
Abstract
Background
The six-transmembrane epithelial antigen of prostate (STEAP) family members are known to be involved in various tumor-related biological processes and showed its huge potential role in tumor immunotherapy.
Methods
Biological differences were investigated through Gene set enrichment analysis (GSEA) and tumor microenvironment analysis by CIBERSORT. Tumor mutation burden (TMB), immunotherapy response and chemotherapeutic drugs sensitivity were estimated in R.
Results
We established a prognostic signature with the formula: risk score = STEAP1 × 0.3994 + STEAP4 × (− 0.7596), which had a favorable concordance with the prediction. The high-risk group were enriched in cell cycle and RNA and protein synthesis related pathways, while the low-risk group were enriched in complement and metabolic related pathways. And the risk score was significantly correlated with immune cell infiltration. Most notably, the patients in the low-risk group were characterized with increased TMB and decreased tumor immune dysfunction and exclusion (TIDE) score, indicating that these patients showed better immune checkpoint blockade response. Meanwhile, we found the patients with high-risk were more sensitive to some drugs related to cell cycle and apoptosis.
Conclusions
The novel signature based on STEAPs may be effective indicators for predicting prognosis, and provides corresponding clinical treatment recommendations for HCC patients based on this classification.
Similar content being viewed by others

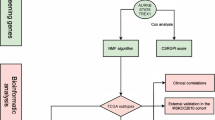

Background
Hepatocellular carcinoma (HCC) is one of the most common cancers with high incidence and mortality [1, 2]. Although therapies have advanced, the mortality of HCC remains high due to the difficulty of diagnosis at early stage [3]. Moreover, it is generally observed that HCC is unfavorable response to radiation and chemotherapy, and the prognosis of patients who receive potentially curative treatment remains poor due to the high rate of recurrence. Therefore, it is of great significance to explore effective prognostic diagnostic models and treatment strategies for HCC.
The six-transmembrane epithelial antigen of prostate (STEAP) family of proteins are located on the cell surface, and it comprises 4 members, named STEAP1, STEAP2, STEAP3, and STEAP4. Cumulative evidence has revealed that STEAPs are abnormally expressed in various cancer tissues and cell lines [4, 5], and are significantly associated with patient prognosis [6, 7]. STEAPs can promote tumorgenesis and development through a variety of biological processess [8,9,10,11,12,13]. Studies have also shown that STEAP mRNA is detectable in serum of patients with different solid tumours [14], suggesting its potential as a tumor detection marker. Moreover, emerging studies have reported that STEAP is closely related to tumor immunity [15,16,17,18]. STEAP is a target of CD8+ T cells [19, 20], which renders STEAP an appealing candidate for tumor immunotherapy. In recent years, STEAP therapeutic peptides and STEAP vaccines [20,21,22] have been used as new methods for tumor treatment, and have been verified to show good effectiveness. Cappuccini et al. found that the combination of STEAP1 vaccine and PD-1 blocking antibody can significantly improved survival of the animals, with 80% of mice remaining tumor-free [23]. Sebastian et al. found that there is a strong antitumor potential of MHC class I-restricted TCR-transgenic CD4+ T cells against a STEAP1-derived peptide in vivo [24]. However, systematic analysis of STEAPs expression profile and function in HCC were still insufficient. Given that, further insight into the function and the role of STEAPs in HCC may provide novel approach for precise treatment and individualized management.
Methods
Study subjects
The RNA-seq profiles and clinical data of HCC patients and normal samples were acquired from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/), and GSE14520 data set stored in the Gene Expression Omnibus database (GEO, https://www.ncbi.nlm.nih.gov/geo/). The TCGA was used as the training set, and contained 371 tumor samples, 50 normal samples, while the GSE14520 as the validation sets and contained 244 tumor samples.
Identification of differentially expressed and prognostic-related STEAPs
The differentially expressed STEAPs were identified by Student’s two-tailed t-test in R 4.0.3. Univariate Cox proportional hazard regression analysis was used to screen the prognostic-related STEAPs. The median survival time and cumulative survival curves were determined by the Kaplan–Meier method. Then, STEAPs with both P ≤ 0.05 were further identified by multivariate Cox proportional hazard regression. Genes with P ≤ 0.05 in multivariate Cox proportional hazard regression analysis was identified as prognostic-related STEAPs for further analysis.
Development and assessment of STEAPs-based prognostic risk model
Risk scores were established using the gene expression values and its prognostic weight coefficients that calculated by the multivariate Cox proportional hazard regression analysis with the following formula:
Based on the formula, the risk score of each patient was calculated, and patients were subdivided into high- or low-risk groups according to the median levels of the risk score. The cumulative survival curves of the grouped patients were determined by the Kaplan–Meier and differences between the groups were analyzed using the log-rank test. P ≤ 0.05 was considered statistically significant. The prognostic performance of the risk score model was assessed by using receiver operating characteristic (ROC) curve analysis within 0.5, 1, and 3 years. Univariate and multivariate Cox hazard regression was used to assess the impact of some prognostic factors.
Building and validation of the nomogram
The clinical characters consisting of age, gender, stage, liver fibrosis, and risk score et al. were selected to construct a prognostic nomogram to help predict the probability of 1-, 3-, and 5-year overall survival rates for HCC patients via the rms R package. The prediction power of the nomogram was graphically displayed via calibration curve.
Gene set enrichment analysis and tumor microenvironment analysis
Differentially expressed genes (DEGs) among the low- and high-risk groups were determined with Student’s two-tailed t-test. Genes with a P ≤ 0.05 and |log2FoldChange| ≥ 1 were defined as a differential gene. Gene set enrichment analysis (GSEA) analysis was implemented to determine the functional pathways enriched by high- and low-risk groups. The KEGG gene set (c2.cp.kegg.v7.0.symbols.gmt) was derived from the website (https://www.gsea-msigdb.org/). The expression of immune cells were evaluated by CIBERSORT algorithm.
Tumor mutation profile, immunotherapy response prediction, and therapeutic drug sensitivity
The original mutation annotation format of each LIHC sample was acquired from TCGA. Then, we calculated the tumor mutation burden (TMB) score according to the somatic mutation data and computed the differences of TMB between the low- and high-risk groups. The analyses were based on R package “maftools”. Subsequently, tumor immune dysfunction and exclusion (TIDE) database (http://tide.dfci.harvard.edu/) were applied to predict the potential immune checkpoint blockade (ICB) response. Finally, we used R package “pRRophetic” to calculate the semi-inhibitory concentration (IC50) values of chemotherapeutic drugs.
Results
Aberrant expression of STEAPs in HCC samples
Based on data from TCGA cohort, expression levels of STEAP1 (P = 9.22E-05) and STEAP2 (P = 7.33E-07) were significantly increased, while STEAP3 (P = 6.73E-26) and STEAP4 (P = 7.91E-09) was significantly decreased in tumors compared with adjacent normal tissues (Fig. 1A).

Identification of differentially expressed and prognostic-related STEAPs in the TCGA cohorts. A Violin plots showed the expression profile of STEAP family in HCC tumor tissues and normal tissues. B Survival analysis of STEAP family related signatures. **P < 0.01
Identification of two prognostic-related STEAPs in HCC samples
In order to further find the prognostic-related STEAPs, we analyzed the relationship between the expression level of STEAPs and overall survival (OS) rate of HCC patients. Following univariate Cox regression analysis, STEAP1, STEAP3, and STEAP4 were obtained for exhibiting significant prognostic correlation with OS (Fig. 1B, Table 1). In addition, Kaplan–Meier survival curves based on the median expression value indicated that the lower expression of STEAP1 had a better prognosis in terms of OS (P = 0.016), while the higher expression of STEAP3 and STEAP4 showed a better prognosis (P = 0.0072 and 0.00033, respectively; Fig. 1B). Subsequently, multivariate Cox regression analysis indicated that STEAP1 (hazard ratio [HR]: 1.49; P = 0.024) and STEAP4 (HR: 0.47; P = 0.0031) exhibited independent prognostic value for HCC (Table 1).
Construction and validation of the STEAP1 and STEAP4 based prognostic risk score
The two prognostic-related genes STEAP1 and STEAP4 were selected to establish a risk score model with the formula as follows: risk score = STEAP1 × 0.3994 + STEAP4 × (− 0.7596). Then, we analyzed the distribution of risk scores (Additional file 1: Fig. S1) and divided all HCC patients in the training and validation groups into high-risk and low-risk groups based on the median risk score. To evaluate the effectiveness of STEAPs-based prognostic risk model, we analyzed the prognosis of the two grouped patients and found that OS of HCC patients in the low-risk group was significantly better than that in the high-risk group in the TCGA training (P = 0.00027; Fig. 2A) and GSE14520 validation cohorts (P = 0.030; Additional file 1: Fig. S2A). Moreover, ROC curve analysis showed that the risk score model had the favorable predictive ability of the 0.5, 1 and 3 year OS, with an area under the ROC curve (AUC) of 0.670, 0.642, and 0.626 in the training cohort (Fig. 2B), and with AUC of 0.641, 0.635, and 0.534 in the validation cohort (Additional file 1: Fig. S2B).

The validation of the prognostic risk model and the nomogram and calibration curve of the model in the TCGA cohorts. A Kaplan–Meier survival curves based on risk score. B 0.5-, 1-, and 3-year ROC curves based on risk score. C Nomogram for predicting overall survival. D The calibration curves for 1-, 3-, and 5-year OS
Construction of nomogram for prognosis evaluation
To verify the independence of the risk score model, we conducted univariate and multivariate COX analysis on the demographic characteristics of all patients in the TCGA and GSE14520 cohorts. The result showed that the risk score was significantly correlated with OS in the TCGA training cohort (HR = 1.89/1.81, p < 0.001) and GSE14520 validation cohort (HR = 1.56/1.27, p = 0.031/0.29; Table 2). Furthermore, the demographic characteristics including risk score were enrolled to construct a nomogram for predicting the 1-, 3-, and 5-year OS incidences (Fig. 2C, Additional file 1: Fig. S2C). The calibration curves exhibited a favorable consistency with the actual observation (Fig. 2D, Additional file 1: Fig. S2D).
Analysis of DEGs and their functional pathways of the prognostic risk groups
A total of 1119 DEGs were identified in TCGA, including 564 upregulated and 555 downregulated (Additional file 2: Table S1). Simultaneously, GSE14520 confirmed 39 DEGs, with 17 upregulated and 22 downregulated (Additional file 3: Table S2). Among them, the expression of AFP——an indicator often used for diagnosis of HCC, showed very significant differences between the high-risk group and the low-risk group both in the training set (P = 0.0041, OR = 4.49) and the validation sets (P = 0.00029, OR = 2.35; Fig. 3A, B). Then, we performed GSEA analyses to further investigate functional pathways associated with the prognostic risk groups. The results showed that genes in the high-risk group were enriched in the pathways of cell cycle and RNA and protein synthesis related pathways (ribosome, spliceosome, DNA replication, proteasome, etc). However, genes in the low-risk group were enriched in the complement and coagulation cascade pathways, as well as pathways related to amino acid, fatty acid, and drug metabolism (Fig. 3C, D, Additional file 4: Table S3, Additional file 5: Table S4).

Differential expression of AFP between two groups and functional enrichment analysis of the two groups. A, B Boxplot showed that the expression of AFP at high-risk group is significantly higher than that at low-risk group in the (A) TCGA and (B) GSE14520 cohorts. C, D The GSEA analysis for TCGA (C) and GSE14520 (D). **P < 0.01
Investigation of the immune microenvironment in risk groups
To explore the immune microenvironment in the prognostic risk groups, we calculated the expression of the tumor-infiltrating immune cell between two risk groups. Patients in the high-risk exhibited a significant decrease in Macrophages M1, Macrophages M2, Mast cells resting, T cells CD4 memory resting and a significant increase in Macrophages M0, T cells CD4 memory activated (Fig. 4A, B).

Immune signature in two groups. A, B discrepancy analysis of tumor-infiltrating immune cells between two groups in the (A) TCGA and (B) GSE14520 cohorts
TMB, TIDE and therapeutic drug sensitivity
Then, we analyzed the variations of the somatic mutations in two risk groups. The highest mutated genes were TP53, TTN, CTNNB1, MUC16, APOB, RYR2, ABCA13, CSMD3, and LRP1B (Fig. 5A). Compared with high-risk group, patients in low-risk group had higher TMB (Fig. 5B). The TIDE score was significantly higher in high-risk group compared with low-risk group (P = 1.90E-08; Fig. 5C). Through drug sensitivity comparison, we found that patients in high-risk group were more sensitive to AS601245, BAY 61–3606, Bortezomib, CGP − 60,474, JNK − 9 L, LFM-A13, RO − 3306, and XMD8–92, while patients in low-risk group were more sensitive to OSI − 027 (Fig. 5D).

TMB analysis, immunotherapy response, and therapeutic drug sensitivity prediction. A OncoPrint of frequently mutated genes in high- and low-risk groups. B TMB difference between high- and low-risk groups. C TIDE score between two groups. D chemotherapeutic drugs with significant IC50 differences between the two groups
Discussion
STEAPs are unique to mammals and were initially identified as important metalloreductases in vivo [25]. They are involved in a wide range of biological processes, such as molecular trafficking in the endocytic and exocytic pathways and control of cell proliferation and apoptosis [26, 27]. Lots of studies have shown that STEAPs are abnormally expressed in a variety of human cancers and play vital role in promoting tumorgenesis and development [28,29,30], making them become potential prognostic biomarker, detection biomarker and therapeutic target [7, 14, 31], However, the clinical significance and role of STEAPs in HCC are still unclear.
In this study, we screened the differentially expressed and prognostic-related STEAPs in the TCGA training set and confirmed two significantly prognostic-related genes (STEAP1 and STEAP4), whose roles in HCC were barely studied. STEAP1 plays an important role in intercellular communication, modulating the transport of small molecules and ions such as Na+, K+ and Ca2+, and releasing soluble cytokines and chemokines [8, 32]. Moreover, STEAP1 is highly expressed in multiple cancer tissues such as prostate, bladder, ovarian, and colon cancer and has the role of promoting invasion of tumor cells [4, 33,34,35]. Several studies have showed that its overexpression inhibits apoptosis and induces epithelial to mesenchymal transition, ultimately contributing to tumor progression and aggressiveness in cancer cells [36,37,38]. In addition, STEAP1 has been considered as an optimal target for T cell-based immunotherapy, with applications in a subset of cancer types nowadays [5, 39]. STEAP4 is also called STAMP2 and TNF-α induce adipose-related protein (TIARP), which can regulate inflammatory reaction, fatty acid metabolism and glucose metabolism [40,41,42]. Besides, studies have demonstrated that STEAP4 is also closely related to tumorigenesis [43,44,45].
Subsequently, we established and validated a risk score model based on the expression of STEAP1 and STEAP4, and divided patients into low- and high-risk groups according to the median values. Studies have shown that certain transcription factors responsible for the holistic progression of fibrosis in HCC are transcriptional regulators of STEAP1 and STEAP4 [46]. To further explore the association between STEAP1 and STEAP4 with the development of HCC, we analyzed the correlation between their expression with some particular etiology (alcohol consumption, hepatitis, non-alcoholic fatty liver disease) and liver fibrosis (including cirrhosis) in HCC. The results showed that STEAP1, STEAP4, and risk score were not correlated with specific etiology, while STEAP1 and risk score were significantly positively correlated with liver fibrosis both in TCGA and GSE14520 (Additional file 1: Fig. S3). These results suggest that STEAP1 and our risk score may have a potentially important role in the progression of liver fibrosis to HCC, but not in the process from pathology to liver fibrosis, which needs to be verified by further experiments.
Numerous studies have shown that immune checkpoint inhibitors are far more effective than chemotherapy in tumor patients with high TMB expression [47, 48]. TIDE algorithm is a method for predicting ICB response in cancer. A higher TIDE score is associated with worse ICB response. Our results suggest that patients in the low-risk group can receive better benefits from clinical immunotherapy. Meanwhile, we used pRRophetic to calculate the IC50 values of chemotherapeutic drugs, and found that patients in high-risk group were more sensitive to AS601245, BAY 61–3606, Bortezomib, CGP − 60,474, JNK − 9 L, LFM-A13, RO − 3306, and XMD8–92. Among them, AS601245, JNK-9 L, and LFM-A13 are selective inhibitors of c-jun-N-terminal kinase (JNK). BAY 61–3606 is an orally available, ATP-competitive, reversible and highly selective Syk inhibitor. Bortezomib is a reversible and selective proteasome inhibitor that effectively inhibits the 20S proteasome by targeting threonine residues. CGP60474 and RO-3306 are potent cyclin-dependent kinase (CDK) inhibitors. XMD8–92 is a potent ERK5 (BMK1)/BRD4 inhibitor. This result shown that all drugs sensitive to high-risk groups with anti-cancer activity by affecting cell cycle and inducing apoptosis [49,50,51,52,53,54,55], which coincides with our result of pathway enrichment in the high-risk group. Our study may provide corresponding clinical treatment recommendations for HCC patients based on this classification.
In the present study, there still exist some limitations. First, the prognostic signature was created and verified based on retrospective data from TCGA and GSE14520 databases. Further large scale prospective clinical studies are required to evaluate its effectiveness and practicability. Besides, more well-designed basic research experiments are warranted to highlight the crucial role of STEAPs and corresponding treatment strategies in the precise treatment of HCC.
Conclusions
Current research indicates that novel signature based on STEAPs may be effective indicators for predicting prognosis, and provides corresponding clinical treatment recommendations for HCC patients based on this classification.
Availability of data and materials
All data used in this study can be acquired from TCGA (http://cancergenome.nih.gov/) and GSE14520 stored in the GEO (https://www.ncbi.nlm.nih.gov/geo) databases.
Abbreviations
- STEAP:
-
Six-transmembrane epithelial antigen of prostate
- TMB:
-
Tumor mutation burden
- TIDE:
-
Tumor immune dysfunction and exclusion
- HCC:
-
Hepatocellular carcinoma
- TCGA:
-
The Cancer Genome Atlas
- OS:
-
Overall survival
- GSEA:
-
Gene set enrichment analysis
- GEO:
-
Gene Expression Omnibus database
- ROC:
-
Receiver operating characteristic
- DEGs:
-
Differentially expressed genes
- ICB:
-
Immune checkpoint blockade
- IC50:
-
Semi-inhibitory concentration
References
Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet (London, England). 2022;400(10360):1345–62.
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Hubert RS, Vivanco I, Chen E, Rastegar S, Leong K, Mitchell SC, Madraswala R, Zhou Y, Kuo J, Raitano AB, et al. STEAP: a prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc Natl Acad Sci U S A. 1999;96(25):14523–8.
Gomes IM, Maia CJ, Santos CR. STEAP proteins: from structure to applications in cancer therapy. Mol Cancer Res. 2012;10(5):573–87.
Ihlaseh-Catalano SM, Drigo SA, de Jesus CM, Domingues MA, Trindade Filho JC, de Camargo JL, Rogatto SR. STEAP1 protein overexpression is an independent marker for biochemical recurrence in prostate carcinoma. Histopathology. 2013;63(5):678–85.
Fang ZX, Li CL, Chen WJ, Wu HT, Liu J. Potential of six-transmembrane epithelial antigen of the prostate 4 as a prognostic marker for colorectal cancer. World J Gastrointest Oncol. 2022;14(9):1675–88.
Grunewald TG, Bach H, Cossarizza A, Matsumoto I. The STEAP protein family: versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol Cell. 2012;104(11):641–57.
Grunewald TG, Diebold I, Esposito I, Plehm S, Hauer K, Thiel U, da Silva-Buttkus P, Neff F, Unland R, Müller-Tidow C, et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res. 2012;10(1):52–65.
Qin DN, Kou CZ, Ni YH, Zhang CM, Zhu JG, Zhu C, Wang YP, Zhu GZ, Shi C, Ji CB, et al. Monoclonal antibody to the six-transmembrane epithelial antigen of prostate 4 promotes apoptosis and inhibits proliferation and glucose uptake in human adipocytes. Int J Mol Med. 2010;26(6):803–11.
Yamamoto T, Tamura Y, Kobayashi J, Kamiguchi K, Hirohashi Y, Miyazaki A, Torigoe T, Asanuma H, Hiratsuka H, Sato N. Six-transmembrane epithelial antigen of the prostate-1 plays a role for in vivo tumor growth via intercellular communication. Exp Cell Res. 2013;319(17):2617–26.
Sun J, Ji G, Xie J, Jiao Z, Zhang H, Chen J. Six-transmembrane epithelial antigen of the prostate 1 is associated with tumor invasion and migration in endometrial carcinomas. J Cell Biochem. 2019;120(7):11172–89.
Ding T, Chen S, Xiao W, Liu Z, Tu J, Yu Y, Dong B, Chen W, Zeng Y. Six-transmembrane epithelial antigen of prostate 3 promotes hepatic insulin resistance and steatosis. J Lipid Res. 2023;64(1):100318.
Valenti MT, Dalle Carbonare L, Donatelli L, Bertoldo F, Giovanazzi B, Caliari F, Lo Cascio V. STEAP mRNA detection in serum of patients with solid tumours. Cancer Lett. 2009;273(1):122–6.
Azumi M, Kobayashi H, Aoki N, Sato K, Kimura S, Kakizaki H, Tateno M. Six-transmembrane epithelial antigen of the prostate as an immunotherapeutic target for renal cell and bladder cancer. J Urol. 2010;183(5):2036–44.
Kobayashi H, Nagato T, Sato K, Aoki N, Kimura S, Murakami M, Iizuka H, Azumi M, Kakizaki H, Tateno M, et al. Recognition of prostate and melanoma tumor cells by six-transmembrane epithelial antigen of prostate-specific helper T lymphocytes in a human leukocyte antigen class II-restricted manner. Cancer Res. 2007;67(11):5498–504.
Liang Y, Xing X, Beamer MA, Swindell WR, Sarkar MK, Roberts LW, Voorhees JJ, Kahlenberg JM, Harms PW, Johnston A, et al. Six-transmembrane epithelial antigens of the prostate comprise a novel inflammatory nexus in patients with pustular skin disorders. J Allergy Clin Immunol. 2017;139(4):1217–27.
Zhao Z, Wang Z, Song Z, Wu Y, Jin Q, Zhao Z. Predictive potential of STEAP family for survival, immune microenvironment and therapy response in glioma. Int Immunopharmacol. 2021;101(Pt A):108183.
Alves PM, Faure O, Graff-Dubois S, Cornet S, Bolonakis I, Gross DA, Miconnet I, Chouaib S, Fizazi K, Soria JC, et al. STEAP, a prostate tumor antigen, is a target of human CD8+ T cells. Cancer Immunol Immunother. 2006;55(12):1515–23.
Chen Q, Bao Y, Burner D, Kaushal S, Zhang Y, Mendoza T, Bouvet M, Ozkan C, Minev B, Ma W. Tumor growth inhibition by mSTEAP peptide nanovaccine inducing augmented CD8(+) T cell immune responses. Drug Deliv Transl Res. 2019;9(6):1095–105.
Garcia-Hernandez Mde L, Gray A, Hubby B, Kast WM. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: a candidate antigen for treating prostate cancer. Cancer Res. 2007;67(3):1344–51.
Herrmann VL, Wieland DE, Legler DF, Wittmann V, Groettrup M. The STEAP1(262–270) peptide encapsulated into PLGA microspheres elicits strong cytotoxic T cell immunity in HLA-A*0201 transgenic mice--A new approach to immunotherapy against prostate carcinoma. Prostate. 2016;76(5):456–68.
Cappuccini F, Stribbling S, Pollock E, Hill AV, Redchenko I. Immunogenicity and efficacy of the novel cancer vaccine based on simian adenovirus and MVA vectors alone and in combination with PD-1 mAb in a mouse model of prostate cancer. Cancer Immunol Immunother. 2016;65(6):701–13.
Schober SJ, Thiede M, Gassmann H, Prexler C, Xue B, Schirmer D, Wohlleber D, Stein S, Grünewald TGP, Busch DH, et al. MHC class I-restricted TCR-transgenic CD4(+) T cells against STEAP1 mediate local tumor control of Ewing sarcoma in vivo. Cells. 2020;9(7):1581.
Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. 2006;108(4):1388–94.
Hasegawa H, Li C, Alba BM, Penny DM, Xia Z, Dayao MR, Li P, Zhang J, Zhou J, Lim D, et al. Membrane cholesterol modulates STEAP2 conformation during dynamic intracellular trafficking processes leading to broad subcellular distribution. Exp Cell Res. 2018;370(2):208–26.
Chen WJ, Wu HT, Li CL, Lin YK, Fang ZX, Lin WT, Liu J. Regulatory roles of six-transmembrane epithelial antigen of the prostate family members in the occurrence and development of malignant tumors. Front Cell Dev Biol. 2021;9:752426.
Xu M, Evans L, Bizzaro CL, Quaglia F, Verrillo CE, Li L, Stieglmaier J, Schiewer MJ, Languino LR, Kelly WK. STEAP1-4 (six-transmembrane epithelial antigen of the prostate 1-4) and their clinical implications for prostate Cancer. Cancers. 2022;14(16):4034.
Chen H, Xu C, Yu Q, Zhong C, Peng Y, Chen J, Chen G. Comprehensive landscape of STEAP family functions and prognostic prediction value in glioblastoma. J Cell Physiol. 2021;236(4):2988–3000.
Liu T, Niu X, Li Y, Xu Z, Chen J, Xu G. Expression and prognostic analyses of the significance of STEAP1 and STEAP2 in lung cancer. World J Surg Onc. 2022;20(1):96.
Rodeberg DA, Nuss RA, Elsawa SF, Celis E. Recognition of six-transmembrane epithelial antigen of the prostate-expressing tumor cells by peptide antigen-induced cytotoxic T lymphocytes. Clin Cancer Res. 2005;11(12):4545–52.
Wu HT, Chen WJ, Xu Y, Shen JX, Chen WT, Liu J. The tumor suppressive roles and prognostic values of STEAP family members in breast Cancer. Biomed Res Int. 2020;2020:9578484.
Maia CJ, Socorro S, Schmitt F, Santos CR. STEAP1 is over-expressed in breast cancer and down-regulated by 17beta-estradiol in MCF-7 cells and in the rat mammary gland. Endocrine. 2008;34(1–3):108–16.
Moreaux J, Kassambara A, Hose D, Klein B. STEAP1 is overexpressed in cancers: a promising therapeutic target. Biochem Biophys Res Commun. 2012;429(3–4):148–55.
Zhao C, Xiong K, Ji Z, Liu F, Li X. The prognostic value and immunological role of STEAP1 in Pan-Cancer: a result of data-based analysis. Oxidative Med Cell Longev. 2022;2022:8297011.
Gomes IM, Rocha SM, Gaspar C, Alvelos MI, Santos CR, Socorro S, Maia CJ. Knockdown of STEAP1 inhibits cell growth and induces apoptosis in LNCaP prostate cancer cells counteracting the effect of androgens. Med Oncol (Northwood, London, England). 2018;35(3):40.
Jiao Z, Huang L, Sun J, Xie J, Wang T, Yin X, Zhang H, Chen J. Six-transmembrane epithelial antigen of the prostate 1 expression promotes ovarian cancer metastasis by aiding progression of epithelial-to-mesenchymal transition. Histochem Cell Biol. 2020;154(2):215–30.
Zhang Z, Hou WB, Zhang C, Tan YE, Zhang DD, An W, Pan SW, Wu WD, Chen QC, Xu HM. A research of STEAP1 regulated gastric cancer cell proliferation, migration and invasion in vitro and in vivos. J Cell Mol Med. 2020;24(24):14217–30.
Dorff TB, Narayan V, Forman SJ, Zang PD, Fraietta JA, June CH, Haas NB, Priceman SJ. Novel redirected T-cell immunotherapies for advanced prostate Cancer. Clin Cancer Res. 2022;28(4):576–84.
Korkmaz CG, Korkmaz KS, Kurys P, Elbi C, Wang L, Klokk TI, Hammarstrom C, Troen G, Svindland A, Hager GL, et al. Molecular cloning and characterization of STAMP2, an androgen-regulated six transmembrane protein that is overexpressed in prostate cancer. Oncogene. 2005;24(31):4934–45.
Scarl RT, Lawrence CM, Gordon HM, Nunemaker CS. STEAP4: its emerging role in metabolism and homeostasis of cellular iron and copper. J Endocrinol. 2017;234(3):R123–r134.
Xue X, Bredell BX, Anderson ER, Martin A, Mays C, Nagao-Kitamoto H, Huang S, Győrffy B, Greenson JK, Hardiman K, et al. Quantitative proteomics identifies STEAP4 as a critical regulator of mitochondrial dysfunction linking inflammation and colon cancer. Proc Natl Acad Sci U S A. 2017;114(45):E9608–e9617.
Liao Y, Zhao J, Bulek K, Tang F, Chen X, Cai G, Jia S, Fox PL, Huang E, Pizarro TT, et al. Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis. Nat Commun. 2020;11(1):900.
Li W, Yin X, Yan Y, Liu C, Li G. STEAP4 knockdown inhibits the proliferation of prostate cancer cells by activating the cGMP-PKG pathway under lipopolysaccharide-induced inflammatory microenvironment. Int Immunopharmacol. 2021;101(Pt B):108311.
Tang Y, Wang Y, Xu X, Sun H, Tang W. STEAP4 promoter methylation correlates with tumorigenesis of hepatocellular carcinoma. Pathol Res Pract. 2022;233:153870.
Mukherjee S, Kar A, Khatun N, Datta P, Biswas A, Barik S. Familiarity Breeds Strategy: In Silico Untangling of the Molecular Complexity on Course of Autoimmune Liver Disease-to-Hepatocellular Carcinoma Transition Predicts Novel Transcriptional Signatures. Cells. 2021;10(8):1917.
Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning-based multi-omics integration robustly predicts survival in liver Cancer. Clin Cancer Res. 2018;24(6):1248–59.
Ricciuti B, Wang X, Alessi JV, Rizvi H, Mahadevan NR, Li YY, Polio A, Lindsay J, Umeton R, Sinha R, et al. Association of High Tumor Mutation Burden in non-small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 2022;8(8):1160–8.
Kuo GH, Deangelis A, Emanuel S, Wang A, Zhang Y, Connolly PJ, Chen X, Gruninger RH, Rugg C, Fuentes-Pesquera A, et al. Synthesis and identification of [1,3,5]triazine-pyridine biheteroaryl as a novel series of potent cyclin-dependent kinase inhibitors. J Med Chem. 2005;48(14):4535–46.
Sunada S, Saito H, Zhang D, Xu Z, Miki Y. CDK1 inhibitor controls G2/M phase transition and reverses DNA damage sensitivity. Biochem Biophys Res Commun. 2021;550:56–61.
Tang JH, Yang L, Chen JX, Li QR, Zhu LR, Xu QF, Huang GH, Zhang ZX, Xiang Y, Du L, et al. Bortezomib inhibits growth and sensitizes glioma to temozolomide (TMZ) via down-regulating the FOXM1-Survivin axis. Cancer Commun (London, England). 2019;39(1):81.
Chen X, Chen Y, Ou Y, Min W, Liang S, Hua L, Zhou Y, Zhang C, Chen P, Yang Z, et al. Bortezomib inhibits NLRP3 inflammasome activation and NF-κB pathway to reduce psoriatic inflammation. Biochem Pharmacol. 2022;206:115326.
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–9.
Kim SY, Park SE, Shim SM, Park S, Kim KK, Jeong SY, Choi EK, Hwang JJ, Jin DH, Chung CD, et al. Bay 61-3606 sensitizes TRAIL-induced apoptosis by downregulating Mcl-1 in breast Cancer cells. PLoS One. 2015;10(12):e0146073.
Tubita A, Lombardi Z, Tusa I, Lazzeretti A, Sgrignani G, Papini D, Menconi A, Gagliardi S, Lulli M, Dello Sbarba P, et al. Inhibition of ERK5 elicits cellular senescence in melanoma via the cyclin-dependent kinase inhibitor p21. Cancer Res. 2022;82(3):447–57.
Acknowledgements
Not applicable.
Funding
We thank financial supports from the National Natural Science Foundation of China (grant no. 82203314) and the Wenzhou Municipal Science and Technology Bureau (grant no. Y20210247).
Author information
Authors and Affiliations
Contributions
DXF, XZ, YZ, and SSH analyzed the data and draft the manuscript. SSH designed the study and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
The histogram of the distribution of risk scores in the TCGA and GSE14520. Fig. S2. The validation of the prognostic risk model and the nomogram and calibration curve of the model in the GSE14520 cohorts. Fig. S3. The correlation between the expression of STEAP1, STEAP4 and risk score with particular etiology and liver fibrosis in the TCGA and GSE14520.
Additional file 2: Table S1.
Differentially expressed genes of the two prognostic risk group in the TCGA.
Additional file 3: Table S2.
Differentially expressed genes of the two prognostic risk group in the GSE14520.
Additional file 4: Table S3.
The GSEA analysis for TCGA.
Additional file 5: Table S4.
The GSEA analysis for GSE14520.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fu, D., Zhang, X., Zhou, Y. et al. A novel prognostic signature and therapy guidance for hepatocellular carcinoma based on STEAP family. BMC Med Genomics 17, 16 (2024). https://doi.org/10.1186/s12920-023-01789-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01789-0