-cervical cancer) (P < 0.05). These variations may affect the integration of HPV16 viral genome into the cell chromosomes.
Xinjiang is a multiethnic region. We included a total of 52 Han ethnic group samples in the 90 samples we studied, including 31 samples in the control groups and 21 samples in the case groups, and recounted several loci with more variations (see Table 8 for details). We found that the missense variation A3115C of the E5 gene was in significantly higher frequency in the case groups than in the control groups (P = 0.02), and the difference was statistically significant. However, as seen in Table 6 above, A3115C did not differ between the case and control groups in the 90 samples, so the A3115C variation may have different effects on different ethnic groups, which needs further validation.
The current study revelaed for the first time sequence variations in HPV16 E4, E5 and L2 genes and in Xinjiang, and the distribution of these variations among different ethnic groups. The sample size of the current study is relatively small and should be increased in future studies, in particular to include samples from more ethnic groups. Based on findings from the current study, variations A3115C, G3377A, G3599A, G3703A and G3757A should be further investigated in cell experiments to determine whether they affect the viral immunity and the integration of viral genome in cell chromosomes.
Conclusion
Phylogenetic tree analysis showed that 87 samples were European strains, 3 cases were Asian strains, there were no other variants, and G4181A was related to Asian strains. HPV16 L2 gene missense variants G3377A, G3599A, G3703A, and G3757A were significantly more frequent in the case groups than in the control groups.
Availability of data and materials
No datasets were generated or analysed during the current study.
References
Sung H, Ferlay J, Siegel R, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Ding X, Jia X, Wang C, Xu J, Gao S. Lu C. A DHX9-lncRNA-MDM2 interaction regulates cell invasion and angiogenesis of cervical cancer. Cell Death Differ. 2019;26(9):1750–65.
Seyoum A, Seyoum B, Gure T, Alemu A, Belachew A, Abeje D, Aseffa A, Howe R, Mulu A, Mihret A. Genotype heterogeneity of high-risk human papillomavirus infection in Ethiopia. Front Microbiol. 2023;14:1116685.
Zhe X, Xin H, Pan Z, Jin F, Zheng W, Li H, Li D, Cao D, Li Y, Zhang C, et al. Genetic variations in E6, E7 and the long control region of human papillomavirus type 16 among patients with cervical lesions in Xinjiang, China. Cancer Cell Int. 2019;19:65.
Fan P, Li X, Feng Y, Cai H, Dong D, Peng Y, Yao X, Guo Y, Ma M, Dong T, et al. PD-1 Expression Status on CD8+ Tumour Infiltrating Lymphocytes Associates With Survival in Cervical Cancer. Front Oncol. 2021;11:678758.
Kaelin E, Skidmore P, Łaniewski P, Holland L, Chase D, Herbst-Kralovetz M, Lim EJ. Cervicovaginal DNA Virome Alterations Are Associated with Genital Inflammation and Microbiota Composition. mSystems. 2022;7(2):e0006422.
Zhou Z, Yang H, Yang L, Yao Y, Dai S, Shi L, Li C, Yang L, Yan Z, Yao Y. Human papillomavirus type 16 E6 and E7 gene variations associated with cervical cancer in a Han Chinese population. Infect Genet Evol. 2019;73:13–20.
Pérez-González A, Cachay E, Ocampo A, Poveda EJM. Update on the epidemiological features and clinical implications of human papillomavirus infection (HPV) and Human Immunodeficiency Virus (HIV) Coinfection. Microorganisms. 2022;10(5):1047.
Wang J, Tang D, Wang K, Wang J, Zhang Z, Chen Y, Zhang X, Ma C. HPV genotype prevalence and distribution during 2009–2018 in Xinjiang, China: baseline surveys prior to mass HPV vaccination. BMC Womens Health. 2019;19(1):90.
Chelimo C, Wouldes TA, Cameron LD, Elwood JM. Risk factors for and prevention of human papillomaviruses (HPV), genital warts and cervical cancer. J Infect. 2013;66(3):207–17.
Burk R, Harari A, Chen Z. Human papillomavirus genome variants. Virology. 2013;445(1–2):232–43.
Pan Z, Song Y, Zhe X, Wang W, Zhu J, Zheng W, Li H, Li D, Cao D, Pan Z, et al. Screening for HPV infection in exfoliated cervical cells of women from different ethnic groups in Yili, Xinjiang, China. Sci Rep. 2019;9(1):3468.
Revathidevi S, Murugan A, Nakaoka H, Inoue I, Munirajan AK. APOBEC: a molecular driver in cervical cancer pathogenesis. Cancer Lett. 2021;496:104–16.
Doorbar J. The E4 protein; structure, function and patterns of expression. Virology. 2013;445(1–2):80–98.
Leeman A, Jenkins D, Marra E, van Zummeren M, Pirog E, van de Sandt M, van Eeden A, van der Schim M, Doorbar J, de Vries H, et al. Grading immunohistochemical markers p16INK4a and HPV E4 identifies productive and transforming lesions caused by low- and high-risk HPV within high-grade anal squamous intraepithelial lesions. Br J Dermatol. 2020;182(4):1026–33.
Kajitani N, Schwartz S. Role of Viral Ribonucleoproteins in Human Papillomavirus Type 16 Gene Expression. Viruses. 2020;12(10):1110.
Scott MLWB, Ulicny J, Raikhy G, Orr AW, Songock WK, Bodily JM. Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance. J Virol. 2020;6(2):e01582–19.
Gutierrez-Xicotencatl L, Pedroza-Saavedra A, Chihu-Amparan L, Salazar-Piña A, Maldonado-Gama M, Esquivel-Guadarrama F. Cellular functions of HPV16 E5 oncoprotein during Oncogenic Transformation. Mol Cancer Res. 2021;19(2):167–79.
Pouyanfard S, Spagnoli G, Bulli L, Balz K, Yang F, Odenwald C, Seitz H, Mariz F, Bolchi A, Ottonello S, et al. Minor Capsid Protein L2 Polytope Induces Broad Protection against Oncogenic and Mucosal Human Papillomaviruses. J Virol. 2018;92(4):e01930–01917.
Aydin I, Villalonga-Planells R, Greune L, Bronnimann M, Calton C, Becker M, Lai K, Campos S, Schmidt M, Schelhaas M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017;13(5):e1006308.
Wang L, Wang F, Fu S, Zhang C, Zhe X, Li H, Li D, Shao R, Pan, Z. Analysis of genetic variation in human papillomavirus type 16 E1 and E2 in women with cervical infection in Xinjiang, China. BMC Med Genomics. 2021;14(1):268.
Plesa A, Anton G, Iancu I, Diaconu C, Huica I, Stanescu A, Socolov D, Nistor E, Popa E, Stoian M, et al. Molecular variants of human papilloma virus 16 E2, E4, E5, E6 and E7 genes associated with cervical neoplasia in Romanian patients. Arch Virol. 2014;159(12):3305–20.
Tsakogiannis D, Ruether I, Kyriakopoulou Z, Pliaka V, Skordas V, Gartzonika C, Levidiotou-Stefanou S, Markoulatos P. Molecular and phylogenetic analysis of the HPV 16 E4 gene in cervical lesions from women in Greece. Arch Virol. 2012;157(9):1729–39.
Hsieh C, Tsao Y, Wang C, Han C, Chang J, Lee J, Chen S. Sequence variants and functional analysis of human papillomavirus type 16 E5 gene in clinical specimens. Arch Virol. 2000;145(11):2273–84.
Tsakogiannis D, Nikolaidis M, Zagouri F, Zografos E, Kottaridi C, Kyriakopoulou Z, Tzioga L, Markoulatos P, Amoutzias G, Bletsa GJV. Mutation profile of HPV16 L1 and L2 genes in different geographic areas. Viruses. 2022;15(1):141.
Acknowledgements
We thank the Yili Friendship Hospital, Kashi People’s Hospital and Xinjiang Uyghur Autonomous Region People’s Hospital for the samples.
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers: 82060518, U1503125) and the International Science and Technology Collaboration Projector of Xinjiang Production and Construction Corps (grant number 2019BC007).
Author information
Authors and Affiliations
Contributions
ZP designed the study. HC and YD carried out the molecular genetic studies, participated in the genetic analysis and wrote the manuscript. LW and XZ carried out specimen collection and DNA extraction. XZ and JT carried out HPV16 identification. DL, HL and RS participated in experimental design and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the ethics committees of the First Affiliated Hospital of Shihezi University (Approval Number: KJ2020-051-01, date: 2020.3.28). Informed consent was obtained from all patients for our research.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cheng, H., Dong, Y., Wang, L. et al. Analysis of human papillomavirus type 16 E4, E5 and L2 gene variations among women with cervical infection in Xinjiang, China. BMC Med Genomics 17, 179 (2024). https://doi.org/10.1186/s12920-024-01926-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01926-3