
Abstract
Various studies showed that the effect of immune activation is pro-atherogenic and coronary heart disease (CHD) should therefore be considered an autoimmune disease. This study aimed to identify potential immune-related biomarkers, pathways, and the potential regulatory networks underlying CHD. Differentially expressed genes (DEGs) between CHD and control samples were determined by analyzing GSE71226 and GSE9128. The overlapping differential expression immune-related genes (DE-IRGs) for CHD were identified by analyzing the ImmPort database and two GEO databases. A total of 384 DE-IRGs were identified. Subsequently, comprehensive enrichment analyses suggested that DE-IRGs were enriched in immune-related pathways, including autoimmune thyroid disease, the intestinal immune network for IGA production, and downstream signaling events of B cell receptors. The signature of DE-IRGs was validated using an external independent dataset GSE20681 (AUC = 0.875). Furthermore, we conducted protein–protein interaction network analysis and identified eight hub genes, which were most enriched in regulation of defense response, NF-κB signaling pathway, regulation of JNK cascade, and regulation of cytokine production. Moreover, networks of miRNAs-mRNAs and transcription factors (TFs)-mRNA underlying the integrated data were established, involving eight miRNAs and 76 TF-targeting hub genes. Ultimately, 17 SNPs in miRNA-mediated gene networks were identified. We screened potential immune-related genes in CHD and constructed miRNA-mRNA-TF and SNP-miRNA networks, which not only provide inspired insights into the occurrence and the molecular mechanisms of CHD but also lay a foundation for targeting potential biomarkers using immunotherapy and for understanding the molecular mechanisms of CHD.
Similar content being viewed by others

Introduction
Coronary heart disease (CHD) is considered a serious threat to human health worldwide, which caused 365,914 deaths in 2017. Approximately 2 in 10 deaths due to CHD are of adults less than 65 years of age [1]. Millions of patients with chest pain symptoms are commonly admitted to hospitals in high-income countries every year. Approximately 50% of them are diagnosed with CHD, including stable angina, unstable angina, and acute myocardial infarction [2]. In China, the number of CHD patients is 11 million in 2017; the mortality rate was higher in rural residents (122.04/100,000) as compared to that in urban residents (115.32/100,000). The mortality rate in males is higher than that in females, showing an upward trend since 2012 [3, 4].
Comprehensive studies on the mechanism of CHD have recognized that the effect of immune activation is pro-atherogenic, and CHD should therefore be considered a type of autoimmune disease [5,6,7]. Various cells and molecules involved in the immune response constitute a complex immune mechanism of cooperation and interaction [8,9,10]. Multiple classic inflammation-related pathways are involved in the survival, proliferation, and migration of immune cells [11,12,13,14,15]. However, there is currently no systematic model to evaluate the immune environment of CHD patients.
CHD was precisely controlled by microRNAs (miRNAs) at the post-transcriptional level and transcription factors (TFs) at the transcriptional level [16, 17]. MiRNAs are a type of endogenous small non-coding RNAs of 20–24 nucleotides in length that are mainly involved in translational repression and/or mRNA destabilization. The widespread availability of high-throughput technologies, including microarray profiling and genome sequencing, has enabled the screening and discovery of an increasing number of differentially expressed (DE) miRNAs that are involved in multiple cardiac transcriptional and signal regulatory pathways [18,19,20]. TFs are fundamental factors in transcriptional and post-transcriptional regulation, which involves physiological and pathological processes of cells by binding to particular DNA sequences [21]. Although an increasing number of miRNAs and TFs are involved in the immune regulation of CHD, potential miRNA biomarkers and molecular mechanisms remain largely unknown.
In our study, we screened differential expression immune-related genes (DE-IRGs) for CHD using bioinformatics analysis of multiple microarray databases. Thereafter, we further revealed the hub genes from a protein–protein interaction (PPI) network that was constructed. Furthermore, we constructed miRNA-TF networks to further clarify the crucial immune mechanisms underlying CHD. The newly established immune-related prognostic model was further validated in an independent Gene Expression Omnibus (GEO) database. Ultimately, we analyzed the SNPs that may be involved in the transcriptional regulation of target miRNAs. This study revealed the integrative network of DE-IRGs genetically related to CHD (Fig. 1), developing a theoretical framework applicable to the systems biology for shedding light on the molecular mechanisms underlying CHD. In the future, CHD therapy may turn to specific molecules and cells involved in the immune system, such as monoclonal antibodies against various cytokines and adhesion factors, and gene therapy, to open new ways for the prevention and treatment of CHD.

Flow chart of the study
Materials and methods
Microarray data download and preprocessing
All analysis steps are performed using R software (version 4.0.3, https://www.r-project.org/). The gene expression profiles GSE71226 [22], GSE9128 [23], and GSE20681 [24, 25] were retrieved from GEO (https://www.ncbi.nlm.nih.gov/geo/) [26] (Table 1). All samples were from the peripheral blood of Homo sapiens. The platform for GSE71226 was GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array, which included three healthy individuals and three CHD patients. The platform for GSE9128 was GPL96 [HG-U133A] Affymetrix Human Genome U133A Array, which collected samples from three healthy individuals and four patients with ischemic cardiomyopathy. The platform for GSE20681 was GPL4133 Agilent-014850 Whole Human Genome Microarray 4 × 44 K G4112F, which contained samples from 4 healthy individuals (CHD prognostic index less than 5) and 36 CHD patients (CHD prognostic index greater than 80) [27]. Series matrix files of GSE71226 and GSE9128 were downloaded to screen and verify differentially expressed genes (DEGs) involved in the coronary subjects.
We further analyzed the original files through background correction and quantile normalization via the "normalize between arrays" function in the limma package (3.52.2) [28]. The hclust package was used for clustering with the default method (“complete”) and “fviz_dend function” in the factoextra package was used for visualization. Next, we checked the batch effect by principal component analysis (PCA) through the "remove batch effect" function in the limma package. The effect of inter-sample correction was shown by the PCA map using the “fviz_pca_ind” function in the FactomineR package [29].
Identification of differentially expressed immune-related genes (DE-IRGs)
We screened the DEGs from two GEO datasets using the limma package, with the threshold criterion of |log2FC|> 1 and adj. p value < 0.05. The volcano plots and the heatmaps of DEGs were plotted using the ggplot2 package [30] and the pheatmap package (1.0.12) [31], respectively. Thereafter, we collected the data on immune-related genes from the Immunology Database and Analysis Portal (ImmPort Database, https://immport.niaid.nih.gov) [32]. The overlapping DE-IRGs were screened from the GEO dataset and the ImmPort database for further analysis, and the online tool Draw Venn Diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/) was used to display these overlaps.
Functional and pathway enrichment analysis
Gene ontology (GO) [33] and Kyoto Encyclopedia of Genes and Genomes analysis (KEGG) [34] enrichment analysis of DE-IRGs were conducted using the clusterProfiler package (4.4.4) [35]. GO analysis includes the following three criteria: molecular function (MF), cellular component (CC), and biological process (BP). The enrichplot package (1.16.1) [36] was used to draw circles, histograms, and bubble diagrams. An adjusted p value (from Benjamin-Hochberg method) of less than 0.05 was defined as statistically significant. The data were visualized via the clusterProfiler plug-in (significance was indicated by p < 0.05 and q-value < 0.05), CluePedia plug-in [37], and ClueGO plug-in [38] in Cytoscape software (http://www.cytoscape.org/) (kappa score ≥ 0.4).
Gene set enrichment analysis (GSEA) and xPierGSEA
The biological pathways that were significantly altered in CHD samples as compared to the control samples were determined using GSEA. GSEA [39] was performed using cluster profiler package, with c2.all.v7.4.symbols.gmt as the reference gene set, and analyzed using the GSEA function from the GSEA database and the gseaplot2 function of the enrichplot package. False discovery rate (FDR) < 0.25 and p value < 0.05 were set as the cut-off criteria. xPierGSEA (2.9.0) was implemented by the Pi package [40], providing us with other alternatives to screen significantly differential biological functions in CHD.
Gene set variation analysis (GSVA)
GSVA [41] was carried out to explore the expression matrix of DE-IRGs using the GSVA package, which does not need to group samples in advance and calculates the enrichment score of the specific gene set in each sample. C2: curated gene sets were selected as the reference gene set, with the t value of GSVA scores 0.5 as the cut-off value standard.
Construction of gene feature selection and random forest (RF) diagnosis model
To improve the performance of the diagnosis model, specific variable genes related to the classification of CHD outcomes were screened by the Boruta feature selection method (18). The RF diagnostic model classifier was implemented using a random forest package (1.18.1) [42], which distinguished patients with and without CHD in the combined data set (GSE71226, GSE1128). DE-IRGs were selected as disease-associated genes for follow-up model building. Next, the average mistranscription rates of all genes were calculated according to the out-of-bag data. The best variable (mtry) of binary trees in the node was set to 2, and 1000 was selected as the best number of trees in the RF. Thereafter, the RF model was built and the importance value was obtained by employing the Gini coefficient method. The accuracy of the RF model was tested by using the tenfold cross model, and the accuracy of the model tended to be stable when the number of genes was confirmed. The pROC package (1.17.0) [43] was used to validate AUC classification performance. External validation of the immune-related signature was conducted using an external dataset (GSE20681).
Construction of PPI network and identification of hub genes
To further study the biological function of DE-IRGs, we constructed a PPI network (https://string-db.org/) [44, 45] and cell landscape (https://metascape.org/) [46] with a combined score ≥ 0.4. Moreover, we used the Cytoscape software [47, 48] to visualize the PPI network. We employed the Molecular Complex Detection (MCODE) [49], a plug-in of Cytoscape, to identify vital modules (MCODE score ≥ 4). We applied the CytoHubba [50], another plug-in of Cytoscape, to indicate essential nodes in the network and hub genes involved in the PPI network.
Construction of a miRNA-DE-IRGs-TF regulatory network
We used NetworkAnalyst (https://www.networkanalyst.ca/) [51] to analyze miRNA databases and predict the interaction between miRNA and DE-IRGs using TargetScan [52] and miRDB [53, 54]. Predicted_Targets_ predictions.txt file was downloaded from the TargetScan official website, and miRDB_v6.0_prediction_result.txt was collected from miRDB. Furthermore, we predicted the interaction of DE-IRGs and transcription factors via the TRRUST database (https://www.grnpedia.org/trrust/) [55]. Ultimately, we visualized miRNA-gene and TF-gene networks using Cytoscape software.
Predictive analysis of SNP affecting miRNA binding to DE-IRGs
We used the miRNASNP database [56] to predict the SNPs in the seed region of the potential candidate miRNAs. We identified genes affected by SNP mutations in the seed region of potential candidate miRNAs and then selected the DE-IRGs to form a list of miRNA-SNP-DE-IRGs combinations. Finally, we constructed the miRNA-SNP-DE-IRGs interaction network according to the list. If miRNA was not completely labeled 3p/5p or two highly homologous miRNAs appeared, both of them were searched.
Results
Identification of DEGs in CHD
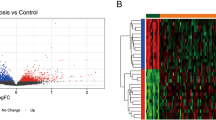
The gene expression profiles of GSE71226 and GSE9128 datasets were merged, and standardized, and the batch effects were removed. Gene expression profiles before and after standardization are displayed in box plots (Fig. 2A, B). Gene expression profiles before and after batch correction are displayed by Cluster dendrogram (Fig. 2C, D) and PCA (Fig. 2E, F), which showed that the control group and CHD group were clustered into two groups after batch correction, indicating the heterogeneity of sample sources. After data were pre-processed, the volcano plot indicated that 4196 DEGs were extracted from two gene expression profiles with 2070 DEGs being up-regulated and 2126 DEGs being down-regulated (Fig. 3A). Then we chose these DEGs with |log2FC|> 4 and adj. p value < 0.001 for visualization in the heatmap (Fig. 3B).

Preprocessing of GSE71226 and GSE9128. A and B Box plots of DEGs before and after standardization of two datasets. C and D Cluster dendrogram of DEGs before and after batch correction. E and F PCA plots of datasets before and after batch correction

Identification of DEGs and DE-IRGs. A Volcano plots of DEGs. 2070 red points represent upregulated genes, 2126 blue points indicate downregulated genes, and 10,002 grey points represent unchanged genes. B Heatmaps of DEGs with |log2FC|> 4 and adj. p value < 0.001. Blue represents the case group and red represents normal control. C Venn Diagram demonstrates the intersection of DEGs and immune-related genes
GO and KEGG enrichment analyses
A total of 348 overlapping DE-IRGs (Fig. 3C), the intersection of DEGs and ImmPort genes, integrated by the Venn Diagram online tool were identified (Fig. 3C). GO term and KEGG enrichment analyses were performed on DE-IRGs. The significantly top five GO terms were positive regulation of cytokine production, positive regulation of leucocyte cell−cell adhesion, external side of the plasma membrane, MHC class II protein complex, and immune receptor activity. For BP, DE-IRGs were significantly enriched in response to molecule of bacterial origin, response to lipopolysaccharide, cell chemotaxis, positive regulation of protein kinase B signaling, positive regulation of cytokine production, and positive regulation of leukocyte cell–cell adhesion. For CC, DE-IRGs were significantly enriched in the external side of the plasma membrane, MHC class II protein complex, secretory granule lumen, cytoplasmic vesicle lumen, and vesicle lumen. For MF, DE-IRGs were significantly enriched in receptor-ligand activity, signaling receptor activator activity, cytokine activity, growth factor activity, and cytokine receptor binding (Fig. 4A, B). The top five significant enrichment pathways indicated by KEGG analysis were Cytokine-cytokine receptor interaction, Chemokine signaling pathway, Viral protein interaction with cytokine and cytokine receptor, T cell receptor signaling pathway, and Rheumatoid arthritis (Fig. 4C, D).

Functional enrichment analyses of DE-IRGs. A GO enrichment analysis with “eGO” function (OrgDb = org.Hs.eg.db, pvalueCutoff = 0.05, qvalueCutoff = 0.25, pAdjustMethod = "BH") in the clusterProfiler package. The results were visualized with the “cnetplot” function in the enrichplot package and the color of the dot marks the logFC of the gene. B GO enrichment analysis. The top 12 results were visualized with the “barplot” function in the enrichplot package. The x-axis and y-axis represent the gene ratio, and GO terms, respectively, and the color represents the corrected p value. C KEGG pathway enrichment analysis with “enrichKEGG” function (organism = "hsa", keyType = "kegg", pvalueCutoff = 0.05, pAdjustMethod = "fdr") in the clusterProfiler package. The top 12 results were visualized with the “dotplot” function in the enrichplot package and the x-axis and y-axis represent the gene ratio and pathway, respectively. D Correlation between KEGG enrichment pathways with the “upsetplot” function in the enrichplot package and the line represents the correlation between pathways
GSEA, xPierGSEA, and GSVA
GSEA indicated that the most significantly enriched gene sets included the intestinal immune network for IgA production and KEGG autoimmune thyroid disease (Fig. 5A–D). The xPierGSEA showed that DE-IRGs play a significant role in the immune mechanism of CHD, especially in the T-cell receptor (TCR) signaling pathway of Na ï CD4 + T cells (FDR = 5.2e−3) (Fig. 5E). GSVA indicated that the same cellular mechanism may occur in multiple diseases other than CHD, such as type II diabetes mellitus, breast cancer basal up, etc. (Fig. 5F).

Enrichment pathway analysis. A GSEA plots. The x-axis represents the proportion of DEGs, the color represents the corrected p value (corrected by Benjamin-Hochberg test), and the dot represents the number of enriched genes. B–D Predictive immune-related pathways in CHD via GSEA. B Autoimmune thyroid disease. C Intestinal immune network for IgA production. D Downstream signaling events of B cell receptor. E xPierGSEA of the top four significant pathways. The leading edge on the left chart reveals leading targets and the bar chart on the right shows the FDR value (numerical value) and NES value. F GSVA (blue: T-score > 0.5, green: T-score < 0.5, gray: not significant)
Diagnosis model construction and analysis of DE-IRGs
Eight hub genes (LTBR, ACKR1, FPR2, PTGS2, JUN, RASGRP1, CYLD, and TNFRSF1A) from the DE-IRGs were screened by Boruta algorithms feature selection (Fig. 6A). The Metascape online analysis demonstrated that the main functions were predominantly enriched in regulation of defense response, NF-kappa B signaling pathway, regulation of JNK cascade, and regulation of cytokine production (Fig. 7C, D). The cluster profiler showed that the main functions were enriched in the NF kappa B signaling pathway and TNF signaling pathway (Fig. 7B).

Hub gene screening, diagnostic model construction, and validation of DE-IRGs. A Box graph of random forest selection. The green marker is an important characteristic gene. B Error rate under the random forest diagnosis model gradually decreases with the increase of ntree, and finally tends to be stable. C The accuracy under tenfold crossover model tends to be stable (> 0.95) when the fourth gene is included. D. Random forest assessment of eight hub genes. FPR2 displays were much more important than other genes. E ROC curve and an area under ROC (AUC) value of GSE71226 data set (AUC = 0.875). F ROC-AUC value of GSE20681 data set (AUC = 0.751)

Functional enrichment analysis of hub genes and miRNA-DE-IRGs-TF regulatory network. A PPI network of DE-IRGs. B Cluster profiler showed that the main functions were enriched in NF-kappa B signaling pathway and TNF signaling pathway. C Metascape network diagram, Metascape functional enrichment correlation analysis of hub gene. D Metascape histogram. The first six functions with the lowest p value were selected. E miRNA-DE-IRGs-TF regulatory network. F miRNA-SNP-DE-IRGs network
In the construction of the random forest diagnosis model, eight hub genes of the Boruta feature selection were recruited to establish the signature (Fig. 6B). The tenfold model verified the accuracy of the random forest model. The accuracy increased concordantly with the growing proportion of gene features. Our model accuracy tended to be stable when the fourth gene was included (> 0.95) (Fig. 6C). According to the results of Mean Decrease Gini, the eight characteristic genes were ranked in decreasing order of importance as FPR2, RASGRP1, LTBR, ACKR1, PTGS2, JUN, TNFRSF1A, and CYLD (Fig. 6D). AUC was used to evaluate the random forest model. The AUC value of GSE71226 is 0.875 (Fig. 6E) and the AUC value of external validation set GSE20681 is 0.751 (Fig. 6F), indicating that the model has high efficiency in predicting the diagnosis and classification of CHD.
PPI network construction and hub gene selection
The PPI network included 18 nodes and 28 edges. The eight colored nodes represented hub genes, and the gray nodes are extended genes with matching confidence cut-off (cut-off score is 0.4) by the String app expand network (Fig. 7A). The results show that FPR2 is at the center of the hub gene and extended gene, revealing that it not only serves as a bridge to communicate with other functional networks but also plays a more important biological role than other key genes in the network.
Construction of miRNA-DE-IRGs-TF network
Based on two miRNA target gene databases (TargetScan and miRcode), eight miRNAs were obtained, which were hsa-miR-411, hsa-miR-371-5p, hsa-miR-292-5p, hsa-miR-186, hsa-miR-495, hsa-miR-543, hsa-miR-340-5p, and hsa-miR-433. TRRUST transcription factor database predicted the targeted DEGs for TFs. Overall, 76 pairs of TF-DE-IRGs interaction groups were found. Among them, PTGS2 and JUN predicted the most TFs, and the STAT family TFs (STAT1, STAT2, STAT3, and STAT6) matched PTGS2 the most (Fig. 7E). FPR2 and TNFRSF1A did not predict matching TFs.
Predictive analysis of SNP-influenced miRNAs and DE-IRGs
Potential candidate miRNAs targeting DE-IRGs were analyzed using miRNASNP to determine how the SNP variants in the seed region of the miRNAs affected their binding to the 3' UTR region of DE-IRGs, and to construct miRNA-SNP-DE-IRGs interaction networks (Fig. 7F). A total of 17 SNP locations were identified (rs1165649618, rs753033941, rs111835650, rs1353902015, rs1185000692, rs1049004182, rs1480794990, rs774879764, rs767556394, rs1292420312, rs749991806, rs1445781653, rs905233879, rs778897748, rs1033402586, rs1177339324, and rs1157324828), which may play regulatory roles in the binding of miRNAs to DE-IRGs. Among them, 13 SNP mutations increased the number of miRNAs that could target the 3'-UTR of the DE-IRGs (gain), whereas 11 SNP mutations had the opposite effect (loss) (Table 2).
Discussion
A growing body of evidence has demonstrated that immune-related genes (IRGs) play significant roles in CHD. Various cellular and molecular-mediated immune mechanisms participate in the occurrence of CHD and promote the development and deterioration of CHD [57, 58]. Although it has been investigated widely, the molecular mechanism and critical regulators involved in IRGs for evaluating CHD patients remain to be further elucidated, which will help to explore the complex pathological process of the formation and development of CHD. In this study, 348 DE-IRGs were identified as significant with respect to CHD by integration analysis of two mRNA microarray datasets and ImmPort database. Next, the intersection of GO term and KEGG pathways enrichment analysis indicated that immune-related terms and pathways were highly enriched. The results suggested that immune-related molecular functions and crucial biological processes are involved in CHD pathogenesis. Also, GSEA enriched pathways are mainly involved in the internal immune network for IgA production and autoimmune theroid disease pathways. The xPierGSEA also showed that DE-IRGs played a vital role in the immune mechanism of CHD, being especially involved in the TCR signaling pathway of naïve CD4 + T cells.
Based on the 348 DE-IRGs, we built a PPI network and obtained eight hub genes using MCODE analysis, including FPR2, RASGRP1, LTBR, ACKR1, PTGS2, JUN, TNFRSF1A, and CYLD to explore the regulatory networks and crucial pathways underlying CHD. Our results demonstrated that FPR2 is at the center of the network including characteristic genes and extended genes, indicating that it might play a potential role in immunoregulation processes. FPR2, a seven-transmembrane G protein-coupled receptor, plays multifaceted functions, such as a role in sensing bacteria and modulation of immune responses [59], colonic epithelial homeostasis, inflammation, and tumorigenesis [60]. PTGS2 was confirmed to suppress cell viability [61] and proliferation [62] of cells by activating the AKT/NF-κB pathway [63]. Overexpression of TNFRSF1A promoted the activation of the NF-κB signaling pathway and plays a potential regulatory role in neuronal cell damage [64]. The variant in TNFRSF1A is involved in Tumor Necrosis Factor Receptor-Associated Periodic Syndrome (TRAPS), and can therefore be used as a novel pathogenic therapeutic target [65]. CYLD is a tumor suppressor protein. Studies showed that the loss of function of CYLD can release the polyubiquitin chain in the ubiquitinated substrate, which can then inhibit the IκBα Kinase complex and then inhibit the activation of the NF-κB signaling pathway, which is a negative regulator of this signaling pathway [66]. The relationship between the abovementioned hub genes and the immune-related molecular mechanism of CHD has not been confirmed before. In this study, several hub genes were found to be involved in the activation or inhibition of the NF-κB and TNF pathways. These findings are consistent with the Metascape analysis enrichment result, which illustrated that the eight hub genes were mainly enriched in the regulation of defense response, NF-κB signaling pathway, regulation of JNK cascade, and regulation of cytokine production. As a key and strictly regulated regulator, the NF-κB family of transcription factors is responsible for immune development, immune responses, and inflammation. The false regulation of NF-κB has been implicated in a wide range of diseases ranging from inflammatory to immune disorders [67]. TNF-α, mainly secreted by macrophages and monocytes, can directly damage vascular endothelial cells, cause a pathological inflammatory reaction, reduce the stability of atherosclerotic plaque, lead to the rupture of atherosclerotic plaque, and indirectly and directly accelerate the formation and development of atherosclerotic plaque [68].
CHD is a complicated disease involving various signaling pathways, protein-coding genes, numerous transcription factors, and post-transcriptional regulation of heart morphogenesis, cardiac development, and cardiac muscle cell differentiation and contractility. It is well known that miRNAs control gene expression mostly at the post-transcriptional level, whereas TFs are involved in the activation or inhibition of transcription at a pre-transcriptional level. Studies have implicated the abnormalities in miRNA expression in CHD [69]: miR-34a protects endothelial function in CHD patients [70]; miR-23a improves restoration of blood flow in the ischemic region [71]; miR-135b and miR-499a induce the proliferation of endothelial cells and vascular smooth muscle cell [72]; miR502 suppresses autophagy [73]; miR-33/33b lowers low-density lipoproteins and raises high-density lipoproteins concentrations [74]; miR-939 in angiogenesis [75]; miR-155 prevents the occurrence and development of atherosclerosis and CHD [76]; miR-126 substantially prevents atherosclerotic cardiovascular diseases [77]; miR-21 prevents the formation of neointimal and restenosis of arteries after angioplasty [78]; miR-146 enhances the collateral coronary circulation in ischemic tissue [79]. In this study, we constructed miRNA-TF co-regulatory networks based on candidate immune-related miRNAs, TFs, and hub genes, and performed network analysis to obtain consensus hub nodes, modules, and motifs involved in CHD. Based on two miRNA target gene databases (TargetScan and miRcode), eight miRNAs were obtained, which were hsa-miR-411, hsa-miR-371-5p, hsa-miR-292-5p, hsa-miR-186, hsa-miR-495, hsa-miR-543, hsa-miR-340-5p, and hsa-miR-433, and they were all target genes. Through the prediction of the trust transcription factor database, 76 pairs of TF-DE-IRGs interaction groups were found. PTGS2 and Jun predicted the most transcription factors, and STAT family transcription factors (STAT1, STAT2, STAT3, and STAT6) matched PTGS2 best. Cluster analysis of the miRNA-DE-IRGs-TF regulatory network revealed that its main functions were promotion or inhibition of the NF-κB signaling pathway and TNF signaling pathway. Among eight miRNAs, miRNA-292-5p [80, 81], which participates in myocardial ischemia–reperfusion injury by activating the peroxisome proliferator-activated receptor-α/-γ (PPARα/PPARγ)-dependent signaling pathway, has been studied since 2018. Recently, several studies have provided evidence that cardiomyocyte-enriched miR-186 contributes to cardiac injury and myocardial infarction as well as the progression of atherosclerosis [82]. The remaining six miRNAs were not retrieved for in-depth study.
Recent studies revealed that miRNA-related Single Nucleotide Polymorphisms (SNPs) might affect disease susceptibility and phenotypes in an SNP-genotype-dependent manner by modifying miRNA regulation [83, 84]. In our study, according to the function enrichment prediction of characteristic genes in Metascape, 17 SNPs were identified. They may play a key role in the binding of miRNAs and DE-IRGs. Most detected SNPs have not been studied yet. Therefore, we recommend combining miRNA expression profiling analysis with genome-wide SNPs to identify potential disease biomarkers for future research.
However, this study has several limitations. Firstly, a comprehensive elucidation of the molecular mechanisms should be conducted to understand the factors underlying the occurrence and development of CHD. Individual samples with varying degrees of CHD need to be collected and studied. Secondly, the low number of patients analyzed in this study likely introduced bias. Larger sample size and comprehensive analysis of the interactions between environmental and genetic factors may better reveal the roles of hub genes and pathways in CHD pathogenesis. Finally, further bioinformatics analyses and validation assays are required for elucidating the gene function and biological mechanisms of the predicted genes in CHD.
Tremendous progress has been made in understanding molecular mechanisms of CHD in vitro, ex vivo, and in vivo. However, we should deepen our understanding of CHD pathogenesis and undertake further investigation to find more feasible treatments. The complex regulatory network involving hub genes, miRNAs, and TFs will be an exciting new field to explore and will shed new light on CHD development. We aimed to discover novel markers that would be helpful in diagnosis, prognosis, prediction, and immune microenvironment detection in CHD patients, and have been successful in finding some very promising leads. Further research in this area will help explore the formation and development of CHD.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. The datasets generated and/or analysed during the current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/.
References
Benjamin EJ, Muntner P, Alonso A, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–528.
Roth GA, Mensah GA, Johnson CO, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020;76(25):2982–3021.
Summary of China cardiovascular health and disease report 2019. Chin J Geriatr Res. 2020;7(04):4-15.
Commission NH. China health statistical yearbook. Beijing: China Union Medical University Press; 2019. p. 284.
Shoenfeld Y, Sherer Y, Harats D. Artherosclerosis as an infectious, inflammatory and autoimmune disease. Trends Immunol. 2001;22(6):293–5.
Hansson GK. Atherosclerosis–an immune disease: the Anitschkov lecture 2007. Atherosclerosis. 2009;202(1):2–10.
Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012;18(28):4266–88.
Hilgendorf I, Gerhardt LM, Tan TC, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114(10):1611–22.
Wang RH, He JP, Su ML, et al. The orphan receptor TR3 participates in angiotensin II-induced cardiac hypertrophy by controlling mTOR signalling. EMBO Mol Med. 2013;5(1):137–48.
Hahn RT, Hoppstädter J, Hirschfelder K, et al. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis. 2014;234(2):391–400.
Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25(10):1576–88.
Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12(1):10–7.
Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5(1):56.
Seldin MM, Meng Y, Qi H, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc. 2016;5(2):e002767.
Linton MF, Moslehi JJ, Babaev VR. Akt signaling in macrophage polarization, survival, and atherosclerosis. Int J Mol Sci. 2019;20(11):2703.
Condorelli G, Latronico MV, Cavarretta E. microRNAs in cardiovascular diseases: current knowledge and the road ahead. J Am Coll Cardiol. 2014;63(21):2177–87.
Murawska M, Brehm A. CHD chromatin remodelers and the transcription cycle. Transcription. 2011;2(6):244–53.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language. Cell. 2011;146(3):353–8.
Olson EN. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci Transl Med. 2014;6(239):239ps3.
Fic P, Kowalczuk K, Grabarska A, Stepulak A. MicroRNA: a new diagnostic tool in coronary artery disease and myocardial infarction. Postepy Hig Med Dosw (Online). 2014;68:410–8.
Meier K, Brehm A. Chromatin regulation: how complex does it get. Epigenetics. 2014;9(11):1485–95.
Li GM, Zhang CL, Rui RP, Sun B, Guo W. Bioinformatics analysis of common differential genes of coronary artery disease and ischemic cardiomyopathy. Eur Rev Med Pharmacol Sci. 2018;22(11):3553–69.
Cappuzzello C, Napolitano M, Arcelli D, et al. Gene expression profiles in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genom. 2009;38(3):233–40.
Elashoff MR, Wingrove JA, Beineke P, et al. Development of a blood-based gene expression algorithm for assessment of obstructive coronary artery disease in non-diabetic patients. BMC Med Genom. 2011;4:26.
Beineke P, Fitch K, Tao H, et al. A whole blood gene expression-based signature for smoking status. BMC Med Genom. 2012;5:58.
Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–7.
O’onnor CM, Gattis WA, Shaw L, Cuffe MS, Califf RM. Clinical characteristics and long-term outcomes of patients with heart failure and preserved systolic function. Am J Cardiol. 2000;86(8):863–7.
Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24(11):1403–5.
Ito K, Murphy D. Application of ggplot2 to pharmacometric graphics. CPT Pharmacomet Syst Pharmacol. 2013;2(10):e79.
Khomtchouk BB, Van Booven DJ, Wahlestedt C. HeatmapGenerator: high performance RNAseq and microarray visualization software suite to examine differential gene expression levels using an R and C++ hybrid computational pipeline. Source Code Biol Med. 2014;9(1):30.
Bhattacharya S, Dunn P, Thomas CG, et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data. 2018;5:180015.
Harris MA, Clark J, Ireland A, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(Database issue):D258–61.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Gaudet P, Dessimoz C. Gene ontology: pitfalls, biases, and remedies. Methods Mol Biol. 2017;1446:189–205.
Bindea G, Galon J, Mlecnik B. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics. 2013;29(5):661–3.
Mlecnik B, Galon J, Bindea G. Comprehensive functional analysis of large lists of genes and proteins. J Proteom. 2018;171:2–10.
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Fang H, De Wolf H, Knezevic B, et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat Genet. 2019;51(7):1082–91.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7.
Svetnik V, Liaw A, Tong C, Culberson JC, Sheridan RP, Feuston BP. Random forest: a classification and regression tool for compound classification and QSAR modeling. J Chem Inf Comput Sci. 2003;43(6):1947–58.
Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77.
Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–8.
Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2.
Chen S, Yang D, Lei C, et al. Identification of crucial genes in abdominal aortic aneurysm by WGCNA. PeerJ. 2019;7:e7873.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
Zhou G, Soufan O, Ewald J, Hancock R, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47(W1):W234–41.
Ritchie W, Flamant S, Rasko JE. Predicting microRNA targets and functions: traps for the unwary. Nat Methods. 2009;6(6):397–8.
Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–52.
Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–31.
Han H, Shim H, Shin D, et al. TRRUST: a reference database of human transcriptional regulatory interactions. Sci Rep. 2015;5:11432.
Liu CJ, Fu X, Xia M, Zhang Q, Gu Z, Guo AY. miRNASNP-v3: a comprehensive database for SNPs and disease-related variations in miRNAs and miRNA targets. Nucleic Acids Res. 2021;49(D1):D1276–81.
Liao J, Wang J, Liu Y, Li J, Duan L. Transcriptome sequencing of lncRNA, miRNA, mRNA and interaction network constructing in coronary heart disease. BMC Med Genom. 2019;12(1):124.
Rada J, Donato M, Penas FN, et al. IL-10-dependent and -independent mechanisms are involved in the cardiac pathology modulation mediated by fenofibrate in an experimental model of Chagas heart disease. Front Immunol. 2020;11:572178.
Alessi MC, Cenac N, Si-Tahar M, Riteau B. FPR2: a novel promising target for the treatment of influenza. Front Microbiol. 2017;8:1719.
Chen K, Liu M, Liu Y, et al. Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J Clin Invest. 2013;123(4):1694–704.
Ruan Z, Wang S, Yu W, Deng F. LncRNA MALAT1 aggravates inflammation response through regulating PTGS2 by targeting miR-26b in myocardial ischemia-reperfusion injury. Int J Cardiol. 2019;288:122.
Saadi E, Sood R, Dromi I, et al. Limited proteolysis of cyclooxygenase-2 enhances cell proliferation. Int J Mol Sci. 2020;21(9):3195.
Tan C, Liu L, Liu X, et al. Activation of PTGS2/NF-κB signaling pathway enhances radiation resistance of glioma. Cancer Med. 2019;8(3):1175–85.
Egusquiaguirre SP, Yeh JE, Walker SR, Liu S, Frank DA. The STAT3 target gene TNFRSF1A modulates the NF-κB pathway in breast cancer cells. Neoplasia. 2018;20(5):489–98.
Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF receptor-associated periodic syndrome (TRAPS): current perspectives. Int J Mol Sci. 2020;21(9):3263.
Deng M, Dai W, Yu VZ, Tao L, Lung ML. Cylindromatosis lysine 63 deubiquitinase (CYLD) regulates NF-kB signaling pathway and modulates fibroblast and endothelial cells recruitment in nasopharyngeal carcinoma. Cancers (Basel). 2020;12(7):1924.
Mitchell S, Vargas J, Hoffmann A. Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med. 2016;8(3):227–41.
Holbrook J, Lara-Reyna S, Jarosz-Griffiths H, McDermott M. Tumour necrosis factor signalling in health and disease. F1000Research. 2019;8:111.
Ali Sheikh MS, Alduraywish A, Almaeen A, et al. Therapeutic value of miRNAs in coronary artery disease. Oxid Med Cell Longev. 2021;2021:8853748.
Tabuchi T, Satoh M, Itoh T, Nakamura M. MicroRNA-34a regulates the longevity-associated protein SIRT1 in coronary artery disease: effect of statins on SIRT1 and microRNA-34a expression. Clin Sci (Lond). 2012;123(3):161–71.
Wang S, He W, Wang C. MiR-23a regulates the vasculogenesis of coronary artery disease by targeting epidermal growth factor receptor. Cardiovasc Ther. 2016;34(4):199–208.
Xu Z, Han Y, Liu J, et al. MiR-135b-5p and MiR-499a-3p promote cell proliferation and migration in atherosclerosis by directly targeting MEF2C. Sci Rep. 2015;5:12276.
Wang J, Pei Y, Zhong Y, Jiang S, Shao J, Gong J. Altered serum microRNAs as novel diagnostic biomarkers for atypical coronary artery disease. PLoS ONE. 2014;9(9):e107012.
Rayner KJ, Esau CC, Hussain FN, et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478(7369):404–7.
Hou S, Fang M, Zhu Q, Liu Y, Liu L, Li X. MicroRNA-939 governs vascular integrity and angiogenesis through targeting γ-catenin in endothelial cells. Biochem Biophys Res Commun. 2017;484(1):27–33.
Zhu J, Chen T, Yang L, et al. Regulation of microRNA-155 in atherosclerotic inflammatory responses by targeting MAP3K10. PLoS ONE. 2012;7(11):e46551.
Zernecke A, Bidzhekov K, Noels H, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2(100):ra81.
Ji R, Cheng Y, Yue J, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100(11):1579–88.
Wang J, Yan Y, Song D, Liu B. Reduced plasma miR-146a Is a predictor of poor coronary collateral circulation in patients with coronary artery disease. Biomed Res Int. 2016;2016:4285942.
Zhu ZD, Ye JY, Niu H, et al. Effects of microRNA-292-5p on myocardial ischemia-reperfusion injury through the peroxisome proliferator-activated receptor-α/-γ signaling pathway. Gene Ther. 2018;25(3):234–48.
García-López J, del Mazo J. Expression dynamics of microRNA biogenesis during preimplantation mouse development. Biochim Biophys Acta. 2012;1819(8):847–54.
Yao Y, Zhang X, Chen HP, et al. MicroRNA-186 promotes macrophage lipid accumulation and secretion of pro-inflammatory cytokines by targeting cystathionine γ-lyase in THP-1 macrophages. Atherosclerosis. 2016;250:122–32.
Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10(6):389–402.
Gong J, Tong Y, Zhang HM, et al. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum Mutat. 2012;33(1):254–63.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Young Natural Science Foundation of Jilin Province of China (2016Q015).
Author information
Authors and Affiliations
Contributions
GW and LZ designed the research. LZ, GL, BL, and GW acquired and analyzed the data. XS, HX, HS, and GW interpreted the results. LZ and BL wrote the manuscript. GW revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was not conducted on human participants or animals by any of the authors. Our data were downloaded directly from public databases and we strictly abided by the publishing guidelines provided by GEO and ImmPort databases; there were no requirements for ethical approvals. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Li, G., Liang, B. et al. Integrative analyses of immune-related biomarkers and associated mechanisms in coronary heart disease. BMC Med Genomics 15, 219 (2022). https://doi.org/10.1186/s12920-022-01375-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01375-w