
Abstract
Background
Bladder cancer (BC) is the 10th most frequent tumor worldwide. Evidence shows an association between elevated risk of BC and various single nucleotide polymorphisms (SNP). BC incidence was the highest in Lebanon according to Globocan 2018 report, but little is known about the genetic susceptibility of Lebanese people to this disease. We aim to evaluate whether this prominent incidence of BC in Lebanon is attributable to known coding genetic variants.
Methods
A case-control study was conducted at Hotel-Dieu de France Hospital, Beirut. A cohort of 51 Lebanese patients with BC were recruited between 2017 and 2020. Whole Exome Sequencing (WES) was performed on peripheral blood samples to detect coding genetic variants in the patients. An in-house database including WES data from 472 Lebanese individuals served as control. Literature review of the genetic predisposition to BC was conducted to establish a database of variants known to influence the risk of BC. In-common SNPs were identified between cases and the aforecited database, and their allelic frequencies was quantified in the former and in controls. Comparative analysis of the allelic frequencies of each in-common SNP was carried out between cases, controls, and the genome aggregation database (gnomAD). Analysis was performed by applying the binomial law and setting the p-value to 10− 10.
Results
484 polymorphisms associated with BC were extracted from the literature review ;151 of which were in-common with the 206 939 variations detected by WES in our cases. Statistically significant differences (p-value < 10− 10) in allelic frequencies was seen in 11 of the 151 in-common SNPs, but none of which corresponds with a higher BC risk. Moreover, rs4986782 variant in the NAT1 gene is not associated with BC in the Lebanese population. `.
Conclusion
This is the first next-generation sequencing (NGS)- based study investigating BC risk in a Lebanese cohort of 51 patients. The majority of known exonic variants in the literature were not associated with BC in our patients. Further studies with larger sample sizes are warranted to explore the association of BC in our population with known non-coding genetic variants, and the remainder of WES-generated private Lebanese variants.
Similar content being viewed by others


Background
Bladder cancer (BC) is the most frequent malignant tumor of the urinary tract and the 10th most frequent cancer worldwide with an annual incidence surpassing half a million new cases in 2018 (424 000 in males and 125 000 in females) from all ages. In terms of mortality, it is ranked the 12th worldwide with 200,000 deaths each year [1]. According to the United Nations, the world population is increasing and is projected to exceed 9.7 billion people by 2050. Thus, the global incidence rate of BC is expected to increase, placing a heavy burden on the healthcare system [1].
Environmental exposures, notably smoking [2], and genetic risk factors impact the incidence of BC [3, 4]. Owing to the advances in genomic analysis, several single center [4, 5] and multi-center case-control studies [6, 7],genome-wide association studies(GWAS) [6, 8,9,10], systematic reviews, and meta-analyses with large pooled samples have shown an elevated risk of BC with different SNPs (Single Nucleotide Polymorphisms) across multiple genetic pathways [11, 12]. Researchers have also identified polymorphisms in both coding and non-coding regions that influence the risk of BC [13, 14]. Additionally, a large body of evidence suggests the presence of population-related genetic polymorphisms that alter the susceptibility to BC [5, 6, 15, 16] such as rs9642880 in individuals of European ancestry [6], and rs10936599 in the Turkish population [5]. Other investigators observed inter-population differences regarding the effect of SNPs on BC, whereby the same genetic variation enhances susceptibility of BC in one population, but mitigates the risk in another group of people [12].Song and companions concluded that Asians have an increased risk of BC with rs699947, whereas Africans exhibit reduced risk of BC with the aforementioned SNP [12].
According to the Globocan 2018 report, Lebanon has the highest global incidence of BC in females and the 2nd highest global incidence in males [17]. However, little is known about the genetic predisposition of the Lebanese people to BC. This underscores the need to investigate the influence of genetic risk factors of BC in a cohort of Lebanese patients.
Admittedly, this study aims to establish a comprehensive database of the variations associated with BC reported in the current literature (known variants) and to evaluate, by Whole Exome Sequencing (WES), whether the high BC incidence in a Lebanese cohort is attributable to a known coding genetic variant. To our knowledge, there are no previous investigations using next generation sequencing (NGS) technology to explore the genetic predisposition of Lebanese people to BC. We hypothesize that novel polymorphisms specific to the Lebanese population contribute to the highly reported incidence of BC in Lebanon.
Materials and methods
Literature search strategy
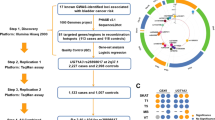
The literature search was conducted on PubMed using the following keywords “Bladder Cancer”, “Genetic predisposition», and “Genetic susceptibility” to collect the articles related to BC susceptibility and genetic association studies between January 2000 and October 2020. The last search was carried out on 10/10/2020 and generated 894 results. Initially, all articles exploring genetic risks for BC between January 2000 and October 2020 were included and all projects unrelated to this subject that appeared in the search results were eliminated after checking each paper manually. After reviewing each article, the papers that targeted hereditary BC, epigenetic, haplotype studies, or various cancer types without emphasizing BC were excluded. The selection process is illustrated in Fig. 1.

Flow Diagram detailing articles’ selection steps
*400 Articles unrelated to the genetic susceptibility of BC are eliminated
** 143 Articles excluded: Hereditary Bladder cancer studies, Epigenetic studies, Haplotype analysis
Lebanese cohort
The studied sample consists of 51 patients with BC from different Lebanese areas, who presented to Hotel-Dieu de France Hospital in Beirut, Lebanon, a tertiary university hospital, between January 2017 and June 2020.
All methods were carried out in accordance with the declaration of Helsinki and performed in accordance with relevant guidelines and regulations. The project was approved by the Ethics Committee of Saint Joseph University, Beirut, Lebanon (Approval number : Tfem-2020-85). All patients signed an informed consent for participation, sample collection, and data publication. Peripheral blood was then collected from each individual enrolled in this study and DNA was extracted using the salting-out method [18].
WES analysis
Exon capture and sequencing: The exome was captured using the SureSelect Human All Exons, reagents (Agilent Inc.® Santa Clara, CA) according to the manufacturer’s standard protocol. The concentration of each library was determined using Agilent’s QPCR NGS Library Quantification Kit (G4880A). Samples were pooled prior to sequencing with a final concentration of each sample equal to 10 nM. Sequencing was performed on the Illumina HiSeq2000 platform using TruSeq v3 chemistry.
Mapping and alignment: Reads files (FASTQ) were generated from the sequencing platform via the manufacturer’s proprietary software. Reads were aligned to the hg19/b37 reference genome using the Burrows-Wheeler Aligner (BWA) package v0.6.1 [19]. Local realignment of the mapped reads around potential insertion/deletion (Indel) sites was carried out with the Genome Analysis Tool Kit (GATK) v1.6 [20]. Duplicate reads were marked using Picard v1.62. Additional BAM file manipulations were performed with Samtools 0.1.18 [21]. Base quality (Phred scale) scores were recalibrated using GATK’s covariance recalibration. SNP and Indel variants are called using the GATK Unified Genotyper for each sample [22]. Variants were called using high stringency settings and annotated with VarAFT software 2.131 [23] containing information from dbSNP147 and GnomAD [https://gnomad.broadinstitute.org]. Finally, bioinformatics analysis of the data was carried out and focused on all the variations detected in the cohort of 51 Lebanese patients that were in common with those included in the list established by our literature review.
Statistical analysis
The allelic frequencies of the variations were calculated in our cohort of 51 Lebanese patients with BC. The frequencies of these variations in the local Lebanese database (sample control of 472 Lebanese individuals) and the international population database (according to the GnomAD database) have been added to Additional file 1. Comparative analysis of the three frequencies for each selected SNP was carried out by applying the binomial law and setting the p-value to 10− 10.
Results
Literature review and association studies
The results of the literature study were grouped into one table (More explicit details can be found in Additional file 2) that includes all the cited polymorphisms: those that are associated with an increased risk for BC, those that present a protective effect, and finally the SNPs that have conflicting associations with BC.
Our search strategy generated 894 records from which 351 articles as shown in Fig. 1. Overall, 484 variants in 269 genes were considered as presenting a positive association with BC. A detailed list of all selected variants with their corresponding references is provided in Additional file 2. Table 1 includes a summary of the types and locations of the selected 484 variations.
Characteristics of the included clinical samples
The studied population consists of 51 Lebanese patients including 9 females and 42 males, from various and different areas across Lebanon. The mean age at BC diagnosis is 67 years in both genders with 68.5 in males and 59.5 in females.
Polymorphisms detected in our cohort of 51 lebanese patients
WES performed in the 51 patients with BC generated a total of 206 939 variations. Among the variants detected in our cohort, a total of 151 were found to be present in the list of variants known to be associated with BC. Those were designated as “in common variations” (More details can be found in Additional file 1). The frequency of each selected SNP was evaluated in our cohort and compared to that in our in-house Lebanese database of 472 controls as well as in the Genome Aggregation Database (GnomAD).
These variations are briefly summarized in Table 2.
In the literature study, 167 (34.5%) variations were intronic, whereas our study enabled the detection of only 7 intronic variations, representing 4.6% of the 151 selected “in common SNPs”. As for the exonic variants, 134 exonic variations were previously reported to be associated with BC risk; of these, 116 variations were detected by WES in our study. Therefore, 86.6% of these exonic variations were covered by WES in our sample.
A thorough analysis of the 151 in common SNPs led to the selection of 11 candidate SNPs based on the difference between their frequencies in the studied Lebanese cohort (A) compared to the local Lebanese database (B) and/or to the GnomAD database (C) (Table 3). Of these, 5 are pyrimidine to pyrimidine changes. Those 11 SNP were grouped into 3 different categories:
-
Group 1: 2 SNPs (rs1707 and rs1063320) showed a statistically significant difference between the cohort of 51 patients (A) and the sample of 472 controls (B) on one hand, and between (A) and the global database / gnomAD (C) on the other hand.
-
Group 2: 6 SNPs (rs9299, rs696, rs1061217, rs1799794, rs1799796, rs1805335) showed only a statistically significant difference between the cohort of 51 patients (A) and the global database / gnomAD (C) but did not show any significant difference with the local database.
-
Group 3: 3 SNPs (rs1710, rs3213239, rs1801320) showed only a statistically significant difference between the cohort of 51 patients (A) and the sample of 472 Lebanese controls (B) but did not show significant differences with the international database.
Evaluation of the candidate SNPs in the NAT1 gene
We compared our results with those obtained by association studies in the Lebanese population published in 2012 and 2013 [28]-[29] which evaluated the following NAT1 variants: rs15561, rs1057126, and rs4986782. Our analysis focused on one variant, the exonic SNP “rs4986782” in NAT1, that is covered by the technique used in our study which is WES. Our findings show a lack of association between the rs4986782 and BC in our Lebanese cohort.
Discussion
BC is the most common malignant tumor of the urinary system. It can be present as an isolated form or as a part of a cancer syndrome. Gene sequencing and GWAS have identified in recent decades several new candidate genes and regions that might modulate BC risk. However, the contribution of genetic factors remains unclear in lots of cases, thus complicating the approach of genetic counseling in these patients and rendering our understanding of the molecular mechanisms driving BC limited to date.
In this project, we focused, initially, on reviewing the literature related to the genetic variants associated with BC. This allowed us to establish a list of 484 polymorphisms located in 269 genes, known to be variants associated with BC. By gathering and summarizing this information, our work provides a comprehensive review related to the genetic predisposition to BC. Our review also highlighted the differences in the association of specific SNPs and BC between populations. It is indeed noted that the same SNP is associated with an increased risk of BC in one population while it is protective or not associated with BC in another population. For example, the TP53 gene’s variant rs1042522 is not associated with BC in the Spanish population [26] while it increases the BC risk in the Bangladeshi population [27] and it decreases this risk in the Brazilian population [28]. The APOBEC3A gene’s variant rs1014971 is associated with an increased risk of BC in the Japanese [29] and Swedish populations [30] but it is not associated with BC in the Taiwanese population [31]. The MMP1 gene’s variant rs1799750 also increases the risk of BC in the Turkish [32] and North Indian populations [33] while it decreases it in the Polish population [34]. Those SNPs were considered as variants with conflicting interpretations.
We then conducted a next-generation sequencing (NGS)-based study on a cohort of 51 Lebanese patients affected with BC. This study allowed us to detect 206 939 SNPs in the studied population. Those were then filtered to select the variants that are included in the list of 484 SNPs known to be associated with BC; in total 151 SNPs were selected in our cohort of 51 Lebanese patients with BC. Subsequently, a thorough analysis was performed to determine candidate SNPs based on the difference in their frequencies between the studied cohort, the Lebanese population database, and the global international database. This subsequent analysis allowed us to narrow the list to 11 candidate SNPs with a possible association with BC.
The selected variants were then classified into three different groups: The first (group 1) includes 2 SNPs in the HLA-G gene (rs1707 and rs1063320) that showed a statistically significant difference between the cohort of 51 patients (A) and the sample of 472 controls (B) on one hand, and between (A) and the global database / gnomAD (C) on the other hand. However, while their frequency in our cohort was higher than that in the Lebanese database, it was lower than that in the international database, which suggests that their association with BC is less likely to be possible. The second group (group 2) includes 6 SNPs (rs9299, rs696, rs1061217, rs1799794, rs1799796, rs1805335) that showed only a statistically significant difference between the cohort of 51 patients (A) and the global database / gnomAD (C) but did not show any significant difference with the local database. Those are likely to be either polymorphisms that are “private” to the Lebanese population in general or variants that are rare in our population. Interestingly, among those 6 SNPs, two SNPs, the rs1061217 in SLAMF1 and rs1805335 in RAD23B showed a high frequency in our cohort compared to the international database, and the Lebanese local database. However, this difference is statistically significant between our cohort and the international database, and not between the cohort and the Lebanese database. This suggests one of the following scenarios: either the cohort size is small and statistical significance between our cohort and the local database will be obtained with bigger sample size or those two variants are « private to the Lebanese population » without being associated with BC.
Last but not least, group 3 consists of 3 SNPs (rs1710, rs3213239, rs1801320) that showed only a statistically significant difference between the cohort of 51 patients (A) and the sample of 472 Lebanese controls (B) but showed a comparable frequency with that reported in international databases (as differences were not statistically significant), thus indicating they are less likely to be contributing to the disease.
Furthermore, our data was then evaluated based on association studies previously performed in the Lebanese population and published in 2012 and 2013 [28]-[29], where three variations in the NAT1 gene, rs4986782, rs1057126, and rs15561 were reported to be associated with an increased risk for BC in the Lebanese population. Our study that is based on WES analysis thus focusing only on the coding SNP, the rs4986782, showed a lack of association between the latter and BC in the Lebanese population. The remaining variants located at the 3’UTR region of NAT1 could not be evaluated by WES. On the other hand, the previous two studies compared the frequency of SNPs to a total of 105 controls individuals, while in our study, the SNPs were compared to a control sample of 472 individuals, considered to be more representative of the Lebanese population. Therefore, our results suggest that NAT1 (rs4986782) is not associated with BC risk in the Lebanese population.
Altogether, the absence of relevant variants that show a statistically significant difference between our cohort, the Lebanese local database, and gnomAD can be due to the possibility that the variants responsible for the high frequency of BC in the Lebanese population were not covered by this study because our approach included only exonic and known SNPs. Another hypothesis would be that the limited size of our cohort did not enable us to reach a statistically significant difference between the frequency of relevant SNPs in our cohort compared to the in-house Lebanese database and the international database, as was mentioned in the case of the two SNPs the rs1061217 in SLAMF1 and rs1805335 in RAD23B. Further studies are thus needed to complement our findings.
Study limitations
WES technique has different limitations; it aims to detect variations mainly in exons and exon/intron junctions to study the acceptor and donor sites of canonical splicing but excludes introns and much of the non-coding 5’ and 3’ UTR regions. This suggests that our findings focused on exonic variants might have missed some intronic variants relevant to BC risk in the Lebanese population.
In conclusion, this project represents the first investigation by NGS of 51 Lebanese patients with BC. In contrast to the literature review, the majority of known exonic variants (86.6%) were not found to be associated with BC in the Lebanese population. Finally, despite the challenging interpretation of WES data, we aim to re-analyze all the obtained data related to the 51 patients with BC to investigate the possible presence of an association between private “Lebanese variants” and BC risk in this population and determine if novel « non-previously reported SNPs » increase BC in the Lebanese population.
On the other hand, one should not ignore the contribution of environmental factors to this type of cancer. Indeed, several studies have described the influence of certain environmental factors on genetic variants and the subsequent epigenetic modification of the risk of BC [35–38]. Many other studies have also shown the effect of intergenic interaction (regions regulatory genes, intergenic sequences, and introns) on BC risk [39, 40]. Therefore, further studies using Whole Genome Sequencing (WGS) or epigenome studies could be needed to explore these factors in all populations in general.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BC:
-
Bladder Cancer
- GWAS:
-
Genome-Wide Association Study
- SNP:
-
Single nucleotide polymorphism
- BWA:
-
Burrows-Wheeler Aligner
- GATK:
-
Genome Analysis Tool Kit
- UTR:
-
Untranslated Regions
- GnomAD:
-
Genome Aggregation Database
- NGS:
-
Next-generation sequencing
- WES:
-
Whole Exome Sequencing
- WGS:
-
Whole Genome Sequencing
References
Richters A, Aben KKH, Kiemeney LALM. The global burden of urinary bladder cancer: an update. World J Urol. 2020 Aug 1;38(8):1895–904.
Freedman ND, Silverman DT, Hollenbeck AR, Schatzkin A, Abnet CC. Association between smoking and risk of bladder cancer among men and women. JAMA. 2011 Aug;17(7):737–45. 306(.
Jung I, Messing E. Molecular mechanisms and pathways in bladder cancer development and progression. Cancer Control. 2000 Aug;7(4):325–34.
Martin C, Leiser CL, O’Neil B, Gupta S, Lowrance WT, Kohlmann W, et al. Familial Cancer Clustering in Urothelial Cancer: A Population-Based Case-Control Study. J Natl Cancer Inst. 2018 01;110(5):527–33.
Polat F, Yilmaz M, Diler SB. The Association of MYNN and TERC Gene Polymorphisms and Bladder Cancer in a Turkish Population. Urol J. 2019 Feb;21(1):50–5. 16(.
Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KKH, Stacey SN, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet. 2008 Nov;40(11):1307–12.
García-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, et al. NAT2 slow acetylation and GSTM1 null genotypes increase bladder cancer risk: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005 Aug;20(9486):649–59. 366(.
Kiemeney LA, Sulem P, Besenbacher S, Vermeulen SH, Sigurdsson A, Thorleifsson G, et al. A sequence variant at 4p16.3 confers susceptibility to urinary bladder cancer. Nat Genet. 2010 May;42(5):415–9.
Rothman N, Garcia-Closas M, Chatterjee N, Malats N, Wu X, Figueroa J, et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat Genet. 2010 Nov;42(11):978–84.
Wu X, Ye Y, Kiemeney LA, Sulem P, Rafnar T, Matullo G, et al. Genetic variation in the prostate stem cell antigen gene PSCA confers susceptibility to urinary bladder cancer. Nat Genet. 2009 Sep;41(9):991–5.
de Maturana EL, Rava M, Anumudu C, Sáez O, Alonso D, Malats N. Bladder Cancer Genetic Susceptibility. A Systematic Review. Bladder Cancer. 4(2):215–26.
Song Y, Yang Y, Liu L, Liu X. Association between five polymorphisms in vascular endothelial growth factor gene and urinary bladder cancer risk: A systematic review and meta-analysis involving 6671 subjects. Gene. 2019 May;25:698:186–97.
Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013 Dec;45(12):1459–63.
Gulìa C, Baldassarra S, Signore F, Rigon G, Pizzuti V, Gaffi M, et al. Role of Non-Coding RNAs in the Etiology of Bladder Cancer. Genes (Basel). 2017 Nov 22;8(11):339.
Zhang Y, Sun Y, Chen T, Hu H, Xie W, Qiao Z, et al. Genetic variations rs11892031 and rs401681 are associated with bladder cancer risk in a Chinese population. Int J Mol Sci. 2014 Oct 24;15(11):19330–41.
Hadami K, Ameziane El Hassani R, Ameur A, Dakka N, Abbar M, Al Bouzidi A, et al. Association between GPX1 Pro189Leu polymorphism and the occurrence of bladder cancer in Morocco. Cell Mol Biol (Noisy-le-grand). 2016 Dec 30;62(14):38–43.
Global cancer statistics 2018. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries - Bray – 2018 - CA: A Cancer Journal for Clinicians - Wiley Online Library [Internet]. [cited 2021 Jan 18]. Available from: https://acsjournals.onlinelibrary.wiley.com/doi/https://doi.org/10.3322/caac.21492.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988 Feb 11;16(3):1215.
Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009 Nov 1;25(21):2744–50.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010 Sep;20(9):1297–303.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009 Jul 15;25(14):1754–60.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011 May;43(5):491–8.
Desvignes J-P, et al., “VarAFT: a variant annotation and filtration system for human next generation sequencing data,” Nucleic Acids Res, 46, no. W1, W545–53, Jul. 2018, doi:https://doi.org/10.1093/nar/gky471.
Yassine IA, Kobeissi L, Jabbour ME, Dhaini HR. N-acetyltransferase 1 (NAT1) genotype: a risk factor for urinary bladder cancer in a Lebanese population. J Oncol. 2012;2012:512976.
Basma HA, Kobeissi LH, Jabbour ME, Moussa MA, Dhaini HR. CYP2E1 and NQO1 genotypes and bladder cancer risk in a Lebanese population. Int J Mol Epidemiol Genet. 2013;4(4):207–17.
Pineda S, et al. Genetic variation in the TP53 pathway and bladder cancer risk. a comprehensive analysis. PLoS ONE. 2014;9(5):e89952. doi:https://doi.org/10.1371/journal.pone.0089952. “,”, , .
Hosen MB, Salam MA, Islam MF, Hossain A, Hawlader MZH, Kabir Y, “Association of TP53 gene polymorphisms with susceptibility of bladder cancer in Bangladeshi population,” Tumour Biol, vol. 36, no. 8, pp. 6369–6374, Aug. 2015, doi: https://doi.org/10.1007/s13277-015-3324-3.
LEM de C Santos, Guilhen ACT, de Andrade RA, Sumi LG, Ward LS, “The role of TP53 PRO47SER and ARG72PRO single nucleotide polymorphisms in the susceptibility to bladder cancer,” Urol Oncol, vol. 29, no. 3, pp. 291–294, Jun. 2011, doi: https://doi.org/10.1016/j.urolonc.2009.03.026.
Matsuda K, Takahashi A, Middlebrooks CD, Obara W, Nasu Y, Inoue K, et al. Genome-wide association study identified SNP on 15q24 associated with bladder cancer risk in Japanese population. Hum Mol Genet. 2015 Feb 15;24(4):1177–84.
Hemminki K, Bermejo JL, Ji J, Kumar R. Familial bladder cancer and the related genes. Curr Opin Urol. 2011 Sep;21(5):386–92.
Chen M, Tsai YT, Chang WS, Shih LC, Shen TC, Lin ML, et al. Association of Caspase-8 Genotypes With Bladder Cancer Risk. Anticancer Res. 2019 Sep;39(9):4767–73.
Tasci AI, Tugcu V, Ozbek E, Ozbay B, Simsek A, Koksal V. A single-nucleotide polymorphism in the matrix metalloproteinase-1 promoter enhances bladder cancer susceptibility. BJU Int. 2008 Feb;101(4):503–7.
Srivastava P, Gangwar R, Kapoor R, Mittal RD. Bladder cancer risk associated with genotypic polymorphism of the matrix metalloproteinase-1 and 7 in North Indian population. Dis Markers. 2010;29(1):37–46.
Wieczorek E, Reszka E, Jablonowski Z, Jablonska E, Krol MB, Grzegorczyk A, et al. Genetic polymorphisms in matrix metalloproteinases (MMPs) and tissue inhibitors of MPs (TIMPs), and bladder cancer susceptibility. BJU Int. 2013 Dec;112(8):1207–14.
Mao F, Niu XB, Gu S, Ji L, Wei BJ, Wang HB. The association between matrix metalloproteinase-7 genetic variant and bladder cancer risk in a Chinese Han population. Clin Exp Med. 2019 Nov;19(4):565–70.
Cai DW, Liu XF, Bu RG, Chen XN, Ning L, Cheng Y, et al. Genetic polymorphisms of MTHFR and aberrant promoter hypermethylation of the RASSF1A gene in bladder cancer risk in a Chinese population. J Int Med Res. 2009 Dec;37(6):1882–9.
Masaoka H, Ito H, Soga N, Hosono S, Oze I, Watanabe M, et al. Aldehyde dehydrogenase 2 (ALDH2) and alcohol dehydrogenase 1B (ADH1B) polymorphisms exacerbate bladder cancer risk associated with alcohol drinking: gene-environment interaction. Carcinogenesis. 2016 Jun;37(6):583–8.
Wang M, Zhang Z, Tian Y, Shao J, Zhang Z. A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter associated with risk and progression of bladder cancer. Clin Cancer Res. 2009 Apr 1;15(7):2567–72.
Wieczorek E, Reszka E, Jablonowski Z, Jablonska E, Krol MB, Grzegorczyk A, et al. Genetic polymorphisms in matrix metalloproteinases (MMPs) and tissue inhibitors of MPs (TIMPs), and bladder cancer susceptibility. BJU Int. 2013 Dec;112(8):1207–14.
Elhawary NA, Nassir A, Saada H, Dannoun A, Qoqandi O, Alsharif A, et al. Combined Genetic Biomarkers Confer Susceptibility to Risk of Urothelial Bladder Carcinoma in a Saudi Population. Dis Markers. 2017;2017:1474560.
Acknowledgements
We express our deepest gratitude and acknowledgment to the patients who accepted to be enrolled in our study and to all individuals who contributed directly or indirectly to this study.
Funding
This work (samples collection, analysis, and interpretation of data) was supported by grants from Saint Joseph University, Beirut, Lebanon.
Author information
Authors and Affiliations
Contributions
HK, BS, CM, NJ, and EC wrote the manuscript.
HK, BS, AM, JZ, NA, NJ, EC, and CM performed data analysis.
HK, JK, EN, NJ, EC, and CM conceived and designed the study.
GS performed the statistical analysis.
NJ conducted the bioinformatics analysis of the raw data.
HK, EN, and JK performed the clinical investigation and the referral of the patients.
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent
All methods were carried out in accordance with relevant guidelines/regulations, and in accordance with the declaration of Helsinki, and approved by the Ethics Committee of Saint Joseph University, Beirut, Lebanon (Approval number: Tfem-2020-85). Signed informed consent to participate in the study was obtained from all patients. Patients’ names were assigned specific codes that were only recognized by the researchers involved in this study, to ensure the anonymity and the confidentiality of the data.
Consent to publish
Not Applicable.
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kourie, H.R., Succar, B., Chouery, E. et al. Genetic susceptibility of bladder cancer in the Lebanese population. BMC Med Genomics 15, 217 (2022). https://doi.org/10.1186/s12920-022-01372-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01372-z