
Abstract
Background
Achondroplasia is a congenital skeletal system malformation caused by missense variant of FGFR3 gene with an incidence of 1 per 20,000–30,000 newborns, which is an autosomal dominant inheritance disease. Despite similar imaging features, the homozygous achondroplasia is absolutely lethal due to thoracic stenosis, whereas heterozygous achondroplasia does not lead to fetal death.
Case presentation
A fetus with progressive rhizomelic short limbs and overt narrow chest was detected by prenatal ultrasound in the second trimester. Gene sequencing results of amniotic fluid sample indicated a rare missense variant NM_000142.4: c.1123G > T(p.Gly375Cys), leading to a glycine to cysteine substitution. Re-sequencing confirmed that it was a heterozygous variant, and thoracic stenosis was then confirmed in the corpse by radiological examination.
Conclusions
We identified a heterozygous variant of the FGFR3 gene as the rare pathogenic variant of severe achondroplasia in a fetus. Heterozygous variants of p.Gly375Cys may have a severe phenotype similar to homozygote. It’s crucial to combine prenatal ultrasound with genetic examination to differentiate heterozygous from homozygous achondroplasia. The p.Gly375Cys variant of FGFR3 gene may serve as a vital target for the diagnosis of severe achondroplasia.
Similar content being viewed by others


Background
Achondroplasia is a congenital skeletal system malformation caused by missense variant of FGFR3 gene with an incidence of 1 per 20,000–30,000 newborns, which is an autosomal dominant inheritance disease [1,2,3]. Variants in the FGFR3 gene lead to hyperactivation of tyrosine kinase, promoting multiple mitosis, such as carcinogenesis and overgrowth of skin, but inhibiting the proliferation and terminal differentiation of chondrocytes. This paradoxical phenomenon may be due to the activation of defense mechanisms that protect mammals from cancer. Because chondrocytes are the most intense target of the FGFR3 gene, the activation of defense mechanisms is particularly severe during chondrocyte activity (this has been thoroughly elaborated in references 1, 2, and 5). The study conducted by Di Rocco F in 2014 demonstrated that FGFR3 greatly affects both endochondral and intramembranous ossification, resulting in impaired development of craniofacial bones and stunted growth as the main clinical manifestations. Therefore, it affects both endochondral and intramembranous ossification [4, 5].
The phenotypic features of affected individuals include disproportionate short stature, rhizomelic shortening of the arms, a prominent forehead, midface hypoplasia, large skull roof, small skull base and spinal cord compression. Additionally, homozygous achondroplasia is absolutely lethal due to thoracic stenosis. Whereas heterozygous achondroplasia does not lead to fetal death. Radiologic images of the skull, spine, chest, and extremities reveal these characteristic features [1, 3, 6]. In this report, we describe a case of heterozygous achondroplasia with thoracic stenosis, which was diagnosed in the second trimester based on ultrasound features and genetic testing.
Case presentation
The case is of term female baby delivered by a gravida 2, parity 0(G2P0) at week 25, who was 29 years old and had an early miscarriage with unknown aetiology. The parents were healthy without family history of genetic diseases or history of infection and medication during the pregnancy. Prenatal ultrasound was firstly performed at 12 + 3w gestational age (GA), the thickness of nuchal translucensy was 0.13 cm and the crown-rump length was in accorded with the clinical gestational week. Non invasice prenatal genetic testing showed a low-risk gestation. At 19w GA, short fetal limbs were found by routine ultrasonography with the femur below -3SD. The biparental and fetal chromosome examination and whole-exon sequencing were then recommended. Ultrasonographic features at 22w GA indicated obviously short limbs, rhizomelic shortening of the hummers, the femur/abdominal circumference and femur/plantar length, which suggested pathogenic skeletal dysplasia. At 24 + 5w GA, the long bones of fetal limbs were obviously short and the condition was progressively aggravated (Tables 1 and 2). Narrow chest was found with a ratio of chest/abdominal circumference less than 0.89. The fetus was finally diagnosed with suspected achondroplasia by ultrasonography.
No significant abnormalities were found in biparental and fetal chromosomes. Since heterozygous achondroplasia with a similarly severe phenotype has never been reported previously, we then collected parental blood and fetal amniotic fluid exfoliated cells to perform whole exome sequencing of FGFR3 gene. As shown in Fig. 1, the sequencing results indicated that a single-base changed from G-to-T at codon 375, which caused a glycine to be replaced by a cysteine.

Sanger sequencing chromatograms showing a missense variant c.1123G > T in the affected fetus in comparison to her unaffected parents
Given the genetic test reports and ultrasound features (Figs. 2 and 3), this case was finally diagnosed with severe achondroplasia. After prenatal consultation, the couple requested to terminate the pregnancy. The physiological characteristics and radiographic evidence of the corpse (Fig. 4) confirmed the final diagnosis.

The sagittal image of narrow chest at 24 + 5w GA

Transverse section of narrow chest and short limbs at 22w GA and 24 + 5w GA

Physiological characteristics and radiographic evidence of the corpse. a Physiological characteristics of the corpse: disproportionate shortening of long bones, frontal bossing, midface hypoplasia, and protuberant abdomen, talipes equinovarus in the right side. b X-ray image showing disproportionate shortening of long bones, large skull roof and small skull base. c Computerized tomography 3D bone reconstruction showing narrow thoracic shape as a bell, flat midface and spine. d X-ray image showing narrow chest and flat vertebrae. e the trident hand
Discussion
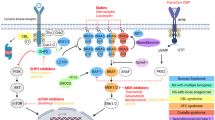
The FGFR3 (fibroblast growth factor receptor 3) have an extracellular ligand-binding domain, a transmembrane domain and an intracellular domain that contains a split tyrosine kinase subdomain [1, 2]. Variants in the FGFR3 gene at different locations result in varying degrees of skeletal deformities, including thanatophoric dysplasia, achondroplasia, and hypochondroplasia (ordered by severity). Thanatophoric dysplasia is typically characterized by a cloverleaf skull, an extremely narrow thorax, or long bones that are extremely short and curved. The phenotypic features of affected achondroplasia individuals include disproportionate short stature, rhizomelic shortening of the arms, a prominent forehead, midface hypoplasia, large skull roof, small skull base and spinal cord compression. In contrast, the symptoms of hypochondroplasia are usually milder, and shortened femur is only occasionally detected prenatally. We have summarized the variant sites that have been reported to cause thanatophoric dysplasia, achondroplasia, or hypochondroplasia (Fig. 5). 98% of achondroplasia patients are caused by the variants of p.Gly380Arg in FGFR3, while the remaining 1% is attributed to other variants. Based on the current reports of achondroplasia, we found none of the heterozygotes showed thoracic stenosis according to the phenotypic analysis. Three cases were reported to be caused by the variant of p.Gly375Cys, but the phenotype was only described in two of them [7,8,9]. These two patients were diagnosed at two years old and four days after birth respectively. They shared typical imaging features and vertebral flattening, but did not have apparent narrow chests [8, 9].

Topology map of FGFR3 with major sites of variant. ACH achondroplasia; TDI thanatophoric dysplasia type I.; HYP hypochondroplasia; TKp/d proximal and distal tyrosine kinase domains; TM transmembrane
Heuertz S. suggested that the cysteine residues in the extracellular domain can cause excess disulfide bond formation, leading to a tertiary structure change of FGFR3, further activating tyrosine kinase, resulting in more severe phenotypes [10]. This mechanism may be one of the possible causes of the severe phenotype in this case, as the variant of p.Gly375Cys (indicated by the red star in Fig. 5) also creates additional cysteine residues.
Traditionally, the diagnosis of achondroplasia is based on genetic examination and radiological features [2]. Prenatal ultrasound serve as a routine repeatable imaging method providing additionally valuable information. Although the surviving achondroplasia fetuses have a low life satisfaction due to abnormal appearance and progressive spinal pain [11, 12], some families are still willing to accept such children with mild symptoms who are expected to have a nearly normal lifespan with short femurs in the third trimester. Therefore, accurate prenatal diagnosis and risk assessment of achondroplasia are important.
In conclusion, it is crucial to combine prenatal ultrasound with genetic examination to fully evaluate the severe phenotype of heterozygous achondroplasia, and the variant of p.Gly375Cys may serve as a vital target for the diagnosis.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- FGFR3 :
-
Fibroblast growth factor receptor 3
- GA:
-
Gestational age
- TDI:
-
Thanatophoric dysplasia type I
References
Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370(9582):162–72.
Hoover-Fong JE, Alade AY, Hashmi SS, et al. Achondroplasia Natural History Study (CLARITY): a multicenter retrospective cohort study of achondroplasia in the United States. Genet Med. 2021;23(8):1498–505.
Patel MD, Filly RA. Homozygous achondroplasia: US distinction between homozygous, heterozygous, and unaffected fetuses in the second trimester. Radiology. 1995;196(2):541–5.
Di Rocco F, Biosse Duplan M, Heuzé Y, Kaci N, Komla-Ebri D, Munnich A, Mugniery E, Benoist-Lasselin C, Legeai-Mallet L. FGFR3 mutation causes abnormal membranous ossification in achondroplasia. Hum Mol Genet. 2014;23(11):2914–25.
Krejci P. The paradox of FGFR3 signaling in skeletal dysplasia: why chondrocytes growth arrest while other cells over proliferate. Mutat Res Rev Mutat Res. 2014;759:40–8.
Shelmerdine SC, Brittain H, Arthurs OJ, Calder AD. Achondroplasia: Really rhizomelic? Am J Med Genet A. 2016;170(8):2039–43.
Ikegawa S, Fukushima Y, Isomura M, Takada F, Nakamura Y. Mutations of the fibroblast growth factor receptor-3 gene in one familial and six sporadic cases of achondroplasia in Japanese patients. Hum Genet. 1995;96(3):309–11.
Nishimura G, Fukushima Y, Ohashi H, Ikegawa S. Atypical radiological findings in achondroplasia with uncommon mutation of the fibroblast growth factor receptor-3 (FGFR-3) gene (Gly to Cys transition at codon 375). Am J Med Genet. 1995;59(3):393–5.
Superti-Furga A, Eich G, Bucher HU, et al. A glycine 375-to-cysteine substitution in the transmembrane domain of the fibroblast growth factor receptor-3 in a newborn with achondroplasia. Eur J Pediatr. 1995;154(3):215–9.
Heuertz S, Le Merrer M, Zabel B, et al. Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. Eur J Hum Genet. 2006;14(12):1240–7.
Hoover-Fong J, Scott CI, Jones MC; COMMITTEE ON GENETICS. Health supervision for people with achondroplasia. Pediatrics. 2020;145(6):e20201010.
Legeai-Mallet L, Savarirayan R. Novel therapeutic approaches for the treatment of achondroplasia. Bone. 2020;141: 115579.
Acknowledgements
The authors thank the family for participating and supporting this study.
Funding
This work was funded by the Chongqing Youth and Middle-aged Medical Advanced Talents Studio (ZQNYXGDRCGZS2021004).
Author information
Authors and Affiliations
Contributions
PL designed the study and revised the manuscript; SC drafted the manuscript; SC, HD, YL and YZ acquired, analyzed, and interpreted the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods were conducted according to relevant guidelines and regulations, in compliance with the "Regulations on Ethical Review of Biomedical Research Involving Human Subjects" (Order No. 11 of the National Health and Family Planning Commission of the People's Republic of China), the "Quality Management Standards for Clinical Trials of Medical Devices" (Order No. 25 of the State Food and Drug Administration and the National Health and Family Planning Commission of the People's Republic of China), the Helsinki Declaration of the World Medical Association, and the ethical principles of the International Ethical Guidelines for Biomedical Research Involving Human Subjects of CIOMS. The study was approved by the Ethics Committee of The First People’s Hospital of Chongqing Liang Jiang New Area. Written informed consent to participate was obtained from the fetal parents.
Consent for publication
Written informed consent for publication of identifying images and other personal or clinical details was obtained from the fetal parents. And the copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, S., Dong, H., Luo, Y. et al. Heterozygous variant in FGFR3 underlying severe phenotypes in the second trimester: a case report. BMC Med Genomics 16, 80 (2023). https://doi.org/10.1186/s12920-023-01517-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01517-8